

Abstract
This paper investigates the early viral dynamics of foot-and-mouth disease (FMD) within infected pigs. Using an existing within-host model, we investigate whether individual variation can be explained by the effect of the initial dose of FMD virus. To do this, we consider the experimental data on the concentration of FMD virus genomes in the blood (viral load). In this experiment, 12 pigs were inoculated with one of three different doses of FMD virus: low; medium; or high. Measurements of the viral load were recorded over a time course of approximately 11 days for every 8 hours. The model is a set of deterministic differential equations with the following variables: viral load; virus in the interstitial space; and the proportion of epithelial cells available for infection, infected and uninfected. The model was fitted to the data for each animal individually and also simultaneously over all animals varying only the initial dose. We show that the general trend in the data can be explained by varying only the initial dose. The higher the initial dose the earlier the development of a detectable viral load.
Keywords: pig, foot-and-mouth disease, dose, differential equation model, within-host model
1. Introduction
Foot-and-mouth disease (FMD) is an infectious viral disease (genus: Aphthovirus, family: Picornaviridae) that affects cloven-hoofed animals, such as cattle, pigs, sheep and goats. It has an almost worldwide distribution with an endemic and occasionally epidemic occurrence. If an outbreak occurs, it can have a significant economic impact on the areas affected. For example, in the UK, the 2001 FMD outbreak was estimated to have caused a loss of £3.1 billion to the agricultural sector and food chain, with a similar amount lost by the tourism industry (Thompson et al. 2002).
The spread of FMD has already been modelled at different levels, in particular farm-to-farm transmissions and within-farm transmissions. A general discussion of these types of models can be found in Woolhouse (2004). One area of FMD dynamics that has received little attention is a mathematical description of the viral dynamics within a host. An understanding of the within-host viral dynamics can be used to aid the study of FMD spreading within a population, providing more accurate information, such as incubation and infectious periods. Quantitative descriptions of FMD viraemia have already been reported (Cottral & Bachrach 1968; Sutmoller & McVicar 1976; Alexandersen et al. 2001, 2003b; Quan et al. 2004), as well as the clearance of virus from the circulation (Sutmoller & McVicar 1976).
Within-host modelling of viral dynamics has previously been directed at human diseases, in particular human immunodeficiency virus (Perelson et al. 1996, 1997; Corfec et al. 2000; Nowak & May 2000), hepatitis B virus (Herz et al. 1996; Nowak et al. 1997; Nowak & May 2000; Lewin et al. 2001; Perelson 2002; Ciupe et al. 2007), hepatitis C virus (Perelson et al. 2005), influenza A virus (Baccam et al. 2006) and human T-cell leukaemia virus (Wodarz et al. 1999). These models have mostly focused on the chronic nature of the diseases, as well as chemotherapeutic interventions. In the models, the life cycle of the viruses is restricted to the circulation or closely associated organs, and there are no divisions between the circulation and organs. There have been no reports on the interaction of virus in the circulation and virus replication within organs. By contrast, pharmacokinetic models of the movement of drugs between different compartments within the body are well established (Riviere 1999).
In this paper, we examine the relationship between the initial dose of FMD virus (FMDV) injected into the blood and the subsequent within-host dynamics. In particular, we question whether individual variation is mostly due to the effect of the initial dose of FMDV. The basis of this study is data from an experiment where 12 pigs were inoculated with one of three different doses of FMD virus: low; medium; or high. Readings of the viral load were recorded over a time course of up to 11 days for every 8 hours. Our model then adds to the simple inspection of, and the statistical analysis of, the raw data in three important ways. First, the model suggests a possible mechanistic explanation for the observed behaviour. Second, it suggests that the wide range of dynamics seen in the experimental data can be largely explained by the differences solely in the initial dose. Third, the model makes testable predictions that can be investigated in subsequent experiments.
The model which we have chosen to use for this study was developed earlier (Quan 2005) to reflect the current knowledge of FMDV biology. We do not consider here alternative and more speculative models of the within-host dynamics, but show that this simple model can be used to explain the patterns shown in the experimental data by varying only the initial dose while also providing estimates of within-host parameters.
2. Material and methods
A more detailed account of the experiment is documented in Quan et al. (2004) and Quan (2005).
