

Abstract
Protein-tyrosine phosphatase 1B (PTP1B) has been implicated in the negative regulation of insulin signaling. We previously demonstrated that light-induced tyrosine phosphorylation of the retinal insulin receptor (IR) results in the activation of phosphoinositide 3-kinase/Akt survival pathway in rod photoreceptor cells. The molecular mechanism behind light-induced activation of IR is not known. We investigated the in vivo mechanism of IR activation and found that PTP1B activity in dark-adapted retinas was significantly higher than in light-adapted retinas. We made a novel finding in this study that the light-dependent regulation of PTP1B activity is signaled through photobleaching of rhodopsin. Conditional deletion of PTP1B in rod photoreceptors by the Cre-loxP system resulted in enhanced IR signaling. Further PTP1B activity negatively regulated the neuroprotective survival signaling in the retina. One of the challenging questions in the retina research is how mutations in human rhodopsin gene slowly disable and eventually disrupt photoreceptor functions. Our studies suggest that a defect in the photobleaching of rhodopsin and mutation in rhodopsin gene enhances the activity of PTP1B, and this activated activity could down-regulate the IR survival signaling. Our studies suggest that PTP1B antagonists could be potential therapeutic agents to treat stress-induced photoreceptor degenerations and provide further evidence that rhodopsin photoexcitation may trigger signaling events alternative to the classic phototransduction.
Keywords: G-proteins/Coupled Receptors, Gene/Knockout, Phosphorylation/Kinases/Tyrosine, Receptors/Photoreceptors, Signal Transduction, Vision/Photoreceptors, Vision/Retina, Insulin Receptor
Introduction
Insulin receptors (IRs)3 and insulin signaling proteins are widely distributed throughout the central nervous system (1). Dysregulation of insulin signaling in the central nervous system has been linked to the pathogenesis of neurodegenerative disorders such as Alzheimer and Parkinson diseases (2, 3). Cells of bovine and rat retinas contain high affinity receptors for insulin (1). IR signaling provides a trophic signal for transformed retinal neurons in culture (4). We recently reported that IR is functionally important for photoreceptor survival (5). IR deletion in photoreceptor cells caused increased sensitivity to light-induced photoreceptor degeneration (5). We previously reported that light induces tyrosine phosphorylation of the retinal IR and that this activation leads to the binding of phosphoinositide 3-kinase (PI3K) to rod outer segment (ROS) membranes (6) and the subsequent activation of Akt (7, 8). More recently, we demonstrated that IR activation is mediated through the photobleaching of the G-protein-coupled receptor, rhodopsin, but not signaling through the phototransduction cascade (9).
The extent of tyrosyl phosphorylation on a given protein is controlled by the reciprocal action of protein-tyrosine kinase and protein-tyrosine phosphatase (PTP) activities. Specific PTPs, including LAR, SHP-2, and PTP1B, have been implicated in the regulation of normal IR signaling (10–23). Of these, PTP1B has received significant attention due to its abundance in all insulin-sensitive tissues (24, 25). PTP1B is an abundant, widely expressed non-receptor tyrosine phosphatase, which is thought to be a key negative regulator of insulin signaling (26, 27). The state of IR dephosphorylation is regulated by PTP1B (26, 27). Previously it was shown that PTP1B overexpression results in the inhibition of the IR and insulin receptor substrate-1 (17, 22, 28) and the introduction of anti-PTP1B antibodies into cells enhances IR signaling (29). Global deletion of PTP1B in mice results in increased systemic insulin sensitivity, enhanced glucose uptake into skeletal muscle, and improved glucose tolerance (30, 31). The increased insulin sensitivity is due to the absence of PTP1B, resulting from failure to dephosphorylate the IR (30, 31). Increased and prolonged tyrosine phosphorylation of the IR was also observed in mice lacking PTP1B (30, 31). Liver-specific deletion of PTP1B has been shown to improve metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress (32). Neuronal PTP1B also regulates body weight, adiposity, and leptin action (33). Furthermore, neuronal PTP1B deficiency results in inhibition of hypothalamic 5′-AMP-activated protein kinase and isoform-specific activation of 5′-AMP-activated protein kinase in peripheral tissues (34). The functional role of PTP1B in retina or photoreceptor cells is not known.
We recently reported that diabetes reduces autophosphorylation of the IR and increases PTP1B activity (35). Further, we found that PTP1B is expressed in rod photoreceptor cells (36); however, the functional importance of this protein as well as its effect on retinal IR signaling is not yet known. In the current study, we demonstrate that photobleaching of rhodopsin regulates PTP1B activity. To determine the functional role of the IR in rod photoreceptor cells, we generated a conditional PTP1B knock-out mouse under the control of the rod opsin promoter by using Cre-loxP technology. Activation of the IR was observed in both light- and dark-adapted PTP1B knock-out mouse retinas and not in wild-type retinas, which had activation only in light. Mice lacking PTP1B were protected from bright light-induced photoreceptor degeneration. Our studies clearly demonstrate that PTP1B regulates the state of IR dephosphorylation in the retina, which in turn regulates the IR/PI3K/Akt survival pathway. These studies suggest that PTP1B antagonists could be potential therapeutic agents to treat stress-induced photoreceptor degenerations and provides further evidence that rhodopsin photoexcitation may trigger signaling events alternative to the classical phototransduction.
EXPERIMENTAL PROCEDURES
Materials
Polyclonal anti-IRβ and anti-PY99 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). An anti-PTP1B antibody and PTP1B substrate (RRLIEDAEPYAARG) were obtained from Upstate Biotechnology (Lake Placid, NY). Anti-Akt, anti-pAkt, and anti-caspase 3-cleaved product antibodies were obtained from Cell Signaling (Danvers, MA). The PTP1B inhibitor 3-(3,5-dibromo-4-hydroxybenzoyl)-2-ethylbenzofuran-6-sulfonicacid-(4-(thiazol-2-ylsulfamyl)phenyl)amide was obtained from Calbiochem. The actin antibody was obtained from Affinity BioReagents (Golden, CO). All other reagents were of analytical grade and from Sigma.
Animals
All animal work was carried out in strict accordance with the NIH Guide for the Care Use of Laboratory Animals and the Association for Research in Vision and Ophthalmology on the Use of Animals in Vision Research. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center and the Dean A. McGee Eye Institute. Mice and rats were born and raised in our vivarium and kept under dim cyclic light (5 lux, 12 h on/off, 7 a.m. to 7 p.m.) before experimentation.
Generation of PTP1B- and RPE65-deficient Mice
The PTP1B-deficient mouse strain used in this study was described previously (30). To identify PTP1B knock-out mice, we used a sense primer 5′-CAG TCT TGG TCT ACA GAG TG-3′, and an antisense primer 5′-ATC TCC TCG AAC TCC TTC TC-3′ to amplify genomic DNA by using PCR. The wild-type allele generates a 650-bp product, and the knock-out allele generates a 350-bp product. Mice lacking retinal pigment epithelium 65 (RPE65−/−) were derived at the National Institutes of Health, Bethesda, MD (37). These mice were kindly provided by Dr. Jing-Xing Ma (Dept. of Medicine and Endocrinology, University of Oklahoma).
