

Abstract
We have used site-directed mutagenesis, EPR spectroscopy, redox potentiometry, and protein crystallography to monitor assembly of the FS0 [4Fe-4S] cluster and molybdo-bis(pyranopterin guanine dinucleotide) cofactor (Mo-bisPGD) of the Escherichia coli nitrate reductase A (NarGHI) catalytic subunit (NarG). Cys and Ser mutants of NarG-His49 both lack catalytic activity, with only the former assembling FS0 and Mo-bisPGD. Importantly, both prosthetic groups are absent in the NarG-H49S mutant. EPR spectroscopy of the Cys mutant reveals that the Em value of the FS0 cluster is decreased by at least 500 mV, preventing its participation in electron transfer to the Mo-bisPGD cofactor. To demonstrate that decreasing the FS0 cluster Em results in decreased enzyme activity, we mutated a critical Arg residue (NarG-Arg94) in the vicinity of FS0 to a Ser residue. In this case, the Em of FS0 is decreased by 115 mV, with a concomitant decrease in enzyme turnover to ∼30% of the wild type. Analysis of the structure of the NarG-H49S mutant reveals two important aspects of NarGHI maturation: (i) apomolybdo-NarGHI is able to bind GDP moieties at their respective P and Q sites in the absence of the Mo-bisPGD cofactor, and (ii) a critical segment of residues in NarG, 49HGVNCTG55, must be correctly positioned to ensure holoenzyme maturation.
Keywords: Bioenergetics/Electron Transport, Bioenergetics/Oxidation/Reduction, Bioenergetics/Respiratory Chain, Electron Transfer, Membrane/Enzymes, Metals/Molybdenum, Methods/Electron Paramagnetic Resonance (EPR)
Introduction
Escherichia coli, when grown anaerobically with nitrate as respiratory oxidant, develops a respiratory chain terminated by a membrane-bound quinol:nitrate oxidoreductase (NarGHI) (1–3). This enzyme is an archetype of the complex iron-sulfur molybdoenzyme (CISM)2 family (4) that includes E. coli formate dehydrogenase N (FdnGHI (5)), E. coli Me2SO reductase (DmsABC (6)), Wolinella succinogenes polysulfide reductase (PsrABC (7, 8)), Salmonella typhymurium thiosulfate reductase (PhsABC (9)), and Salmonella enterica tetrathionate reductase (TtrABC (10)). These enzymes and their close relatives contribute significantly to the remarkable metabolic diversity of bacteria.
NarGHI and the other CISM archetypes comprise a mononuclear molybdenum cofactor (molybdo-bis(pyranopterin guanine dinucleotide) (Mo-bisPGD))-containing catalytic subunit, e.g. NarG; a four-cluster protein subunit, e.g. NarH; and a membrane anchor protein, e.g. NarI. Each catalytic subunit contains a [4Fe-4S] cluster in addition to Mo-bisPGD, and many of the membrane anchor subunits are also diheme cytochromes b. The five [Fe-S] clusters are referred to as FS0–FS4, with increasing distance from Mo-bisPGD, and the entire electron transfer relay (ETR) in NarGHI, including the two hemes, spans a distance of almost 100 Å (2).
In NarGHI, Mo-bisPGD provides the site of nitrate reduction at its redox-active molybdenum atom. The molybdenum atom is coordinated by two pyranopterin guanine dinucleotide (PGD) moieties via a bis-dithiolene linkage (11–13). These PGD groups are referred to as the P- and Q-pterins, and they are proximal and distal, respectively, to the FS0 [4Fe-4S] cluster. The bis-dithiolene coordination is supplemented by one oxo group and two oxygens of NarG-Asp222 (2). The arrangement of the P-pterin and the FS0 cluster may facilitate electron entry to the molybdenum active site via the unsaturated ring system of the P-pterin. The Q-pterin of NarG and that of the catalytic subunit (EbdA) of Aromatoleum aromaticum ethylbenzene dehydrogenase (EbdABD) (14) both have a bicyclic molybdopterin structure rather than the tricyclic pyranopterin structure found in other mononuclear molybdoenzymes studied to date (15–17).
In NarG, the molybdenum atom has midpoint potentials (Em) of ∼100 and 200 mV for the Mo(IV/V) and Mo(V/VI) couples, respectively (18, 19). The nearby FS0 cluster has an Em of approximately −55 mV (18). During catalysis, electrons flow from FS1 with an Em value of +130 mV to FS0, with this step presenting a barrier of ∼17.9 kJ mol−1. Thus, the FS0 Em value may play a critical role in regulating electron flow to the Mo-bisPGD cofactor.
FS0 plays a critical role in CISM enzymes because it is in direct redox equilibrium with Mo-bisPGD, either being the first [Fe-S] cluster in the ETR of the dehydrogenase enzymes (e.g. FdnGHI) or the final [Fe-S] cluster in the ETR of the reductase enzymes (e.g. NarGHI and DmsABC). FS0 is coordinated by a ferredoxin-like Cys group with the consensus sequence (CA/HA)x2–3CBx3CCx27–34CDx(K/R) (20). Enzymes with two residues between the first two Cys residues (CA and CB) and a Lys following the fourth Cys residue are referred to as Type I enzymes and include CISM archetypes such as FdnGHI (5) and PsrABC (8), as well as enzymes that contain a catalytic subunit but that are not archetypes of the CISM family (4). These include the FdhF subunit of the E. coli formate:hydrogen lyase complex (21) and the NapA subunit of the periplasmic nitrate reductase (NapAB) (22, 23). NarG and DmsA are examples of enzymes that have a CA-CB spacing of three residues and an Arg following CD, and NarG has a His residue at the CA position. These are referred to as Type II enzymes.