2.1. Experiment overview
Twelve female white Landrace pigs, weighing 20–30 kg, were split into three groups of four animals. The first group was housed in two separate boxes, each box containing two animals that were physically separated from each other by double, solid, watertight, wooden partitions (30 cm apart), so that direct contact between the animals was not possible (Alexandersen et al. 2002a,b). The group was inoculated with a low dose of FMDV (106.1 FMDV genomes).
The second group was treated in the same way as the first except for receiving a medium inoculation (107.1 FMDV genomes).
The third group received a high inoculation (108.1 FMDV genomes), and the pigs were all housed in the same box without any separation between animals. This was due to the limitation of space in the isolation unit. Each animal had serum collected every 8 hours for up to 11 days.
2.2. Isolation unit
The animals were housed in a secure isolation unit located at the Institute for Animal Health, Pirbright Laboratory. The unit consisted of five boxes in a row connected by a ‘clean’ corridor on one side and a ‘dirty’ corridor on the other. The personnel entrance to each box was from the clean corridor through the double doors with a shower unit between the doors. Before leaving the box, the personnel underwent disinfection with FAM (Evans Vanodine International, Preston, UK), followed by showering. Each box was self-contained with its own high-efficiency particulate air filtration, so that cross contamination was not possible.
2.3. Inoculation of animals
The animals were inoculated intravenously in the anterior vena cava/brachiocephalic vein with 0.5 ml diluted inoculum, either 1 in 10, 1 in 100 or 1 in 1000. This was equal to 108.1, 107.1 or 106.1 FMDV genomes, respectively. The inoculum used was isolated from a pig with FMDV strain O UKG 34/01 at Cheale Meats Abattoir, Brentwood, Essex, on 20 February 2001 (Alexandersen & Donaldson 2002; Alexandersen et al. 2002a,b, 2003a,c). The inoculum contained 107.2 TCID50 in the primary bovine thyroid (BTY) cells or 109.4 FMDV genomes ml−1, and was diluted in HEPES tissue culture medium just before use. After inoculation, the dilutions were tested for genome quantity and infectivity using fluorogenic reverse transcription polymerase chain reaction (RT-PCR) and viral titration in the BTY cells.
2.4. Collection of samples
Serum was collected by venipuncture (anterior vena cava/brachiocephalic vein) into plain serum tubes with no additive (Vacutainer; Becton Dickinson, Plymouth, UK). Samples were taken before (to act as controls) and then approximately 5 min after inoculation to estimate the true nature of the initial dose of FMDV. Thereafter, the samples were taken every 8 hours: between 7.00 and 08.00, 15.00 and 16.00, and 23.00 and 24.00.
The virus was taken to be well mixed within the blood after 5 min. Taking the blood volume to be 1/13 of the pig weight (Chang et al. 2002) gives a volume of 1.923 l for a 25 kg pig. The mean cardiac output of a typical pig is 4.5 l min−1 (Åberg et al. 2004). This gives an estimate of (1.923/4.5)×60=25.6 s for the blood to circulate.
2.5. Processing of samples
A detailed description of RNA isolation, reverse transcription, PCR and quantitation of the serum samples from the experiment can be found in the previous work (Quan et al. 2004).
2.6. Model assumptions
The main assumptions in the model are as follows.
— There is a homogeneous mixing within each compartment of the model.
— The rate of exit of virus from each compartment can be approximated as a constant exit rate. This includes all immune-mediated clearance of virus in the blood and virus lost to the epithelial cells. Although the exit rate of virus will vary in practice due to a varying immune response, here we are only modelling the early dynamics of the virus spread.
— In some replicates, there is a clear peak in the viral load. Here, we are more concerned with the dynamics up to this peak than explaining the peak itself. In the model, a constraint on virus replication rates is provided only by reduced availability of cells to infect and not, for example, due to an increasing immune response.
— There is a reduced rate of infection of epithelial cells at low concentrations of virus in the interstitial space, Y. This is consistent with the experimental data from Ohlmann et al. (1995), showing a reduced ability of FMDV to produce viral proteins at low concentrations. To achieve this, we use a term that converges to 0 as Y tends to 0, and converges to 1 as Y increases. We used a standard function in biological modelling, the sigmoidal function Y2/(m2 + Y2), where m is the value at which the infection rate is at half efficiency. The exponent of this function was set to a constant as it did not contribute to fitting the model. The surplus parameter was avoided to obtain the best fit for this simple model against the small dataset available. The value 2 was chosen as this was the most commonly used value.