Generation of Photoreceptor-specific PTP1B Knock-out mice
PTP1B-floxed mice were generated as described previously (33). The PTP1B-floxed homozygous mice were bred with opsin-driven Cre mice, who have a 0.2-kb opsin-Cre promoter. The resultant mice were genotyped for Cre, and floxed PTP1B and were heterozygous for the PTP1B floxed allele. To create photoreceptor-specific PTP1B knock-out mice, the floxed PTP1B mice carrying the Cre transgene were bred with the PTP1B-floxed homozygous mice (backcross). The genotype of the photoreceptor-specific PTP1B knock-out mice (i.e. animals carrying the Cre transgene and homozygous for the PTP1B-floxed allele) was confirmed by using PCR analysis of tail DNA. To identify rhodopsin-Cre, PCR was done with 1 μl of genomic DNA and the sense primer 5′-AGG TGT AGA GAA GGC ACT TAG C-3′, and the antisense primer: 5′-CTA ATC GCC ATC TTC CAG CAG G-3′ to amplify a 411-bp product. To identify PTP1B-floxed mice, we used the sense primer 5′-TGC TCA CTC ACC CTG CTA CAA-3′, and the antisense primer 5′-GAA ATG GCT CAC TCC TAC TGG-3′. The wild-type allele generates a 206-bp product, and the floxed allele generates 327-bp product.
PTP1B Activity Assay
The in vitro PTP activity assay was conducted based on a previously published protocol (38). Retinas were lysed in solubilization buffer (phosphate-buffered saline) containing 1% Nonindet P-40, 1% deoxycholate, 5 mm EDTA, 1 mm EGTA, 2 mm phenylmethylsulfonyl fluoride, 0.1 mm leupeptin, 2 μg/ml N-acetyl-leucinyl-leucinyl-norleucinal. The lysates were centrifuged for 10 min at 4 °C in a microcentrifuge, and supernatants were collected for immunoprecipitation. Before immunoprecipitation, cell lysates were subjected to preclearing with non-immune serum and protein A-Sepharose for 15 min at 4 °C. Equal quantities of each sample (750 μg of total protein) were then subjected to immunoprecipitation with anti-PTP1B antibody (PTP1B, Upstate Biotechnology) overnight at 4 °C. PTP1B immunocomplexes were precipitated with protein A-Sepharose at 4 °C for an additional 2 h. Immunoprecipitates were washed in PTP assay buffer (100 mm HEPES (pH 7.6), 2 mm EDTA, 1 mm dithiothreitol, 150 mm NaCl, 0.5 mg/ml bovine serum albumin). The peptide RRLIEDAEPYAARG (Upstate Biotechnology) was added to a final concentration of 200 μm in total reaction volume of 60 μl in PTP assay buffer, and the reaction was allowed to proceed for 1 h at 30 °C. At the end of the reaction, 40-μl aliquots were placed in 96-well plate, 100 μl of Malachite Green Phosphatase reagent (Upstate Biotechnology) was added, and absorbance was measured at 630 nm.
Insulin Receptor Kinase Activity
Mice were dark-adapted overnight, and then half of the animals were exposed to normal room light (300 lux) for 30 min as described previously (9). Retinas were harvested and lysed in lysis buffer (39) (1% Nonidet P-40, 20 mm HEPES (pH 7.4) and 2 mm EDTA containing phosphatase inhibitors (100 mm sodium fluoride, 10 mm sodium pyrophosphate, 1 mm NaVO3, and 1 mm molybdate) and protease inhibitors (10 μm leupeptin, 10 μg/ml aprotinin, and 1 mm phenylmethylsulfonyl fluoride)). Insoluble material was removed by centrifugation at 17,000 × g for 20 min, and the solubilized proteins were pre-cleared by incubation with 40 μl of protein A-Sepharose for 1 h at 4 °C with mixing. The supernatant was incubated with anti-IRβ antibody overnight at 4 °C and subsequently with 40 μl of protein A-Sepharose for 2 h at 4 °C. After centrifugation at 14,000 rpm for 1 min, immunocomplexes were washed three times with wash buffer (50 mm HEPES (pH 7.4) containing 118 mm NaCl, 100 mm sodium fluoride, 2 mm NaVO3, 0.1% (w/v) sodium dodecyl sulfate and 1% (v/v) Triton X-100). The kinase reaction was performed at room temperature in kinase assay buffer (50 mm HEPES, pH 7.4, 12 mm MgCl2, and 5 mm MnCl2) containing 100 μm ATP, 3 mg/ml poly-Glu:Tyr peptide (Sigma) and 10 μCi/ml [γ-32P]ATP for 60 min. The reaction was briefly centrifuged at 14,000 rpm for 1 min, and 15 μl of supernatant was spotted on Whatman p81 phosphocellulose paper discs. Filter paper discs were washed three times for 5 min in 0.75% O-phosphoric acid and once for 5 min in acetone before counting the radioactivity in a Beckman LS 6000SC Scintillation Counter (Beckman Instruments, Fullerton, CA).
Exposure of Animals to Light Stress
Animals were born and raised in dim cyclic (5 lux) light. Pigmented mice were exposed to constant light for 7 days at an illuminance level of 14,000 lux using white fluorescent light bulbs that are suspended 50 cm above the floor of the cage. During light exposure, animals were maintained in transparent polycarbonate cages with stainless-steel wire bar covers. Drinking water was supplied by a bottle attached to the side of the cage, and food was placed on bedding in the bottom of the cage so that there was no obstruction between the light and the animal. Mice were maintained in 100-lux cyclic light for 7 days after light exposure. Morphologic and functional analyses in mice were then completed.
Intravitreal Injection of PTP1B Inhibitor
Two microliters of either vehicle (DMSO) or PTP1B (200 μm) inhibitor were injected into the vitreous body of the rat eye using a Hamilton syringe. Rats were deeply anesthetized with a single intraperitoneal injection of xylazine (7 mg/g) and ketamine (40 mg/g) before the injection. Two days after injection, animals were subjected to light stress at 2700 lux for 6 h. At the end of light stress, the rats were killed with CO2 asphyxiation, and the retinas were harvested. Retinal proteins were either subjected to immunoprecipitation with anti-IRβ antibody followed by immunoblot analysis with anti-PY99 antibody or directly subjected to immunoblot analysis with the anti-pAkt, anti-Akt, anti-caspase 3-cleaved product, and anti-actin antibodies. Some mice were maintained in 100-lux cyclic light for 7 days after light exposure. Morphologic and functional analyses in mice were then completed.
Statistical Procedures
All values are reported as the mean ± S.D. Un-paired t-tests were used to compare groups. For the light stress experiments we performed an analysis of variance followed by a Scheffe's post hoc test. Significance was established at probability values of <0.05.
Other Methods
The preparation of retinal tissue sections, the methods of outer nuclear layer (ONL) thickness measurement, and immunohistochemistry were described previously (40). Flash electroretinograms (ERGs) were recorded by using a Ganzfeld-type ERG recording system (UTAS-E3000, LKC Technologies Inc., Gaithersburg, MD) as reported previously (41).