The role of FS0 in NarGHI maturation has yet to be clearly established. In a ΔmobAB strain that is unable to add GMP to the molybdenum-pyranopterin precursor (24), NarGHI lacks Mo-bisPGD but retains FS0–FS4 and the two hemes. NarGHI is encoded by the narGHJI operon (25, 26), of which only the NarG, NarH, and NarI proteins appear in the functional holoenzyme. The fourth gene product, NarJ, is a system-specific chaperone that has been suggested to hold NarGH in a cofactor-binding competent conformation (27–29). FS0 does not appear to assemble in the absence of NarJ. Li and Turner (30) have recently demonstrated an interaction between NarJ and a peptide comprising the first 50 amino acid residues of NarG, which has significant sequence similarity to a range of twin-arginine translocation (tat) pathway leader peptides (29) but lacks the critical pair of Arg residues essential for translocation to the periplasmic compartment via the tat export pathway (31–34).
In this work, we have investigated the role of the NarG FS0 cluster in NarGHI maturation using a combination of site-directed mutagenesis, EPR spectroscopy, and protein crystallography. Our results demonstrate that FS0 assembly is an important prerequisite for Mo-bisPGD cofactor insertion during holoenzyme maturation.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Mutagenesis
E. coli LCB79 (araD139 Δ(lacIPOZYA-argF) rpsL, thi ϕ79(nar-lac)) (35) was used as the host for all experiments described herein. NarGHI was expressed from plasmid pVA700 (36). NarG-H49S, NarG-H49C, and NarG-R94S mutants were prepared as previously described (19).
Growth of Cells
E. coli LCB79/pVA700 was grown overnight in 5-liter batches with 1% inoculum in a B. Braun Biostat B fermenter at 30 °C in the presence of 100 μg ml−1 ampicillin and 100 μg ml−1 streptomycin. The growth medium contained 12 g liter−1 Tryptone, 24 g liter−1 yeast extract, 5 g liter−1 NaCl, 4 ml liter−1 glycerol, and 0.1 mm ammonium molybdate. NarGHI overexpression was induced at A600 = 2.0 by the addition of 0.2 mm isopropyl-1-thio-β-d-galactopyranoside, after which the cultures were grown for an additional 10–11 h. Cells were harvested by centrifugation, washed in a buffer containing 100 mm MOPS and 5 mm EDTA (pH 7.0), frozen in liquid nitrogen, and stored at −70 °C prior to use.
Isolation of Membrane Fractions and Purified Preparations
Crude membrane vesicles were prepared from E. coli cells by French pressure cell lysis and differential centrifugation (37). Enriched inner membrane vesicles were prepared from these crude membranes by sucrose step centrifugation as previously described (38). All membrane preparation steps were carried out in a buffer containing 100 mm MOPS and 5 mm EDTA (pH 7.0). Excess sucrose was removed by resuspension and recentrifugation in this buffer. For EPR experiments, membranes were resuspended and recentrifuged in a buffer containing 100 mm Tricine and 5 mm EDTA (pH 8.0). Membrane samples were flash-frozen in liquid nitrogen and subsequently stored at −70 °C prior to use.
Purification, Crystallization, Data Collection, and Structure Determination of NarGHI Mutants
Purified NarG-H49S, NarG-H49C, and NarG-R94S mutant enzymes were prepared by anion exchange chromatography and subsequently crystallized as previously described (2, 18, 39). The crystals obtained were isomorphous with the native enzyme (C2221, a = 154.00 Å, b = 241.28 Å, c = 140.39 Å). Data were integrated and scaled with the HKL suite of programs (40). The structure was determined using difference Fourier techniques and was refined following rigid body refinement of the native 1.9-Å resolution model (2) with the CNS program (41). Additional cycles of manual rebuilding with Xfit (42) and refinement with CNS resulted in final models for the mutant forms of NarGHI with refinement parameters as described in Table 1.
TABLE 1.
Data collection and refinement statistics for the structures of NarGH49CHI, NarGH49SHI, and NarGR94SHI
| Mutant | NarGH49CHI | NarGH49SHI | NarGR94SHI |
|---|---|---|---|
| PDB code | 3IR5 | 3IR6 | 3IR7 |
| Data collection | |||
| Resolution (Å) | 30.0-2.3 | 30.0-2.8 | 25.0-2.5 |
| Unique reflections | 114,119 | 63,564 | 89,737 |
| Completeness (%)a | 99.6 (98.6) | 99.8 (96.7) | 99.8 (99.8) |
| Rmerge (%)a,b | 10.1 (44.0) | 10.5 (46.0) | 8.7 (40.3) |
| I/σ | 14.7 | 14.5 | 13.3 |
| Refinement | |||
| Rwork (%)c | 20.0 | 18.8 | 20.2 |
| Rfree (%)d | 23.7 | 24.9 | 24.3 |
| Average B factor (Å2) | 33.6 | 40.6 | 36.7 |
| r.m.s.d. bond lengthe (Å) | 0.007 | 0.008 | 0.007 |
| r.m.s.d. bond angles (°) | 1.5 | 1.4 | 1.4 |
a Values in parentheses are for the highest-resolution shell.
b Rmerge = S|(Ihkl)-<I>|/S(Ihkl), where Ihkl is the integrated intensity of a given reflection.
c Rwork = (S|Fo−Fc|)/(SFo), where Fo and Fc are observed and calculated structure factors.
d 6% of reflections were excluded from the refinement to calculate Rfree.
e r.m.s.d., root mean square deviation.