2.7. The model
The model is a set of differential equations that describe the interactions between the virus concentration in the blood, X, and interstitial space, Y, and the proportion of available epithelial cells for infection that are either infected, F, or uninfected, U. The proportion of cells varies from 0 to 1; to obtain the number of cells either infected or not, the variables U and F must be multiplied by the total number of epithelial cells available for infection. A schematic of the model is shown in figure 1 and the variables X, Y, U and F are defined in table 1. The model differential equations are
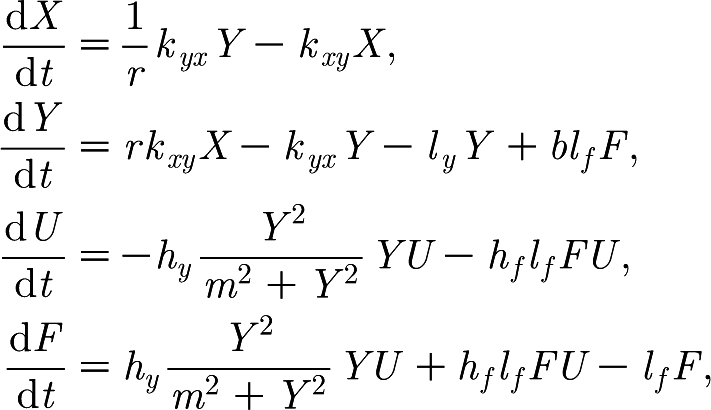 |
where the initial condition X(0) is a fitted parameter, Y(0)=0, U(0)=1 and F(0)=0.
Figure 1.

Compartment model of FMDV within host, where X is the blood; Y is the interstitial space; U is the proportion of available epithelial cells for infection; and F is the proportion of infected epithelial cells. Arrows indicate movement of FMDV genomes or infection of cells.
Table 1.
Description of model variables, parameters and constants. (Note, ‘cell’ unit refers to proportion of available cells for infection.)
| description | units | value | |
|---|---|---|---|
| X | concentration of FMDV in the blood | genomes ml−1 | variable |
| Y | concentration of FMDV in the interstitial space | genomes ml−1 | variable |
| U | proportion of available epithelial cells for infection | cell | variable |
| F | proportion of infected epithelial cells | cell | variable |
| X(0) | initial concentration of FMDV in the blood | genomes ml−1 | parameter |
| Y(0) | initial concentration of FMDV in the interstitial space | genomes ml−1 | 0 |
| U(0) | initial proportion of available epithelial cells for infection | cell | 1 |
| F(0) | initial proportion of infected epithelial cells | cell | 0 |
| ly | rate of loss of FMDV genomes in the interstitial space lost | h−1 | parameter |
| hy | rate uninfected cells are infected per FMDV genome ml−1 serum in the interstitial space | genomes−1 ml h−1 | parameter |
| m | FMDV concentration in the interstitial space at which the rate of cell infection is half the maximum | genomes ml−1 | parameter |
| hf | proportion of cells infected per proportion of burst infected cells | cell−1 | parameter |
| kxy | rate of FMDV genomes in blood lost to the interstitial space | h−1 | parameter |
| kyx | rate of FMDV genomes in interstitial space lost to the blood | h−1 | parameter |
| r | ratio of volume of the blood to interstitial space | 0.53 | |
| lf | rate at which infected cells die | h−1 | 0.33 |
| b | number of FMDV genomes released ml−1 of interstitial space per proportion of burst infected cells | genomes ml−1 cell−1 | parameter |
2.7.1. Constant values.
The ratio of volume of the blood to interstitial space, parameter r, was calculated from the volume of plasma (6.91% body weight) to interstitial space (13.09% body weight) (Pond & Hought 1978). Therefore, parameter r was fixed at a value of 0.53 in all our fits.
The lifespan of an infected cell, lf−1, was calculated as the approximate time for cell attachment and intracellular infection time of FMDV between 2.5 and 3.5 hours (Cartwright et al. 1957). For our fits, we chose to fix lf at 0.33, so that lf−1 is approximately 3.03 hours.
2.7.2. Model dynamics.
Initially, the proportion of infected cells, F(0), is zero. The initial concentration of the FMDV genomes in the interstitial space, Y(0), is also zero. The initial concentration of the FMDV genomes in the blood, X(0), is a non-zero value given by intravenous inoculation, and is treated as a parameter due to the variable nature of the procedure. The virus then spreads as follows.