RESULTS
Light Regulates the PTP1B Activity in Vivo
To determine whether light regulates PTP1B activity, we immunoprecipitated PTP1B from lysates of dark- and light-adapted mouse retinas and measured the PTP1B activity. The PTP1B activity was significantly greater in dark-adapted retinas than in the light-adapted retinas (Fig. 1A). To determine whether this greater PTP1B activity was due to increased protein expression in the dark-adapted retinas, we subjected the proteins from dark- and light-adapted retinas to immunoblotting with anti-PTP1B antibody (Fig. 1B). No significant differences in the expression of PTP1B was found between the dark- and light-adapted mouse retinas (Fig. 1B), suggesting that light regulates PTP1B activity in vivo.
FIGURE 1.

PTP1B activity and expression in dark- and light- adapted wild-type mouse retinas. Retinas from each mouse were immunoprecipitated with anti-PTP1B antibody and measured for PTP1B activity (A). Mean (S.D.) PTP1B activity in dark-adapted retinas was significantly greater than the mean PTP1B activity in the light-exposed retinas (n = 4 for each group, p < 0.05). 20 μg of retinal proteins from dark- and light-adapted mouse retinas was immunoblotted with anti-PTP1B (B) and anti-actin (C) antibodies.
Photoactivation of Rhodopsin Regulates the PTP1B Activity in Vivo
To determine whether the light-induced inhibition of PTP1B is signaled through bleachable rhodopsin, we examined PTP1B activity in the retinas from retinal pigment epithelium 65 knock-out (RPE65) mice. RPE65−/− mice have opsin in their ROS but do not form photobleachable rhodopsin due to the absence of regeneration of chromophore 11-cis-retinal (37). Significantly greater PTP1B activity was found in the RPE65−/− mouse retinas than in the wild-type retinas under light-adapted conditions (Fig. 2A), suggesting that photoactivation of rhodopsin is necessary for the regulation of PTP1B activity. The increase in PTP1B activity in the RPE65−/− mouse retinas was not due to increased PTP1B (Fig. 2B) expression or increased protein loading (Fig. 2C). Densitometric analysis of PTP1B Western blots was performed in the linear range of detection, and absolute values were then normalized to actin (Fig. 2D). Collectively, these data suggest that photoactivation of rhodopsin post-translationally regulates the activity of PTP1B.
FIGURE 2.

PTP1B activity and expression in wild-type and RPE65−/− mouse retinas. Wild-type and Rpe65−/− mouse retinas were lysed and immunoprecipitated with anti-PTP1B antibody and measured for PTP1B activity (A). Mean (S.D.) PTP1B activity was significantly greater in the RPE65−/− retinas than in the wild-type retinas (n = 4 for each group, p < 0.001). 20 μg of retinal proteins from wild-type and RPE65−/− mice was immunoblotted with anti-PTP1B (B) and anti-actin (C) antibodies. Densitometric analysis of PTP1B immunoblots was performed in the linear range of detection, and absolute values were then normalized to actin (D). PTP1B activity in wild-type and VPP transgenic mice is shown. Wild-type and VPP mouse retinas were lysed and immunoprecipitated with anti-PTP1B antibody and measured for PTP1B activity (E). Values are mean (S.D.), n = 5, p < 0.05. 10 μg of retinal proteins from wild-type and VPP mice was immunoblotted with anti-opsin (F). G, densitometric analysis of opsin levels between wild-type and VPP mice. Significance, p < 0.001.
Increased PTP1B Activity in Rhodopsin Mutant Mice
VPP mice, which possess a mutant transgene for opsin (V20G, P23H, and P27L), exhibit a progressive rod degeneration that resembles one form of human autosomal dominant retinitis pigmentosa (42). In rhodopsin mutant VPP-transgenic mice (42), the PTP1B activity was significantly higher compared with wild-type mice (Fig. 2E). Due to retinal degeneration the opsin content in VPP-transgenic mice were significantly reduced compared with wild-type mice (Fig. 2, F and G). These studies suggest that mutant rhodopsin or its signaling may result in increased PTP1B activity in VPP mice.
Generation of Photoreceptor-specific Conditional Knock-out Mice
We previously reported that light induces activation of the retinal IR (6). Under these conditions we found little activity of PTP1B (Fig. 1A). These observations led to the hypothesis that light-induced IR phosphorylation is due to reduced PTP1B activity and that the reduced IR phosphorylation in darkness is due to increased PTP1B activity. To investigate the effect of PTP1B on the state of IR dephosphorylation we disrupted PTP1B gene specifically in mouse rod photoreceptors using Cre-loxP technology and examined the state of IR dephosphorylation under dark- and light-adapted conditions.
To investigate the functional significance of PTP1B on IR activation in rods, we generated a rod photoreceptor-specific conditional PTP1B knock-out mouse model. For this purpose, mice expressing the floxed PTP1B (33) allele were bred with rod-expressing Cre driven by 0.2-kb mouse opsin promoter-controlled Cre recombinase mouse line (43). We previously used the same Cre recombinase mouse line for the generation of photoreceptor-specific conditional IR knock-out mice (5). With this line we were able to delete >60% of IR gene from rod photoreceptors (5). We were unable to assess the deletion of PTP1B in mouse rod photoreceptors, because commercially available PTP1B antibodies are not suitable for immunocytochemistry. We utilized indirect approach and assessed Cre recombinase protein expression and cellular localization in the retinas of wild-type and knock-out littermates by immunoblotting and immunofluorescence microscopy using anti-Cre antibody. The Cre recombinase expression was localized to rod photoreceptor nuclei in the knock-out retinas, whereas Cre stain was not detected in wild-type controls (Fig. 3A). Previously, Cre mice were characterized by having normal rod distribution and function as late as 8 months of age (43). We also assessed Cre recombinase protein expression in the retinas of wild-type and knock-out littermates by immunoblotting with anti-Cre antibody. The results indicate Cre protein expression observed in knock-out retinal lysates was absent in wild-type controls (Fig. 3B). Actin expression was used as loading control (Fig. 3C). The proper localization of Cre in rod photoreceptors indirectly confirms the deletion of PTP1B in rod photoreceptors.
FIGURE 3.

Generation of rod-specific PTP1B conditional knock-out mouse model. Rod photoreceptor-specific deletion of PTP1B was done by cross-breeding floxed PTP1B mice to mice that express Cre recombinase under the control of the mouse rod opsin promoter. A, immunohistochemical analysis of Cre recombinase expression in knock-out and wild-type control retinas harvested from litter mates. Immunoblot analysis of duplicate knock-out and wild-type control retinal extracts was used to determine the expression of Cre recombinase (B), and actin levels were used as a loading control (C). IR kinase activity was measured from retinas harvested from dark- and light-adapted wild-type and PTP1B−/− mice (D). The IR immunoprecipitates (E) were subjected to IR kinase activity employing poly-Glu:Tyr peptide as substrate. Actin levels were used as loading control (F).
Light-dependent Activation of IR Is PTP1B-dependent
Wild-type and PTP1B knock-out mice were dark-adapted overnight, and then half of the mice were exposed to normal room light (300 lux) for 30 min as described previously (9). Retinal proteins were prepared and subjected to immunoprecipitation with anti-IRβ antibody, and the immunocomplexes were used for the measurement of IR kinase activity. We detected a light-dependent IR kinase activity in wild-type mice (Fig. 3D). In the absence of PTP1B, the IR kinase activity in dark-adapted retinas was similar to the IR kinase activity found in the light-adapted wild-type mouse retinas (Fig. 3D). A similar finding was also observed with global PTP1B knock-out mouse retinas (data not shown). This differential IR kinase activity was not due to the differences in the IR protein, because we recovered similar amounts of the IR protein in both wild-type and PTP1B knock-out mouse retains (Fig. 3E). Actin expression was used as loading control (Fig. 3F). These data clearly indicate that PTP1B regulates the IR dephosphorylation in rod photoreceptor cells.