Redox Potentiometry and EPR Spectroscopy
Redox titrations were carried out under argon at 25 °C as previously described (38, 43) in 100 mm Tricine and 5 mm EDTA (pH 8.0). The protein concentration used was ∼30 mg ml−1. The following redox mediators were used at a concentration of 50 μm: quinhydrone, 2,6-dichloroindophenol, 1,2-naphthoquinone, toluylene blue, phenazine methosulfate, thionine, duroquinone, methylene blue, resorufin, indigo trisulfonate, indigo disulfonate, anthraquinone-2-sulfonic acid, phenosafranine, benzyl viologen, and methyl viologen. All samples were prepared in 3-mm internal diameter quartz EPR tubes, rapidly frozen in liquid nitrogen-chilled ethanol, and stored under liquid nitrogen until used. EPR spectra were recorded using a Bruker Elexys spectrometer equipped with a Bruker SHQE cavity and an Oxford Instruments ESR-900 flowing helium cryostat. Spectra were recorded under conditions of temperature and microwave power as described in the individual figure legends. Estimates of Em were obtained from n = 1 fits of potentiometric spectral data to the Nernst equation. For studies of the Mo(V) signal of NarGHI, a Bruker ESP300E spectrometer equipped with a TE102 microwave cavity and a Bruker liquid nitrogen-evaporating cryostat were used (Bruker ER4111 VT variable temperature unit). In this case, spectra were recorded at 150 K.
Protein Assays
Protein concentrations were assayed by the Lowry method (44), modified by the inclusion of 1% (w/v) sodium dodecyl sulfate in the incubation mixture to solubilize membrane proteins (45).
Enzyme Assays
Quinol:nitrate oxidoreductase activity assays were carried out using reduced lapachol (LPCH2) as quinol analog substrate (2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinol) (46). Assays were carried out in a degassed MOPS/EDTA buffer containing 4 mm KNO3 and 300 μm quinol. Enzyme activities were measured using enriched membranes prepared as described above. Activities were also measured using reduced benzyl viologen as substrate (47, 48).
RESULTS AND DISCUSSION
NarG-His49 and NarG-Arg94 Are Critical for Enzyme Activity
Fig. 1 illustrates the coordination environment of the FS0 [4Fe-4S] cluster in wild-type NarGHI. The cluster is coordinated by NarG-His49, NarG-Cys53, NarG-Cys57, and NarG-Cys92. NarG-Arg94 is sandwiched between FS0 and the proximal pterin of the Mo-bisPGD cofactor. Membranes enriched in NarGHI, NarG-H49CHI, NarG-H49SHI, and NarG-R94SHI have nitrate reductase-specific activities of 4.2, 0.1, 0.0, and 1.4, respectively (in units of μmol of lapachol oxidized per min (mg of protein)−1). The elimination of activity in the NarG-His49 mutants indicates that this residue plays a critical role in either holoenzyme maturation or catalytic turnover. We also used reduced benzyl viologen as electron donor in nitrate reductase assays (47, 48), which donates electrons nonspecifically to NarGHI (49). Membranes enriched in NarGHI, NarG-H49CHI, NarG-H49SHI, and NarG-R94SGH exhibit nitrate reductase activities with benzyl viologen of 45, 0.4, 0.1, and 37, respectively (in units of μmol of benzyl viologen oxidized per min (mg of protein)−1). Both assays indicate loss of activity in the NarG-His49 mutants, but the NarG-R94S mutant retains almost wild-type levels of activity in the benzyl viologen assay and only ∼30% of wild-type activity in the lapachol assay. The distribution of activities in the mutant mirrors that found in a mutant of the equivalent residue in E. coli DmsA (DmsA-R61S) (50). In this case, quinol:Me2SO oxidoreductase activity is completely eliminated, but benzyl viologen:Me2SO activity is retained.
FIGURE 1.

Structure and potential electron transfer routes between FS0 and the Mo-bisPGD cofactor of NarGHI. Two views of the FS0- and Mo-bisPGD-coordinating region of NarG are shown, with a rotation on the vertical axis of ∼180° between the two panels (A and B). Shown in the figure is the position of the two residues mutated in this work (NarG-His49 and NarG-Arg94), as well as putative electron transfer pathways between FS0 and the proximal pyranopterin (PPT-P). Distances within these putative pathways are indicated in the figure. In this figure, iron, sulfur, molybdenum, oxygen, carbon, and phosphorus atoms are rendered in red, yellow, blue, red, green, and orange, respectively.
FS0 Is Absent in the NarG-H49S Mutant
Previous work on the NarG-His49 mutants indicated that the cofactor is present in the NarG-H49C mutant but absent in the NarG-H49S mutant (19). To determine how the Mo-bisPGD site is perturbed in these mutants, we subjected both to analysis by protein crystallography. Fig. 2A shows the structure of the NarG-H49C mutant around the FS0 cluster and demonstrates that the Cys residue introduced by mutagenesis is clearly able to provide sulfur coordination to FS0. Also shown is the sequence of residues 49CGVNCTG55, which is unresolved in the structure of the NarG-H49S mutant (see below; Fig. 2B). Significantly, this overlaps with the 51VNCT54 sequence that is unresolved in the structure of apomolybdo-NarGHI (Protein Data Bank code 1SIW (18)).
FIGURE 2.