(i) There is a natural flow in both directions between the concentration of FMDV in the blood, X, and the interstitial space, Y, governed by the parameters kxy, kyx and the volume ratio, r, between the two compartments.
(ii) Once there is a concentration of FMDV genomes in the interstitial space, Y, the proportion of uninfected cells, U, changes to the proportion of infected cells, F, at a rate given by the parameter hy. (The term Y2/(m2 + Y2) acts as a delay mechanism, so that the cell infection rate for low values of Y is very low. The higher the value of m the greater the delay.)
(iii) FMDV is lost from the interstitial space, Y, at a constant rate given by the fitted parameter ly. This is the only exit point from the model of FMDV in the blood or interstitial space and therefore models all removal of viruses from the system. This includes loss into the epithelial cells and any kind of immune-mediated clearance.
(iv) The proportion of infected cells, F, bursts at a constant rate lf. When the cells burst, FMDV genomes are released back into the interstitial space, increasing the concentration of FMDV by b per proportion of total cells. Furthermore, some of the FMDV genomes that are released infect the surrounding uninfected cells at a rate given by the parameter hf.
The modelled dynamics are illustrated in figure 2. Sensitivity analysis of each parameter was investigated by scaling the parameter with different factors and plotting the resultant graphs.
Figure 2.

A graph showing typical behaviour of the model, where the proportion of cells is scaled to a maximum of 1.25×1010 (where parameters ly=20.0, hy=10−11, hf=1.56×1010, m=10, b=4.789×1020, kxy=1.48, kyx=0.26, X(0)=105). Solid line, X; short dashed line, Y; dotted line, U; long dashed line, F.
2.8. Solution of differential equations
The system of differential equations was solved using a fourth-order Runge–Kutta method. An adaptive step-size control was used to ensure the accuracy of the solution, which was necessary due to the rapid changes in the derivative of the solution (Press et al. 1986).
2.9. Model fitting methods
Parameters were estimated using maximum-likelihood methods, assuming lognormal errors in the data and left-censoring observations below a minimum detection threshold of 102 genomes ml−1. The likelihood for the data, D, and parameter set, P, is given by
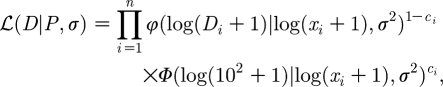 |
where Di is the observed concentration; xi is the expected concentration given by the model; and φ and Φ are the probability and cumulative density functions for a normal distribution, respectively. The number of data points is given by n. The observations are left censored as indicated by ci, where ci is 0 if Di>102 and 1 otherwise. The standard deviation of the measurement error, σ, was unfortunately not available, and so was estimated by considering it as a parameter that must be fitted (Stewart et al. 1998).
The likelihood surface was sampled using Markov chain Monte Carlo with 200 000 iterations after a 200 000-iteration burn-in period (see Gilks et al. 1995). Proposal distributions were given by normal distributions, where the standard deviations were adjusted before the burn-in period to achieve good acceptance rates using a Metropolis–Hastings acceptance rule. The sample with the highest likelihood was taken as the estimate of the maximum likelihood. The Nelder–Mead downhill simplex method (Press et al. 1986) was used to search the parameter space to find suitable regions in the parameter space to initialize the chain. The 95% CI of a parameter was taken as the range between the 2.5 and 97.5 percentiles of the estimated cumulative distribution function of that parameter. The model parameter analysis was calculated on a Unix platform using GNU-complied C++ code.
2.9.1. Model fitting.
The data from each animal were fitted to the model individually. The model was also fitted simultaneously to all the 12 animals, where the parameters are constant across all animals, except for the initial dose of FMDV inoculated, X(0), which was fitted to each animal individually.
The likelihood for the common fit, ℒcom, is therefore given by
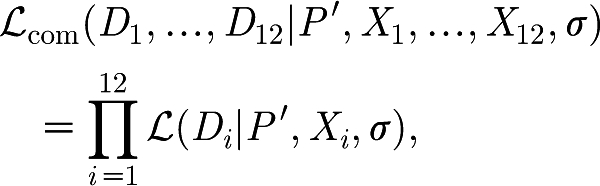 |
where Di and Xi are the data and the initial dose, Xi(0), for animal i, respectively, and P′ denotes the model parameters without the initial dose.
3. Results
In our study, we wished to explore the early within-host dynamics of FMDV in pigs using a within-host model. We can make some observations of the data, as shown in table 2, before considering the model.
Table 2.