Characterization of Global PTP1B Knock-out Mice
The generation of PTP1B knock-out mice was described previously (30). Retinal proteins from wild-type and PTP1B knock-out mice were subjected to immunoblotting with anti-PTP1B antibody (Fig. 4A). The blot was stripped and reprobed with anti-actin antibody to ensure equal amounts of protein in each lane (Fig. 4B). PTP1B protein was present in wild-type retinas and not in knock-out retinas (Fig. 4A). We also measured the PTP1B activity in the wild-type and PTP1B knock-out mouse retinas immunoprecipitated with anti-PTP1B antibody. PTP1B activity was not found in knock-out retinas but was found in the wild-type retinas (Fig. 4C). It is important to note that complex breeding is involved to generate conditional PTP1B knockouts, and also the number of knockouts in each breeding is not sufficient (in terms of age- and sex-matched controls) to carry out the functional studies. Therefore, global PTP1B knock-out mice were used to study the retinal morphology and function under normal and light stress conditions as described below.
FIGURE 4.
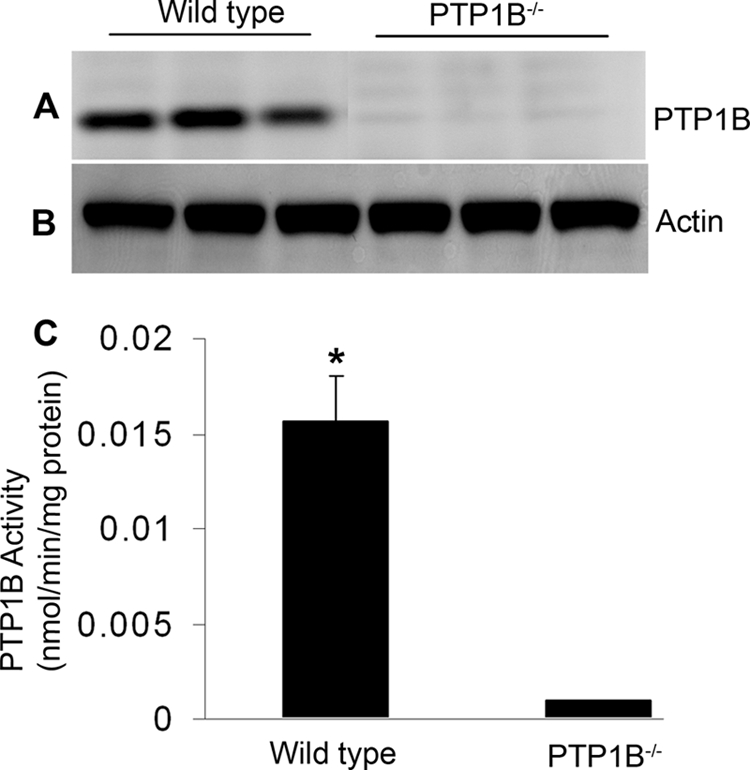
PTP1B expression and activity in wild-type and PTP1B knock-out mouse retinas. 20 μg of retinal proteins from wild-type and PTP1B knock-out mice were immunoblotted with anti-PTP1B (A) and anti-actin (B) antibodies. Retinal proteins from wild-type and PTP1B knock-out mice were immunoprecipitated with anti-PTP1B antibody and measured for PTP1B activity (C). Data are mean ± S.D., n = 3 for each group, p < 0.001.
Effect of Loss of PTP1B on Retinal Morphology and Function
To confirm the percentage of protein deletion in heterozygous in comparison to knock-out and wild-type, we examined the PTP1B protein expression with anti-PTP1B (Fig. 5A). Actin expression was used as a loading control (Fig. 5B). The results indicate that >95% of protein was deleted in homozygous PTP1B, whereas in heterozygote, >50% of PTP1B was deleted in comparison to wild type. Light microscopic examination of the retinas from wild-type, heterozygous, and homozygous PTP1B knock-out mice at 6–8 weeks of age showed no difference in retinal structure among the three groups when each group was maintained in dim cyclic light (Fig. 5C). The retinas appeared normal, and ROS appeared to be well organized (Fig. 5C). There were 11 to 12 rows of photoreceptor nuclei in the ONL, the number usually found in rodents without retinal degeneration (44). Quantitative analysis of the superior and inferior regions of the ONL layer showed no significant differences among the three groups in the average ONL thickness measured at 0.25-mm intervals from the ONL to the inferior and superior ora serrata (Fig. 5D), indicating that rod photoreceptor viability was not different among these mice. Thus, mice lacking PTP1B did not exhibit any structural phenotype when maintained in dim cyclic light.
FIGURE 5.

Morphological analysis of PTP1B knock-out mice born and raised in dim cyclic light. Retinal proteins from wild-type, heterozygous, and homozygous PTP1B knock-out mice were subjected to immunoblot analysis with anti-PTP1B (A) and anti-actin (B) antibodies. C, hematoxylin and eosin-stained retinal sections of the retina from the eyes of wild-type, heterozygous, and homozygous PTP1B mice at 6–8 weeks of age. D, quantification of morphological changes. Plots of total retinal thickness in the inferior and superior regions of the retinas of wild-type, heterozygous, and homozygous PTP1B mice are shown. Values are mean (S.D.), n = 2 (wild-type), 3 (heterozygous), and 3 (homozygous). Examination of retinas from each group revealed no structural differences in any of the retinal cells at the light microscope level. RPE, retinal pigment epithelium; ROS, rod outer segments; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
ERG was used to evaluate photoreceptor function in PTP1B knock-out and wild-type mice. No differences among the animals were found in the amplitude of the a-wave of the scotopic ERG, which measures the response of rod photoreceptor to light stimuli, or the b-wave, which measures the response of the inner retinal cells. Amax and Bmax, the maximal values of the a- and b-wave amplitudes at saturating light intensities, respectively, were calculated for each group, and no significant differences were found among the groups, indicating that the absence of PTP1B did not adversely affect the function of the retinas of mice born and raised in a relatively dim cyclic light environment (see Fig. 7, B and C). These results show that PTP1B knock-out mice do not exhibit any functional phenotype when they are maintained in dim cyclic light.
FIGURE 7.

Functional assay of PTP1B knock-out mice after light damage. The ERG amplitudes were recorded for wild-type, heterozygous, and homozygous PTP1B knock-out mice after light exposure at 14,000 lux for 7 days. A, representative ERG tracing from wild-type, heterozygous, and homozygous PTP1B knock-out mice before and after light stress. Average a- (B) and b-wave (C) amplitudes of scotopic ERG were measured from wild-type, heterozygous, and homozygous PTP1B knock-out mice. The a-wave amplitude was measured from the resting level to the peak of the cornea-negative deflection, and the b-wave amplitude was measured from the trough of the a-wave to the crest of the cornea-positive response. Data are mean (S.D.), and the number of animals used in this study were 6 (wild-type), 6 (heterozygous), and 5 (homozygous). *, p < 0.05 and **, p < 0.01 by one-way analysis of variance followed by Scheffe's posthoc test.