Structure of the NarG-H49C (A) and NarG-H49S (B) variants in the vicinity of FS0 and the Mo-bisPGD cofactor. In addition to the FS0-coordinating residues, the position of the 49CGVNCTG55 segment is shown in the NarG-H49C variant (A) that is unresolved in the structure of the NarG-H49S variant (B). FS0 and Mo-bisPGD are missing in the structure of the NarG-H49S mutant. A water molecule is coordinated by the NarG-Asp222 residue that coordinates the molybdenum atom in the NarG-H49C and wild-type enzymes. A single iron atom is coordinated by the sulfur atoms of NarG-Cys57 and NarG-Cys92.
The NarG-H49S mutant lacks molybdenum as well as the pterin components of the cofactor but retains the GDP moieties (Fig. 2B). The structure in this region differs from that of apomolybdo-NarGHI (Protein Data Bank code 1SIW) in that it lacks FS0 but retains a single iron atom coordinated by the sulfur atoms of NarG-Cys57 and NarG-Cys92. These results are consistent with correct assembly of FS0 being a prerequisite for Mo-bisPGD assembly.
Further structural analysis (Fig. 3) reveals that the unresolved sequence (49CGVNCTG55) of the NarG-H49S mutant is involved in a number of potentially critical interactions with the P-pyranopterin and the Q-guanine moieties. These include hydrogen bonds between the amide nitrogen of NarG-Asn52 and dithiolene sulfurs of both pterins and a hydrogen bond between the carbonyl oxygen of NarG-Cys53 and the N-10 secondary amine of the P-pterin. Finally, the carbonyl oxygen of the NarG-Gly50 amide is ∼3.4 Å from the N-7 of the purine bicycle of the guanine nucleotide attached to the Q-pterin. Based on these observations, it is likely that correct positioning of 49CGVNCTG55 is critical for insertion of the Mo-bisPGD cofactor during NarGHI maturation.
FIGURE 3.

Unresolved residues in the NarG-H49S mutant. Residues comprising NarG-Ser49 to NarG-Gly55 are not observed in the NarG-H49S mutant. The location of these residues in the NarG-H49C mutant is shown in dark gray. The carbonyl oxygen of the NarG-Gly50 amide is ∼3.4 Å from the N-7 of the purine bicycle. The amine nitrogen of NarG-Asn52 is 3.0 Å from a Q-pterin dithiolene sulfur and 3.2 Å from a P-pterin dithiolene sulfur. The carbonyl oxygen of NarG-Cys53 is 3.6 Å from the N-10 secondary amine of the P-pterin.
Analysis of the apomolybdo-NarGHI structures indicates that GDP moieties can be inserted into positions corresponding to GDP-P and GDP-Q in the NarG-H49S mutant (Fig. 2B) and into the GDP-Q position of the wild-type apomolybdoenzyme (18). These observations suggest that GDP can occupy its two binding pockets within NarG when the mature Mo-bisPGD cofactor is unavailable. The estimated intracellular concentration of GDP is ∼0.68 mm (51), a concentration that hypothetically could competitively inhibit Mo-bisPGD insertion. The final steps of cofactor insertion are orchestrated by the MobAB, MoeA, MogA, and NarJ proteins (52), and it is likely that these steps prevent inhibition by cytoplasmic GDP.
Potential Electron Transfer Pathways Are Retained in the NarG-R94S Mutant
The overall conservation of protein structure around FS0 and Mo-bisPGD in the NarG-H49C mutant prompted us to speculate that its lack of activity is due to a large shift being elicited on its Em. To test this hypothesis, we solved the structure of the NarG-R94S mutant, which should eliminate a positive charge in the vicinity of FS0, thus lowering its Em. A Lys or Arg residue is highly conserved at this position in all known CISM catalytic subunits and, in the Ralstonia eutropha NapA mutagenesis of a Lys to an Arg at this position, results in decreased enzyme activity (to ∼23% of wild type) (53). As already indicated above, in E. coli Me2SO reductase (DmsABC), mutation of an Arg to a Ser at this position eliminates quinol-dependent Me2SO reductase activity (50). To establish that a significant shift in the FS0 Em value is responsible for the observed decrease in enzyme activity, it is important to verify that the NarG-R94S mutant introduces no gross structural changes in the vicinity of either FS0 or the Mo-bisPGD cofactor.
Fig. 4A shows potential electron transfer routes from FS0 to the P-pterin. NarG-Arg94 could contribute to electron transfer via its interaction with the amine attached to the C-8 position of the P-pterin. Other possible routes include the interaction between the carbonyl oxygen of NarG-Cys53 and N-10 of the P-pterin. Finally, a conserved Asn (NarG-Asn52) provides an interaction between its carboximide amine side chain and two dithiolene sulfurs, one from each pterin. Fig. 4B shows the structure of the NarG-R94S mutant. The side chain oxygen of the Ser variant is ∼5.9 Å from the amine nitrogen attached to C-8 of the P-pterin and 4.5 Å from the side chain sulfur of NarG-Cys92. This compares with 3.4 and 3.7 Å for the equivalent distances in wild-type NarGHI (Fig. 1). Although the importance of defined electron transfer conduits between ETR components has been proposed (54–56), the presence of three potential pathways between FS0 and the Mo-bisPGD cofactor indicates that disruption of one is unlikely to significantly decrease intercenter electron transfer rates in the absence of other factors. The most likely candidate is a significant shift in the midpoint potential of FS0 that would render the FS0 → Mo-bisPGD electron transfer step unfavorable. A similar shift could also explain the lack of enzyme activity in the NarG-H49C mutant.
FIGURE 4.

Structure of the NarG-R94S variant. A, wild-type structure showing the three putative electron transfer pathways from FS0 to Mo-bisPGD. B, structure of the NarG-R94S variant.