The FMDV (log) genomes ml−1 of serum for given experiment times. (a, morning; b, afternoon; c, evening. Inoculation occurred at 0 hours. A ‘n.d.’ indicates that the value was below the detection threshold of 102.)
| low dose (106.1) | medium dose (107.1) | high dose (108.1) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| day | hour | UQ10 | UQ11 | UQ12 | UQ13 | UQ14 | UQ15 | UQ16 | UQ17 | UQ18 | UQ19 | UQ20 | UQ21 |
| 0 | 0.10 | 2.00 | 1.43 | 2.41 | 3.54 | 4.81 | 4.25 | 4.25 | 3.77 | 5.09 | 5.27 | 5.27 | 5.43 |
| 0b | 7.00 | n.d. | n.d. | 1.75 | n.d. | n.d. | n.d. | n.d. | n.d. | 4.75 | 5.02 | n.d. | 3.41 |
| 0c | 15.00 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 3.63 | 4.84 | n.d. | n.d. |
| 1a | 23.50 | n.d. | n.d. | n.d. | n.d. | n.d. | 2.75 | n.d. | n.d. | 7.08 | 6.50 | 4.50 | 3.55 |
| 1b | 31.00 | n.d. | n.d. | n.d. | 2.93 | 2.10 | 3.55 | n.d. | n.d. | 7.08 | 6.65 | 4.41 | 3.62 |
| 1c | 39.00 | n.d. | n.d. | 3.65 | 4.04 | 4.68 | 2.74 | n.d. | n.d. | 7.92 | 6.96 | 5.70 | 5.02 |
| 2a | 47.50 | n.d. | 1.97 | 5.05 | 5.00 | 5.64 | 3.85 | 2.81 | n.d. | 9.00 | 7.44 | 7.59 | 6.79 |
| 2b | 55.00 | n.d. | n.d. | 5.44 | 6.00 | 6.51 | 4.47 | 4.17 | n.d. | 9.59* | 7.65 | 8.37 | 6.79 |
| 2c | 63.00 | 3.59 | n.d. | 6.45 | 6.48 | 7.64 | 4.63 | 4.71 | 3.72 | 8.30 | 8.56 | 6.84 | |
| 3a | 71.50 | n.d. | n.d. | 6.71 | 7.14 | 8.53 | 4.25 | 5.08 | n.d. | 8.66* | 8.31* | 7.52 | |
| 3b | 79.00 | n.d. | n.d. | 6.31 | 7.02 | 8.18 | 4.27 | 5.95 | n.d. | 8.08 | |||
| 3c | 87.00 | 3.38 | n.d. | 6.82 | 7.43 | 8.83 | 3.76 | 6.90 | n.d. | 8.68 | |||
| 4a | 95.50 | 5.14 | n.d. | 7.92 | 7.80 | 8.99 | 3.49 | 7.66 | n.d. | 8.72* | |||
| 4b | 103.00 | 5.68 | n.d. | 8.22 | 7.69 | 7.72 | 5.66 | 7.50 | n.d. | ||||
| 4c | 111.00 | 6.28 | n.d. | 8.67* | 7.11* | 6.92* | 6.14 | 7.61 | n.d. | ||||
| 5a | 119.50 | 6.94 | 3.42 | 6.59 | 7.74* | n.d. | |||||||
| 6a | 143.50 | 8.22 | n.d. | 7.24 | 5.30 | ||||||||
| 7a | 167.50 | 6.95 | n.d. | 5.99 | 7.48 | ||||||||
| 8a | 191.50 | 4.07 | 5.02 | 4.34 | 8.09* | ||||||||
| 9a | 215.50 | 3.63 | 6.01 | n.d. | |||||||||
| 10a | 239.50 | n.d. | 6.73 | n.d. | |||||||||
| 11a | 263.50 | n.d.* | 6.43* | n.d.* | |||||||||
The last reading before the animal died or was killed.
The reading recorded at 0.1 hours gives an indication of the effective dose that each animal received, since this was taken shortly after inoculation. First, consider the low-dose animals, UQ10–UQ13. Animal UQ11 had a smaller reading at time 0.1 hours, which subsequently has a late rise in the viral load. Second, in the medium-dose animals, UQ14–UQ17, animal UQ17 has a lower reading at time 0.1 hours than any other animals, which again shows a late rise in the viral load. Interestingly, this initial value is still higher than all the readings from the low-dose animals. The high-dose animals, UQ18–UQ21, all have high readings at time 0.1 hours.