Effect of PTP1B on Retinal Structure after Light Stress
Fig. 3 shows that PTP1B regulates the state of IR dephosphorylation. Further, IR activation has shown to rescue retinal neurons from apoptosis in culture through PI3K/Akt activation (4), and we recently reported that deletion of IR from rod photoreceptors resulted in stress-induced photoreceptor degeneration (5). Therefore, we hypothesized that deletion of PTP1B in the retina could be neuroprotective. To test our hypothesis, we exposed wild-type, heterozygous, and homozygous PTP1B knock-out mice to light stress (14,000 lux) for 7 days. After a 7-day recovery period, we measured the extent of photoreceptor cell loss. Control mice were maintained in dim cyclic light. The ONL thickness in mice from each group reduced after light exposure, indicating that the number of rod photoreceptors reduced (Fig. 6A). The greatest reduction was apparent in the retinas of the wild-type mice. Quantitative analysis of the superior and inferior regions of the ONL layer of unexposed control, heterozygous, and homozygous PTP1B knock-out mice indicated no significant differences in the average ONL thickness measured at 0.25-mm intervals from the ONL to the inferior and superior ora serrata, indicating that there was no difference in rod photoreceptor viability between the three groups under dim-rearing conditions (similar to Fig. 5B). When exposed to 14,000 lux, wild-type mice had significantly fewer rod photoreceptors in the superior and inferior regions than the heterozygous and homozygous PTP1B knock-out mice (Fig. 6, B and C). It appears that heterozygous and homozygous PTP1B knock-out mice were protected from light stress while the wild-type mice were not.
FIGURE 6.

Morphological analysis of PTP1B knock-out mice after light stress. Eyes of dim or light-stressed wild-type, heterozygous, and homozygous 6- to 8-week-old PTP1B knock-out mice were fixed, embedded in paraffin, and stained with hematoxylin and eosin (A). Plots of ONL thickness at 0.25-mm intervals from the optic nerve head (ONH) along the vertical meridian in the superior and inferior regions of the PTP1B knock-out retinas after light stress at 14,000 lux (B) for 7 days are shown. C, quantification of morphological changes. The average ONL thickness was calculated for the superior and inferior regions of the eyes from wild-type, heterozygous, and homozygous PTP1B knock-out mice. Values are mean (S.D.), n = 6 (wild-type), 6 (heterozygous), and 5 (homozygous). *, p < 0.05 and **, p < 0.01 between +/+ and −/−, and †, p < 0.05 between +/+ and +/−, by one-way analysis of variance followed by Scheffe's post hoc test. RPE, retinal pigment epithelium; ROS, rod outer segments; ONL, outer nuclear layer; INL, inner nuclear layer; and GCL, ganglion cell layer.
Functional Evaluation of Photoreceptor Cells in PTP1B Knock-out Mice after Light Damage
Seven days after light exposure, we evaluated the retinal function by ERG. Fig. 7A shows typical wave forms from wild-type, heterozygous, and homozygous PTP1B knock-out mice before and after light stress. Mean amplitudes of a- and b-waves from all mice tested per group, are plotted in Fig. 7 (B and C), respectively. No differences in ERG amplitudes between the wild-type, heterozygous, or homozygous PTP1B knock-out groups were found when the mice were maintained in dim cyclic light (Fig. 7, B and C). There was a reduction in the amplitudes of both waves from all groups as a result of light stress; the greatest reduction occurred in the wild-type mice. Both a- and b-wave amplitudes were lower in the wild-type mice than in either heterozygous or homozygous PTP1B knock-out mice that were exposed to 14,000 lux (Fig. 7, B and C). These data suggest that the absence of PTP1B renders the photoreceptors of these knock-out mice less sensitive to light damage than those of wild-type mice.
PTP1B Inhibitor Protects the IR Phosphorylation and Prevents the Cleavage of Caspase 3 during Light Stress
Our results suggest that PTP1B knock-out mouse retinas do not degenerate, but the wild-type retinas are more susceptibility to light stress-induced photoreceptor generation. Therefore, it is difficult to study the effect of PTP1B on IR dephosphorylation in wild-type retinas. To address the PTP1B effect on IR signaling in light stress we took advantage of a PTP1B inhibitor approach. PTP1B inhibitors have already been recognized as potential therapeutics in the treatment of type-2 diabetes and obesity (45–47). However, the role of PTP1B in retinal diseases has not been investigated. We examined the effect of PTP1B inhibitor on IR signaling during light stress. Retinas were harvested from light stress-exposed (2700 lux for 6 h) and unexposed rats from DMSO or PTP1B inhibitor-injected groups. Retinal proteins were subjected to immunoblot analysis with anti-pAkt, anti-Akt, anti-caspase 3-cleaved product, and anti-actin antibodies or immunoprecipitated with anti-IRβ antibody followed by immunoblot analysis with anti-PY99 antibody. The blot was stripped and reprobed with anti-IRβ antibody to ensure equal amounts of protein were recovered in each immunoprecipitate. The DMSO-treated light stress group had no detectable IR phosphorylation, whereas phosphorylation was preserved in the PTP1B inhibitor-treated light stress group (Fig. 8A). Akt phosphorylation was higher in the PTP1B inhibitor-treated, light stress group (Fig. 8, C and E). The total Akt levels remained constant among all treatments (Fig. 8D). Increased caspase 3-cleaved product formation was observed in the DMSO-treated, light stress group, and this cleavage product formation was protected in the PTP1B inhibitor-treated, light stress group (Fig. 8F). Actin levels were used to verify equal protein loading (Fig. 8G). These data suggest that inhibition of PTP1B results in the inhibition of caspase activation through IR-mediated Akt signaling when light-stressed. Seven days after light exposure, we also evaluated the retinal function by ERG. After exposure to light, compared with control (un-exposed DMSO or PTP1B inhibitor-treated rats) DMSO-treated animals, there was a 59% loss of ERG a-wave, whereas PTP1B inhibitor-treated rats had a 35% loss of ERG a-wave amplitudes, respectively (Fig. 8H). Compared with DMSO-treated rats, ERG a-wave amplitudes in light-exposed eyes were higher in PTP1B inhibitor-treated rats. These results suggest that pharmacological inhibition of PTP1B provides functional protection against light stress. Structural protection was also observed in PTP1B inhibitor-treated rat retinas (data not shown).
FIGURE 8.

Effect of PTP1B inhibitor on IR signaling during light stress. Rats were intravitreally injected with either DMSO or PTP1B inhibitor as described under “Experimental Procedures.” Retinal proteins were subjected to either immunoprecipitation with anti-IRβ antibody (B) followed by immunoblot analysis with anti-PY99 antibody (A) or immunoblotting with anti-pAkt (C), anti-Akt (D), anti-caspase 3-cleaved product (F), and anti-actin (G) antibodies. Densitometric analysis of pAkt (C) immunoblots was performed in the linear range of detection, and absolute values were then normalized to total Akt (D). Data are mean (S.D.) (n = 4 for each group). LS, light stress. The ERG amplitudes were recorded from 6 rats from each of 4 groups: DMSO dark, DMSO LS, PTP1B inhibitor dark, and PTP1B inhibitor LS. Average a- and b-wave (H) amplitudes of scotopic ERG were measured from 4 groups. Percentage of ERG functional loss was calculated from respective dark-adapted DMSO or PTP1B inhibitor control (set as 100%).