EPR Analyses of the NarG Mutants
High-spin [4Fe-4S] clusters with an S = 3/2 ground state exhibit EPR spectra around g = 5.0 (57, 58). Fig. 5 shows EPR spectra in this region of membrane samples enriched in wild-type and mutant NarGHI. The wild-type enzyme exhibits peaks at g = 5.02 and g = 5.55, as previously reported (18, 28, 59). No signals corresponding to a reduced high-spin [4Fe-4S] cluster were observed in either the NarG-H49C or the NarG-H49S variant. The NarG-R94S variant exhibited a modified high-spin spectrum with two almost merged peaks at g = 5.25 and g = 5.57. These features both exhibited temperature dependences similar to that of the g = 5.55 feature of the wild-type spectrum (with peak signal intensities occurring at <5 K; data not shown), so it is likely that both arise from different subpopulations undergoing ΔS = 3/2 transitions.
FIGURE 5.
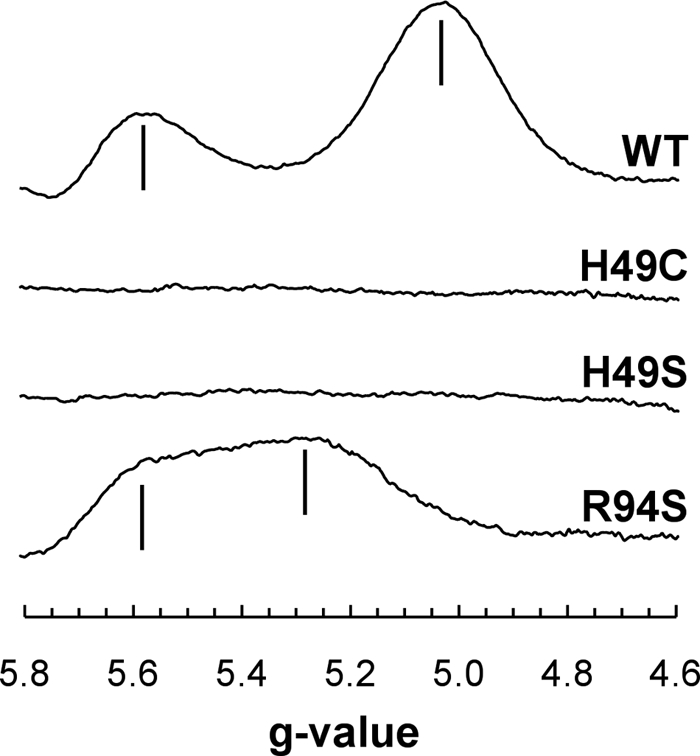
EPR spectra of the reduced wild-type and mutant enzymes. EPR conditions were as follows: temperature, 9 K; microwave power, 20 milliwatts at 9.381 GHz; and modulation amplitude (peak to peak), 20 Gpp at 100 KHz. Spectra are of membrane samples normalized to a protein concentration of ∼30 mg ml−1. WT, wild type.
FS0 in the NarG-R94S mutant titrates with an Em of −170 mV, a decrease of 115 mV compared with its value in the wild-type enzyme (Fig. 6) (18). Given the overall lack of structural perturbation observed in the NarG-R94S mutant (Fig. 4), it is likely that the ΔEm of −115 mV is elicited by removal of the positive charge of the Arg residue. Also, because two of the three putative electron transfer routes remain unaltered in the NarG-R94S variant, we conclude that its decreased enzyme activity results from the greater thermodynamic barrier presented by its FS0 cluster. This increases from 17.9 kJ mol−1 in the wild-type enzyme to 29.0 kJ mol−1 in the NarG-R94S mutant. Similar phenomena have been observed in other enzymes. For example, in E. coli succinate dehydrogenase (SdhCDAB), a mutant that decreases the Em of an intermediate ETR [4Fe-4S] (FS2) by ∼125 mV (an SdhB-I150H mutant) also elicits a significant decrease in succinate:quinone oxidoreductase activity (60).
FIGURE 6.

Potentiometric titration of FS0 in the NarG-R94S variant. Shown is a plot of the intensity of the FS0 signal in wild-type (WT) NarGHI (squares) and the NarG-R94S variant (triangles) versus Eh. Data were fit to single n = 1.0 components with Em values of −55 mV (wild-type) and −170 mV (NarG-R94S), respectively.
Table 2 summarizes the effects of the mutants described herein on the Mo(IV/V/VI), FS0, and FS1 redox chemistry based on potentiometric titrations followed by EPR spectroscopy (18, 61). No significant effects were elicited on the Mo(IV/V/VI) Em values in the NarG-H49C and NarG-R94S mutants. As previously reported by us (18, 61), shifts in the Em values of FS0 (or the absence thereof) elicit a negative ΔEm on FS1 (Table 2).
TABLE 2.
Effects of the NarG-H49 and NarG-R94 mutants on the NarGHI molybdenum and [Fe-S] cluster Em values (mV at pH 8.0)
The absence of an FS0 signal in the NarG-H49C mutant is surprising, because FS0 and the Mo-bisPGD cofactor are clearly visible in the structure (Fig. 2). Possible explanations for this are (i) that the cluster no longer has a high-spin ground state and (ii) that its Em is decreased to a value that prevents catalytic turnover. To address this question, we examined the potentiometric behavior of these two mutants at lower potentials. In the NarG-H49C mutant, we estimate that FS2 and FS3 have Em values of −420 and −80 mV, and in the NarG-H49S mutant, we estimate that FS2 and FS3 have potentials of −420 and −40 mV, respectively. Thus, the most likely explanation for the lack of activity in the NarG-H49C mutant is that the Em of FS0 is decreased below potentials readily accessible in potentiometric titrations (approximately −550 mV at pH 8.0). We therefore estimate that in a NarG-H49C mutant, the FS0 cluster undergoes a ΔEm of at least −500 mV and that this is the explanation for the lack of enzyme activity in this mutant.