The early, smaller peak in the data for the animal UQ15 indicates an early round of viral replication and is frequently seen in naturally infected animals. In this case, FMDV replicates locally before disseminating systematically. In this animal, it is possible that epithelial cells were infected directly when the needle was pushed and pulled out of the skin. However, we do not attempt to model this feature with our simple model, which aims to still capture the basic dynamics.
The viral load readings could not be reliably detected for the values below a threshold of 102 FMDV genomes ml−1. This creates some uncertainty in the data, in particular with animals UQ11 and UQ17. The viral load readings for these animals are mostly low; therefore, the dynamics of the virus reproduction are mostly unobservable. The best-fit parameters in terms of the maximum likelihood for UQ11 are perhaps unrealistic, having a large delay in effective virus production and an ineffective clearance of virus after the peak. This fit is due to two factors in the data: first, the large number of values below the threshold of 102, including the initial value; second, the high later values with no clear peak. The model fit of UQ17 shows a delayed peak, which is likely to be the true behaviour of UQ11, which is supported by the model fitted simultaneously to all datasets.
3.1. Model fits
The viral load measurements for each animal (UQ10–UQ21) over the time course of the experiment are reported in table 2, and are also shown graphically in figures 3 and 4.
Figure 3.

Graphs showing the FMDV genomes ml−1 of serum against time (h) for the model fitted individually to each animal. Values below the threshold of 102 FMDV genomes ml−1 are marked with a dash. (a) UQ10, (b) UQ11, (c) UQ12, (d) UQ13, (e) UQ14, (f) UQ15, (g) UQ16, (h) UQ17, (i) UQ18, (j) UQ19, (k) UQ20 and (l) UQ21.
Figure 4.

Graphs showing the FMDV genomes ml−1 of serum against time (h) for the common fit where the initial dose is fitted individually. Values below the threshold of 102 FMDV genomes ml−1 are marked with a dash. (a) UQ10, (b) UQ11, (c) UQ12, (d) UQ13, (e) UQ14, (f) UQ15, (g) UQ16, (h) UQ17, (i) UQ18, (j) UQ19, (k) UQ20 and (l) UQ21.
Unfortunately, there are no data over the full time course of the experiment for every animal. Many animals were killed or died before the end of the experiment. Some animals clearly reached peak viral load: UQ10; UQ14; and UQ15. The data for other animals suggest that the peak viral load may have been reached: UQ13; UQ16; UQ20; and UQ21. The results for the remaining animals are more uncertain.
3.1.1. Model fitted individually.
The best-fit parameter values for each animal are shown in table 3 and the corresponding 95% CIs in figures 5 and 6, calculated as described in §2.9. The resultant viral load curves of the best-fit parameters for each animal are illustrated in figure 3.
Table 3.
Maximum-likelihood estimates of model parameters for the individual fits to each animal (UQ10–UQ21) and the common fit to all animal data. (For the common fit, the parameters were fitted simultaneously to all animals, except for X(0), which was allowed to vary for each animal.)
| animal | ly (h−1) | hy (genomes ml−1) | hf (cell−1) | M (genomes ml−1) |
|---|---|---|---|---|
| UQ10 | 2.07×102 | 4.29×10−10 | 1.38 | 2.55×102 |
| UQ11 | 5.63×10−4 | 5.97×10−8 | 2.39 | 7.04×1013 |
| UQ12 | 5.28×102 | 1.42×10−11 | 8.62×10−1 | 2.83×10−6 |
| UQ13 | 3.27×101 | 1.13×10−10 | 1.65 | 1.33×10−15 |
| UQ14 | 1.39×102 | 1.18×10−11 | 1.98 | 3.99×103 |
| UQ15 | 1.86×102 | 2.08×10−9 | 1.21 | 8.30×104 |
| UQ16 | 2.98×102 | 4.24×10−12 | 1.67 | 1.81×10−16 |
| UQ17 | 6.63×101 | 7.40×10−8 | 2.27 | 7.49×1013 |
| UQ18 | 2.30×102 | 2.23×10−11 | 1.49 | 2.27×10−16 |
| UQ19 | 6.65 | 2.32×10−10 | 5.61×10−1 | 6.70×10−20 |
| UQ20 | 2.99×102 | 6.21×10−9 | 2.14 | 6.65×1011 |
| UQ21 | 3.36×102 | 4.61×10−11 | 1.65 | 6.02×10−11 |