DISCUSSION
The central observation of this study was that photoactivation of rhodopsin regulates PTP1B activity, which in turn regulates the state of IR dephosphorylation in the retina. Our results suggest that photoreceptors possess a novel IR cell survival pathway that uses rhodopsin-regulated PTP1B activity to connect rhodopsin signaling with a tyrosine kinase signal transduction pathway. Further PTP1B activity negatively regulates the neuroprotective potential of the retina.
We hypothesized that the light-induced tyrosine phosphorylation of the IR that we reported previously (6) could be due to the inhibition of PTP1B. We found an increase in PTP1B activity in the dark-adapted mouse retinas and not in the light-adapted mouse retinas, supporting our hypothesis. Photoreceptor-specific deletion of PTP1B in rods resulted in constitutive activation of PTP1B regardless of light exposure, suggesting that loss of IR activation in the dark is due to increased activity of PTP1B. Our current study suggests that light regulates the activity of PTP1B and protects the IR dephosphorylation.
We have previously reported that photoactivation of rhodopsin but not transducin signaling is required for IR phosphorylation in the retina (9), because we failed to observe the retinal IR phosphorylation in RPE65−/− mouse retinas (9). These animals have opsin in their ROS but do not have a bleachable rhodopsin due to the absence of the chromophore (37). In this study we found increased PTP1B activity in light-adapted RPE65−/− mouse retinas, which can explain the lack of IR phosphorylation in RPE65−/− mouse retinas that we reported previously (9). Collectively, these data suggest that photoactivation of rhodopsin regulates PTP1B activity. Interestingly, the higher PTP1B activity observed in dark-adapted and RPE65−/− mouse retinas are not correlated with its protein expression. These results suggest that photobleaching of rhodopsin may post-translationally regulate the activity of PTP1B. PTP1B has recently been shown to be modified by sumoylation (48). Sumoylation plays an important role in rod photoreceptor development (49). Sumoylation is also shown to control the protein stability and the biological function of the photoreceptor-specific protein, phosducin (50). We previously found that light induces the activation of Akt in rod photoreceptor cells (7). Akt phosphorylates substrates at the consensus motif RXRXX(S/T). PTP1B contains this motif (RYRDVS50), which can be phosphorylated by Akt on Ser50 (51). It has also been shown that phosphorylated PTP1B has an impaired ability to dephosphorylate the IR (51).
The potential contribution of other photosensitizers to the activation of PTP1B cannot be ruled out. Although the response in the RPE65−/− mice suggests that rhodopsin is important in the regulation of PTP1B, it does not entirely exclude the contribution of other photosensitizers. For example, melanopsin, the photopigment found in specialized photosensitive ganglion cells of the retina, when impacted by light, generates reactive oxygen species (52) that could reduce the activity of phosphatases in the retina, including PTP1B. Similarly, Lipofuscin acts as a photosensitizer within the retina and could have a similar effect as melanopsin (53). Further, light activation of cytochrome c oxidase, which reproducibly releases reactive oxygen species (54), could also reduce the PTP1B activity. The most common regulator of PTP1B is reactive oxygen species (55). These reactive oxygen species cause the oxidative inactivation of catalytic cysteine, which is in the active site of the PTP1B (56). These positive responses to cellular reactive oxygen species may seem “paradoxical,” because chronic exposure to relatively high levels of reactive oxygen species leads to several diseases, including retinal degenerations (57). It is very clear that mild concentrations of reactive oxygen species are shown to inhibit the activity of PTP1B in vivo (55). Further studies, however, are required to confirm this phenomenon.
We previously reported that the G-protein-coupled receptor rhodopsin regulates the phosphorylation of the IR, which leads to the activation of downstream neuronal survival pathway in rods (6, 7). The significance of this pathway is underscored by the observation that deletion of the IR from rods resulted in stress-induced photoreceptor degeneration (5). Several studies have clearly indicated that the state of IR dephosphorylation is tightly regulated by PTP1B (26, 27). Mice lacking PTP1B shows sustained IR phosphorylation in muscle and hepatic tissues (31). We also observed a constitutive activation of IR in both light- and dark-adapted conditions in the conditional PTP1B knock-out mouse retinas and not in the wild-type mouse retinas, which is activated only in light (9). These studies suggest that IR activation is under the control of the rhodopsin-regulated PTP1B activity whose deletion results in light-independent activation of IR.
Bright light exposure causes rod photoreceptor death by apoptosis (58, 59). IR activation reduced the activation of caspase-3 in R28 retinal neurons, and this effect was mediated through PI3K/Akt activation (4). We recently reported on the light stress-induced activation of caspase-3 in IR knock-out mouse retinas (5). These studies suggest that IR regulates the activation of caspase-3 through the PI3K/Akt pathway, and our studies clearly indicate the reduced recruitment of PI3K/Akt to ROS membranes of IR knock-out mice (5). Akt inhibits many pro-apoptotic targets by phosphorylating caspase-9, glycogen synthase kinase, BCL-2-associated death promoter, and members of the forkhead family transcriptional factors (60–67). Caspase-9 is the master player of caspase protease cascade (61). Caspase-3 activation may be due to impaired IR activated Akt to phosphorylate caspase-9 (4). These results suggest that reduction of IR activation could lead to apoptosis mediated by caspase-3 activation in rod photoreceptors presumably through the dephosphorylation of IR by PTP1B. Intravitreal injection of a PTP1B inhibitor (68) protects against photoreceptor degeneration through activation of IR-mediated caspase-3 inhibition. These studies suggest that PTP1B promotes cell death through dephosphorylation of the IR, which in turn down-regulates the PI3K/Akt pathways that result in the activation caspase-3.
Retinitis pigmentosa (RP) is one of the most common forms of inherited retinal degeneration (69). This disorder is characterized by the progressive loss of photoreceptor cells and may eventually lead to blindness (70). There are multiple genes when mutated that can cause the RP phenotype (69). More than 100 mutations have been found in this gene, accounting for 15% of all types of retinal degeneration. Studies show that mutations in rhodopsin gene are responsible for ∼25% of autosomal dominant forms of RP (69, 71). Recently, it was reported that the IR signaling pathway was found to be important for cone cell survival and that down-regulation of this pathway has been reported in RP (72). Systemic administration of insulin has shown to delay the cone cell death in RP (72), but the role of PTP1B in RP has not been studied. One of the challenging questions remaining to be answered in the field is how mutations in rhodopsin gene slowly disable and eventually disrupt photoreceptor functions. Our studies suggest that a defect in the photobleaching of rhodopsin/mutations in the rhodopsin gene enhances the activity of PTP1B, and this activated activity could down-regulate the IR signaling pathway. It is tempting to speculate that mutations in rhodopsin gene may trigger the activation of PTP1B, and perhaps reducing or blocking the activation of PTP1B could be beneficial to protect the dying retinal cells in rhodopsin mutants.
In summary, our results demonstrate that PTP1B regulates the state of IR dephosphorylation in rod photoreceptor cells. Deletion of PTP1B results in enhanced IR-mediated signaling pathway in rod photoreceptor cells and attenuates light stress-induced photoreceptor degeneration. Our results also demonstrate that signaling proteins, which are not directly involved in phototransduction, can be regulated by rhodopsin photoexcitation and that rhodopsin may trigger signaling events additional to classic phototransduction.