The increased thermodynamic barrier presented by FS0 explains the inability of the ETR to transfer electrons from quinol to nitrate. In and of itself, it does not explain the lack of activity with the soluble, nonspecific donor benzyl viologen. In the equivalent mutant of E. coli DmsABC (DmsA-R61S), quinol:Me2SO oxidoreductase activity is also eliminated, but activity with benzyl viologen as donor is retained (50). Comparison of the active site funnels of NarGHI and an enzyme related to DmsABC (Rhodobacter Me2SO reductase) reveals that the former enzyme has an unusually narrow active site funnel (4) and that this may prevent electron donation directly to the Mo-bisPGD cofactor.
Assembly of FS0 Is a Prerequisite for NarGHI Maturation
The data presented herein provide a structural facet to an emerging model for NarGHI maturation. Turner et al. (29, 30) observed significant sequence similarity between the N terminus of NarG and that of the consensus tat leader. This group also demonstrated that NarJ interacts with a peptide comprising the first 50 amino acids of NarG in a tat-dependent manner (30, 62). Phylogenic analyses have clearly indicated an evolutionary link between NarG and a family of enzymes that includes Rhodovulum sulfidophilum ethylbenzene dehydrogenase (EbdABC), Thauera selenatis selenate reductase (SerABC), and Ideonella dechloratans chlorate reductase (ClrABC). Each of these enzymes is directed to the periplasmic compartment by a tat leader (4, 63), and as a group, they may represent a step in the evolution of NarGHI. The hypothesis that the N terminus of NarG is related to a tat leader is supported by the crystal structure of NarGHI, in which the first 40 amino acids of NarG extend away from the five domains of its core structure toward the membrane-intrinsic domain of the enzyme (2). Sequence analyses (29, 64) indicate that the tat recognition sequence, (S/T)RRXF(X/L)K, is replaced by the sequence 5DRFRYFK11 in NarG and that the first ∼40 amino acids of NarG represent a tat pseudoleader. It is notable that NarG-Arg6 interacts with the phosphate moiety of a phosphatidylglycerol in the structure, and it can be speculated that such an interaction occurs in other tat leaders prior to their cleavage from their respective holoenzyme precursors. Lanciano et al. (28) recently proposed a model in which FS0 and Mo-bisPGD insertion is coordinated with NarJ dissociation from the NarGH dimer during enzyme maturation. This model will require further clarification by mutagenesis of critical residues in the NarG tat pseudoleader, including NarG-Arg6. Crystallographic studies will also be necessary to assess the effects of a ΔnarJ mutant on the structure of the apoenzyme around the Mo-bisPGD/FS0-binding pocket. Such studies are in progress in our laboratories.
An additional unresolved question raised by the structures presented herein and elsewhere (18) is the ability of the apoenzyme to assemble GDP moieties into their respective P- and Q-binding sites. This raises the question of the possible role of NarJ in preventing GDP from competitively inhibiting holoenzyme maturation. Finally, although it is known that nucleotide addition occurs only after the proximal pterin has acquired a molybdenum atom (65), it is still not known whether the entire Mo-bisPGD cofactor is inserted into apomolybdoenzymes or whether the two halves are inserted sequentially with the loss of one of the molybdenum atoms (66). Future studies in our laboratory will resolve this issue by mutating residues surrounding the individual PGD-binding pockets.
Conclusions
Overall, we have generated structural data that indicate FS0 assembly into NarGHI is a prerequisite for Mo-bisPGD insertion. We have also demonstrated that a critical sequence of amino acids 49HGVNCTG55 is unresolved in the apomolybdo-NarG-H49SHI enzyme and may play a role in stabilizing Mo-bisPGD during maturation. The absence of FS0 does not prevent assembly of the holoenzyme to the cytoplasmic membrane. In NarG-H49CHI, the midpoint potential of FS0 is decreased to an extent that it is no longer able to redox-cycle in catalytic turnover. We validated the effect of decreasing FS0 Em on enzyme turnover by characterizing NarG-R94SHI, in which the potential of FS0 is decreased by 115 mV.
This work was supported, in whole or in part, by National Institutes of Health Grant GMO68451-01. This work was also supported by Canadian Institutes of Health Research Grant MOP15292, the Canada Foundation for Innovation, and the Alberta Heritage Foundation for Medical Research.
The atomic coordinates and structure factors (codes 3IR7, 3IR5, and 3IR6) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- CISM
- complex iron-sulfur molybdoenzyme
- Mo-bisPGD
- molybdo-bis(pyranopterin guanine dinucleotide)
- PGD
- pyranopterin guanine dinucleotide
- ETR
- electron transfer relay
- MOPS
- 4-morpholinepropanesulfonic acid
- Tricine
- N-tris(hydroxymethyl)methylglycine.