Acknowledgments
We thank Dr. Yun Le (Dept. of Medicine and Endocrinology, University of Oklahoma Health Sciences Center) for providing rod opsin-Cre mice. The technical assistance of Yu Lee, Dustin Allen, and Ivana Ivanovic is highly acknowledged. We thank Ashok Dilly and Jiangang Wang for help in intravitreal injections.
This work was supported, in whole or in part, by National Institutes of Health Grants EY016507 and EY00871. This work was also supported by NEI, NIH Core Grant P30-EY12190, NCRR COBRE core modules P20-RR17703, and Research to Prevent Blindness Inc.
- IR
- insulin receptor
- PTP1B
- protein-tyrosine phosphatase 1B
- PI3K
- phosphoinositide 3-kinase
- ROS
- rod outer segments
- ONL
- outer nuclear layer
- ERG
- electroretinogram
- RP
- retinitis pigmentosa
- ONH
- optic nerve head.
REFERENCES
- 1.Havrankova J., Roth J., Brownstein M. (1978) Nature 272, 827–829 [DOI] [PubMed] [Google Scholar]
- 2.Takahashi M., Yamada T., Tooyama I., Moroo I., Kimura H., Yamamoto T., Okada H. (1996) Neurosci. Lett. 204, 201–204 [DOI] [PubMed] [Google Scholar]
- 3.Frölich L., Blum-Degen D., Bernstein H. G., Engelsberger S., Humrich J., Laufer S., Muschner D., Thalheimer A., Türk A., Hoyer S., Zöchling R., Boissl K. W., Jellinger K., Riederer P. (1998) J. Neural Transm. 105, 423–438 [DOI] [PubMed] [Google Scholar]
- 4.Barber A. J., Nakamura M., Wolpert E. B., Reiter C. E., Seigel G. M., Antonetti D. A., Gardner T. W. (2001) J. Biol. Chem. 276, 32814–32821 [DOI] [PubMed] [Google Scholar]
- 5.Rajala A., Tanito M., Le Y. Z., Kahn C. R., Rajala R. V. (2008) J. Biol. Chem. 283, 19781–19792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajala R. V., McClellan M. E., Ash J. D., Anderson R. E. (2002) J. Biol. Chem. 277, 43319–43326 [DOI] [PubMed] [Google Scholar]
- 7.Li G., Rajala A., Wiechmann A. F., Anderson R. E., Rajala R. V. (2008) J. Neurochem. 107, 1382–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajala R. V. (2010) J. Lipid Res. 51, 4–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajala A., Anderson R. E., Ma J. X., Lem J., Al-Ubaidi M. R., Rajala R. V. (2007) J. Biol. Chem. 282, 9865–9873 [DOI] [PubMed] [Google Scholar]
- 10.Ahmad F., Azevedo J. L., Cortright R., Dohm G. L., Goldstein B. J. (1997) J. Clin. Invest. 100, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmad F., Considine R. V., Bauer T. L., Ohannesian J. P., Marco C. C., Goldstein B. J. (1997) Metabolism 46, 1140–1145 [DOI] [PubMed] [Google Scholar]
- 12.Ahmad F., Considine R. V., Goldstein B. J. (1995) J. Clin. Invest. 95, 2806–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad F., Goldstein B. J. (1995) Metabolism 44, 1175–1184 [DOI] [PubMed] [Google Scholar]
- 14.Bleyle L. A., Peng Y., Ellis C., Mooney R. A. (1999) Cell. Signal. 11, 719–725 [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto N., Feener E. P., Zhang W. R., Goldstein B. J. (1992) J. Biol. Chem. 267, 13811–13814 [PubMed] [Google Scholar]
- 16.Kenner K. A., Hill D. E., Olefsky J. M., Kusari J. (1993) J. Biol. Chem. 268, 25455–25462 [PubMed] [Google Scholar]
- 17.Kenner K. A., Anyanwu E., Olefsky J. M., Kusari J. (1996) J. Biol. Chem. 271, 19810–19816 [DOI] [PubMed] [Google Scholar]
- 18.Kuhné M. R., Pawson T., Lienhard G. E., Feng G. S. (1993) J. Biol. Chem. 268, 11479–11481 [PubMed] [Google Scholar]
- 19.Maegawa H., Hasegawa M., Sugai S., Obata T., Ugi S., Morino K., Egawa K., Fujita T., Sakamoto T., Nishio Y., Kojima H., Haneda M., Yasuda H., Kikkawa R., Kashiwagi A. (1999) J. Biol. Chem. 274, 30236–30243 [DOI] [PubMed] [Google Scholar]
- 20.Myers M. G., Jr., Mendez R., Shi P., Pierce J. H., Rhoads R., White M. F. (1998) J. Biol. Chem. 273, 26908–26914 [DOI] [PubMed] [Google Scholar]
- 21.Seely B. L., Staubs P. A., Reichart D. R., Berhanu P., Milarski K. L., Saltiel A. R., Kusari J., Olefsky J. M. (1996) Diabetes 45, 1379–1385 [DOI] [PubMed] [Google Scholar]
- 22.Byon J. C., Kusari A. B., Kusari J. (1998) Mol. Cell Biochem. 182, 101–108 [PubMed] [Google Scholar]
- 23.Goldstein B. J., Ahmad F., Ding W., Li P. M., Zhang W. R. (1998) Mol. Cell Biochem. 182, 91–99 [PubMed] [Google Scholar]
- 24.Frangioni J. V., Beahm P. H., Shifrin V., Jost C. A., Neel B. G. (1992) Cell 68, 545–560 [DOI] [PubMed] [Google Scholar]
- 25.Woodford-Thomas T. A., Rhodes J. D., Dixon J. E. (1992) J. Cell Biol. 117, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simoncic P. D., McGlade C. J., Tremblay M. L. (2006) Can. J. Physiol. Pharmacol. 84, 667–675 [DOI] [PubMed] [Google Scholar]
- 27.Tonks N. K. (2003) FEBS Lett. 546, 140–148 [DOI] [PubMed] [Google Scholar]
- 28.Byon J. C., Kenner K. A., Kusari A. B., Kusari J. (1997) Proc. Soc. Exp. Biol. Med. 216, 1–20 [DOI] [PubMed] [Google Scholar]
- 29.Ahmad F., Li P. M., Meyerovitch J., Goldstein B. J. (1995) J. Biol. Chem. 270, 20503–20508 [DOI] [PubMed] [Google Scholar]
- 30.Klaman L. D., Boss O., Peroni O. D., Kim J. K., Martino J. L., Zabolotny J. M., Moghal N., Lubkin M., Kim Y. B., Sharpe A. H., Stricker-Krongrad A., Shulman G. I., Neel B. G., Kahn B. B. (2000) Mol. Cell. Biol. 20, 5479–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A. L., Normandin D., Cheng A., Himms-Hagen J., Chan C. C., Ramachandran C., Gresser M. J., Tremblay M. L., Kennedy B. P. (1999) Science 283, 1544–1548 [DOI] [PubMed] [Google Scholar]
- 32.Delibegovic M., Zimmer D., Kauffman C., Rak K., Hong E. G., Cho Y. R., Kim J. K., Kahn B. B., Neel B. G., Bence K. K. (2009) Diabetes 58, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bence K. K., Delibegovic M., Xue B., Gorgun C. Z., Hotamisligil G. S., Neel B. G., Kahn B. B. (2006) Nat. Med. 12, 917–924 [DOI] [PubMed] [Google Scholar]
- 34.Xue B., Pulinilkunnil T., Murano I., Bence K. K., He H., Minokoshi Y., Asakura K., Lee A., Haj F., Furukawa N., Catalano K. J., Delibegovic M., Balschi J. A., Cinti S., Neel B. G., Kahn B. B. (2009) Mol. Cell. Biol. 29, 4563–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajala R. V., Wiskur B., Tanito M., Callegan M., Rajala A. (2009) Invest. Ophthalmol. Vis. Sci. 50, 1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajala R. V., Chan M. D., Rajala A. (2005) Biochemistry 44, 15461–15471 [DOI] [PubMed] [Google Scholar]
- 37.Redmond T. M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J. X., Crouch R. K., Pfeifer K. (1998) Nat. Genet. 20, 344–351 [DOI] [PubMed] [Google Scholar]
- 38.Taghibiglou C., Rashid-Kolvear F., Van Iderstine S. C., Le-Tien H., Fantus I. G., Lewis G. F., Adeli K. (2002) J. Biol. Chem. 277, 793–803 [DOI] [PubMed] [Google Scholar]
- 39.Li G., Anderson R. E., Tomita H., Adler R., Liu X., Zack D. J., Rajala R. V. (2007) J. Neurosci. 27, 203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanito M., Agbaga M. P., Anderson R. E. (2007) Free Radic. Biol. Med. 42, 1838–1850 [DOI] [PubMed] [Google Scholar]
- 41.Tanito M., Kaidzu S., Anderson R. E. (2007) Invest. Ophthalmol. Vis. Sci. 48, 1864–1872 [DOI] [PubMed] [Google Scholar]
- 42.Naash M. I., Hollyfield J. G., al-Ubaidi M. R., Baehr W. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 5499–5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Y. Z., Zheng L., Zheng W., Ash J. D., Agbaga M. P., Zhu M., Anderson R. E. (2006) Mol. Vis. 12, 389–398 [PubMed] [Google Scholar]
- 44.Rapp L. M., Smith S. C. (1992) Invest. Ophthalmol. Vis. Sci. 33, 3367–3377 [PubMed] [Google Scholar]
- 45.Goldstein B. J. (2001) Curr. Drug Targets Immune Endocr. Metabol. Disord. 1, 265–275 [DOI] [PubMed] [Google Scholar]
- 46.Goldstein B. J. (2002) J. Clin. Endocrinol. Metab. 87, 2474–2480 [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z. Y., Lee S. Y. (2003) Expert. Opin. Investig. Drugs 12, 223–233 [DOI] [PubMed] [Google Scholar]
- 48.Dadke S., Cotteret S., Yip S. C., Jaffer Z. M., Haj F., Ivanov A., Rauscher F., 3rd, Shuai K., Ng T., Neel B. G., Chernoff J. (2007) Nat. Cell Biol. 9, 80–85 [DOI] [PubMed] [Google Scholar]
- 49.Onishi A., Peng G. H., Hsu C., Alexis U., Chen S., Blackshaw S. (2009) Neuron 61, 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klenk C., Humrich J., Quitterer U., Lohse M. J. (2006) J. Biol. Chem. 281, 8357–8364 [DOI] [PubMed] [Google Scholar]
- 51.Ravichandran L. V., Chen H., Li Y., Quon M. J. (2001) Mol. Endocrinol. 15, 1768–1780 [DOI] [PubMed] [Google Scholar]
- 52.Osborne N. N., Li G. Y., Ji D., Mortiboys H. J., Jackson S. (2008) J. Neurochem. 105, 2013–2028 [DOI] [PubMed] [Google Scholar]
- 53.Terman A., Kurz T., Navratil M., Arriaga E., Brunk U. (2010) Antioxid. Redox. Signal. 12, 503–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Z. X., Pervaiz S. (2009) Cell Death. Differ. 17, 408–420 [DOI] [PubMed] [Google Scholar]
- 55.Goldstein B. J., Mahadev K., Kalyankar M., Wu X. (2005) Diabetes 54, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., Barford D. (2003) Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 57.Chen J., Patil S., Seal S., McGinnis J. F. (2006) Nat. Nanotechnol. 1, 142–150 [DOI] [PubMed] [Google Scholar]
- 58.Organisciak D. T., Darrow R. M., Barsalou L., Darrow R. A., Kutty R. K., Kutty G., Wiggert B. (1998) Invest. Ophthalmol. Vis. Sci. 39, 1107–1116 [PubMed] [Google Scholar]
- 59.Krishnamoorthy R. R., Crawford M. J., Chaturvedi M. M., Jain S. K., Aggarwal B. B., Al-Ubaidi M. R., Agarwal N. (1999) J. Biol. Chem. 274, 3734–3743 [DOI] [PubMed] [Google Scholar]
- 60.Datta S. R., Brunet A., Greenberg M. E. (1999) Genes Dev. 13, 2905–2927 [DOI] [PubMed] [Google Scholar]
- 61.Cardone M. H., Roy N., Stennicke H. R., Salvesen G. S., Franke T. F., Stanbridge E., Frisch S., Reed J. C. (1998) Science 282, 1318–1321 [DOI] [PubMed] [Google Scholar]
- 62.Hetman M., Cavanaugh J. E., Kimelman D., Xia Z. (2000) J. Neurosci. 20, 2567–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M. E. (1997) Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 64.Datta K., Franke T. F., Chan T. O., Makris A., Yang S. I., Kaplan D. R., Morrison D. K., Golemis E. A., Tsichlis P. N. (1995) Mol. Cell. Biol. 15, 2304–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kops G. J., de Ruiter N. D., Vries-Smits A. M., Powell D. R., Bos J. L., Burgering B. M. (1999) Nature 398, 630–634 [DOI] [PubMed] [Google Scholar]
- 66.Nakae J., Park B. C., Accili D. (1999) J. Biol. Chem. 274, 15982–15985 [DOI] [PubMed] [Google Scholar]
- 67.Rena G., Guo S., Cichy S. C., Unterman T. G., Cohen P. (1999) J. Biol. Chem. 274, 17179–17183 [DOI] [PubMed] [Google Scholar]
- 68.Wiesmann C., Barr K. J., Kung J., Zhu J., Erlanson D. A., Shen W., Fahr B. J., Zhong M., Taylor L., Randal M., McDowell R. S., Hansen S. K. (2004) Nat. Struct. Mol. Biol. 11, 730–737 [DOI] [PubMed] [Google Scholar]
- 69.Hartong D. T., Berson E. L., Dryja T. P. (2006) Lancet 368, 1795–1809 [DOI] [PubMed] [Google Scholar]
- 70.Farrar G. J., Kenna P. F., Humphries P. (2002) EMBO J. 21, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berson E. L., Rosner B., Sandberg M. A., Dryja T. P. (1991) Arch. Ophthalmol. 109, 92–101 [DOI] [PubMed] [Google Scholar]
- 72.Punzo C., Kornacker K., Cepko C. L. (2009) Nat. Neurosci. 12, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]