REFERENCES
- 1.Blasco F., Guigliarelli B., Magalon A., Asso M., Giordano G., Rothery R. A. (2001) Cell. Mol. Life Sci. 58, 179–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertero M. G., Rothery R. A., Palak M., Hou C., Lim D., Blasco F., Weiner J. H., Strynadka N. C. (2003) Nat. Struct. Biol. 10, 681–687 [DOI] [PubMed] [Google Scholar]
- 3.Jormakka M., Richardson D., Byrne B., Iwata S. (2004) Structure 12, 95–104 [DOI] [PubMed] [Google Scholar]
- 4.Rothery R. A., Workun G. J., Weiner J. H. (2008) Biochim. Biophys. Acta 1778, 1897–1929 [DOI] [PubMed] [Google Scholar]
- 5.Jormakka M., Törnroth S., Byrne B., Iwata S. (2002) Science 295, 1863–1868 [DOI] [PubMed] [Google Scholar]
- 6.Weiner J. H., Rothery R. A., Sambasivarao D., Trieber C. A. (1992) Biochim. Biophys. Acta 1102, 1–18 [DOI] [PubMed] [Google Scholar]
- 7.Krafft T., Bokranz M., Klimmeck O., Schröder I., Fahrenholz F., Kojro E., Kröger A. (1992) Eur. J. Biochem. 206, 503–510 [DOI] [PubMed] [Google Scholar]
- 8.Jormakka M., Yokoyama K., Yano T., Tamakoshi M., Akimoto S., Shimamura T., Curmi P., Iwata S. (2008) Nat. Struct. Mol. Biol. 15, 730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinzinger N. K., Fujimoto S. Y., Clark M. A., Moreno M. S., Barrett E. L. (1995) J. Bacteriol. 177, 2813–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinsley A. P., Berks B. C. (2002) Microbiology 148, 3631–3638 [DOI] [PubMed] [Google Scholar]
- 11.Hille R. (1996) Chem. Rev. 96, 2757–2816 [DOI] [PubMed] [Google Scholar]
- 12.Hille R. (2002) Met. Ions Biol. Syst. 39, 187–226 [PubMed] [Google Scholar]
- 13.Moura J. J., Brondino C. D., Trincão J., Romão M. J. (2004) J. Biol. Inorg. Chem. 9, 791–799 [DOI] [PubMed] [Google Scholar]
- 14.Kloer D. P., Hagel C., Heider J., Schulz G. E. (2006) Structure 14, 1377–1388 [DOI] [PubMed] [Google Scholar]
- 15.Hilton J. C., Rajagopalan K. V. (1996) Arch. Biochem. Biophys. 325, 139–143 [DOI] [PubMed] [Google Scholar]
- 16.Johnson J. L., Rajagopalan K. V. (1982) Proc. Natl. Acad. U.S.A. 79, 6856–6860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson J. L., Bastian N. R., Rajagopalan K. V. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 3190–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothery R. A., Bertero M. G., Cammack R., Palak M., Blasco F., Strynadka N. C., Weiner J. H. (2004) Biochemistry 43, 5324–5333 [DOI] [PubMed] [Google Scholar]
- 19.Magalon A., Asso M., Guigliarelli B., Rothery R. A., Bertrand P., Giordano G., Blasco F. (1998) Biochemistry 37, 7363–7370 [DOI] [PubMed] [Google Scholar]
- 20.Trieber C. A., Rothery R. A., Weiner J. H. (1996) J. Biol. Chem. 271, 4620–4626 [DOI] [PubMed] [Google Scholar]
- 21.Gladyshev V. N., Boyington J. C., Khangulov S. V., Grahame D. A., Stadtman T. C., Sun P. D. (1996) J. Biol. Chem. 271, 8095–8100 [DOI] [PubMed] [Google Scholar]
- 22.González P. J., Rivas M. G., Brondino C. D., Bursakov S. A., Moura I., Moura J. J. (2006) J. Biol. Inorg. Chem. 11, 609–616 [DOI] [PubMed] [Google Scholar]
- 23.Jepson B. J., Mohan S., Clarke T. A., Gates A. J., Cole J. A., Butler C. S., Butt J. N., Hemmings A. M., Richardson D. J. (2007) J. Biol. Chem. 282, 6425–6437 [DOI] [PubMed] [Google Scholar]
- 24.Palmer T., Santini C. L., Iobbi-Nivol C., Eaves D. J., Boxer D. H., Giordano G. (1996) Mol. Microbiol. 20, 875–884 [DOI] [PubMed] [Google Scholar]
- 25.Blasco F., Pommier J., Augier V., Chippaux M., Giordano G. (1992) Mol. Microbiol. 6, 221–230 [DOI] [PubMed] [Google Scholar]
- 26.Blasco F., Iobbi C., Giordano G., Chippaux M., Bonnefoy V. (1989) Mol. Gen. Genet. 218, 249–256 [DOI] [PubMed] [Google Scholar]
- 27.Blasco F., Dos Santos J. P., Magalon A., Frixon C., Guigliarelli B., Santini C. L., Giordano G. (1998) Mol. Microbiol. 28, 435–447 [DOI] [PubMed] [Google Scholar]
- 28.Lanciano P., Vergnes A., Grimaldi S., Guigliarelli B., Magalon A. (2007) J. Biol. Chem. 282, 17468–17474 [DOI] [PubMed] [Google Scholar]
- 29.Turner R. J., Papish A. L., Sargent F. (2004) Can. J. Microbiol. 50, 225–238 [DOI] [PubMed] [Google Scholar]
- 30.Li H., Turner R. J. (2009) Can. J. Microbiol. 55, 179–188 [DOI] [PubMed] [Google Scholar]
- 31.Weiner J. H., Bilous P. T., Shaw G. M., Lubitz S. P., Frost L., Thomas G. H., Cole J. A., Turner R. J. (1998) Cell 93, 93–101 [DOI] [PubMed] [Google Scholar]
- 32.Berks B. C., Sargent F., Palmer T. (2000) Mol. Microbiol. 35, 260–274 [DOI] [PubMed] [Google Scholar]
- 33.Berks B. C. (1996) Mol. Microbiol. 22, 393–404 [DOI] [PubMed] [Google Scholar]
- 34.Ize B., Coulthurst S. J., Hatzixanthis K., Caldelari I., Buchanan G., Barclay E. C., Richardson D. J., Palmer T., Sargent F. (2009) Microbiology 155, 3992–4004 [DOI] [PubMed] [Google Scholar]
- 35.Pascal M. C., Burini J. F., Ratouchniak J., Chippaux M. (1982) Mol. Gen. Genet. 188, 103–106 [DOI] [PubMed] [Google Scholar]
- 36.Guigliarelli B., Magalon A., Asso M., Bertrand P., Frixon C., Giordano G., Blasco F. (1996) Biochemistry 35, 4828–4836 [DOI] [PubMed] [Google Scholar]
- 37.Rothery R. A., Weiner J. H. (1991) Biochemistry 30, 8296–8305 [DOI] [PubMed] [Google Scholar]
- 38.Rothery R. A., Blasco F., Magalon A., Asso M., Weiner J. H. (1999) Biochemistry 38, 12747–12757 [DOI] [PubMed] [Google Scholar]
- 39.Bertero M. G., Rothery R. A., Boroumand N., Palak M., Blasco F., Ginet N., Weiner J. H., Strynadka N. C. (2005) J. Biol. Chem. 280, 14836–14843 [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 41.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 42.McRee D. E. (1999) J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 43.Rothery R. A., Blasco F., Weiner J. H. (2001) Biochemistry 40, 5260–5268 [DOI] [PubMed] [Google Scholar]
- 44.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 45.Markwell M. A., Haas S. M., Bieber L. L., Tolbert N. E. (1978) Anal. Biochem. 87, 206–210 [DOI] [PubMed] [Google Scholar]
- 46.Rothery R. A., Chatterjee I., Kiema G., McDermott M. T., Weiner J. H. (1998) Biochem. J. 332, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sambasivarao D., Turner R. J., Simala-Grant J. L., Shaw G., Hu J., Weiner J. H. (2000) J. Biol. Chem. 275, 22526–22531 [DOI] [PubMed] [Google Scholar]
- 48.Morpeth F. F., Boxer D. H. (1985) Biochemistry 24, 40–46 [DOI] [PubMed] [Google Scholar]
- 49.Buc J., Santini C. L., Blasco F., Giordani R., Cárdenas M. L., Chippaux M., Cornish-Bowden A., Giordano G. (1995) Eur. J. Biochem. 234, 766–772 [DOI] [PubMed] [Google Scholar]
- 50.Trieber C. A., Rothery R. A., Weiner J. H. (1994) J. Biol. Chem. 269, 7103–7109 [PubMed] [Google Scholar]
- 51.Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., Rabinowitz J. D. (2009) Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vergnes A., Gouffi-Belhabich K., Blasco F., Giordano G., Magalon A. (2004) J. Biol. Chem. 279, 41398–41403 [DOI] [PubMed] [Google Scholar]
- 53.Hettmann T., Siddiqui R. A., von Langen J., Frey C., Romão M. J., Diekmann S. (2003) Biochem. Biophys. Res. Commun. 310, 40–47 [DOI] [PubMed] [Google Scholar]
- 54.Daizadeh I., Medvedev D. M., Stuchebrukhov A. A. (2002) Mol. Biol. Evol. 19, 406–415 [DOI] [PubMed] [Google Scholar]
- 55.Wittekindt C., Schwarz M., Friedrich T., Koslowski T. (2009) J. Am. Chem. Soc. 131, 8134–8140 [DOI] [PubMed] [Google Scholar]
- 56.Moser C. C., Chobot S. E., Page C. C., Dutton P. L. (2008) Biochim. Biophys. Acta 1777, 1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hagen W. R. (1992) Adv. Inorg. Chem. 38, 165–222 [Google Scholar]
- 58.Duderstadt R. E., Brereton P. S., Adams M. W., Johnson M. K. (1999) FEBS Lett. 454, 21–26 [DOI] [PubMed] [Google Scholar]
- 59.Lanciano P., Savoyant A., Grimaldi S., Magalon A., Guigliarelli B., Bertrand P. (2007) J. Phys. Chem. B 111, 13632–13637 [DOI] [PubMed] [Google Scholar]
- 60.Cheng V. W., Ma E., Zhao Z., Rothery R. A., Weiner J. H. (2006) J. Biol. Chem. 281, 27662–27668 [DOI] [PubMed] [Google Scholar]
- 61.Rothery R. A., Magalon A., Giordano G., Guigliarelli B., Blasco F., Weiner J. H. (1998) J. Biol. Chem. 273, 7462–7469 [DOI] [PubMed] [Google Scholar]
- 62.Chan C. S., Howell J. M., Workentine M. L., Turner R. J. (2006) Biochem. Biophys. Res. Commun. 343, 244–251 [DOI] [PubMed] [Google Scholar]
- 63.Watts C. A., Ridley H., Dridge E. J., Leaver J. T., Reilly A. J., Richardson D. J., Butler C. S. (2005) Biochem. Soc. Trans. 33, 173–175 [DOI] [PubMed] [Google Scholar]
- 64.Sargent F. (2007) Microbiology 153, 633–651 [DOI] [PubMed] [Google Scholar]
- 65.Temple C. A., Rajagopalan K. V. (2000) J. Biol. Chem. 275, 40202–40210 [DOI] [PubMed] [Google Scholar]
- 66.Neumann M., Mittelstädt G., Seduk F., Iobbi-Nivol C., Leimkühler S. (2009) J. Biol. Chem. 284, 21891–21898 [DOI] [PMC free article] [PubMed] [Google Scholar]