| common | 4.28×102 | 2.39×10−9 | 1.18 | 2.43×101 |
| animal | kxy (h−1) | kyx (h−1) | X(0) (genomes ml−1) | b (genomes ml−1 cell−1) |
| UQ10 | 9.54×101 | 2.12×101 | 4.30×102 | 1.59×1011 |
| UQ11 | 7.13×101 | 3.54 | 1.67×10−40 | 1.94×106 |
| UQ12 | 6.18 | 2.23×101 | 1.76×103 | 5.18×1012 |
| UQ13 | 2.29×101 | 9.45 | 3.69×104 | 4.87×1011 |
| UQ14 | 7.80×101 | 5.44 | 7.42×104 | 2.30×1011 |
| UQ15 | 8.99×101 | 5.20×101 | 2.95×105 | 3.67×1010 |
| UQ16 | 8.34×101 | 1.04×101 | 3.91×104 | 7.88×1010 |
| UQ17 | 3.77×101 | 6.67 | 1.36×102 | 5.46×1011 |
| UQ18 | 8.08×101 | 1.01×101 | 4.86×105 | 3.38×1012 |
| UQ19 | 5.65×101 | 1.34×101 | 2.54×105 | 2.70×1010 |
| UQ20 | 2.77×101 | 9.32×101 | 8.06×107 | 9.36×1011 |
| UQ21 | 2.32×101 | 6.95×10−1 | 7.52×104 | 5.47×1010 |
| common | 5.19×101 | 4.58 | 5.83×1010 |
Figure 5.

Initial dose, X(0) genomes ml−1 and 95% CIs for each animal UQ_. (a) Individual fits and (b) common fit. Animals UQ11 and UQ17 have lower bounds that are almost zero for the individual fit and UQ11 is almost zero for the common fit.
Figure 6.

Parameter 95% CIs for parameter estimates for each animal UQ_, and the common fit denoted by ‘com’. (a) Interstitial space virus clearance rate, ly (h−1), (b) interstitial-to-cell infection rate, hy (genomes−1 ml h−1), (c) cell-to-cell infection, hf (cell−1), (d) reduced infection, m (genomes ml−1), (e) blood-to-interstitial space rate, kxy (h−1), (f) interstitial space-to-blood rate, kyx (h−1) and (g) virus released cell−1 (proportion of total), b (genomes ml−1 cell−1).
3.1.2. Common fit model.
The model was fitted to all the 12 animals simultaneously, where the initial dose of FMDV was fitted to each animal, as described in §2.9. The common maximum-likelihood parameter estimates found are listed in table 3.
The fitted initial values of X for each animal are shown in figure 5b. The 95% CIs are illustrated with the individually fitted parameters in figure 6 (rightmost value). The viral load curves for the common fit are shown in figure 4. Note that the general shape of the curves is identical only to the timing of the peak differing.
Although there will be differences between the animals (such as weight), the model fitted over all animals simultaneously shows that the overall trend can be represented quite accurately by only varying the initial dose of FMDV (figure 4). Some compromise must be made between the individual model fits in order to obtain a good overall fit to all datasets. Therefore, the common fit of the model for each animal will not be as accurate as the model fitted individually. Nevertheless, the common fit captures the basic dynamics of each animal correctly, which is discussed further within §3.2.
The two model-fitting approaches were compared using Akaike's ‘An Information Criterion’ with the second-order information correction (AICc) (Sugiura 1978; Hurvich & Chih-Ling 1989, 1995), showing that the common fit model is the better approach, because it is using only 20 parameters compared with 96 parameters of the individual fits. This test accounts for the large difference in the number of parameters, as well as considering the small amount of data. (The individual fits have an AICc value of 825.7 and the common fit a value of 640.1.)
3.2. Model parameter analysis
In this section, we consider each model parameter in turn: the role they have in the model dynamics and how they differ when fitted to different animals. The manner in which the model dynamics alter when a parameter is scaled is shown in figure 7.
Figure 7.

Graphs of the variation in the model dynamics when a parameter is multiplied by a factor. Parameters are given by the common fit with ly=20 and X(0)=1448 (table 3). (a) Blood-to-interstitial space rate, kxy (h−1), (b) interstitial space-to-blood rate, kyx (h−1), (c) blood-to-interstitial space volume ratio, r, (d) interstitial space virus clearance rate, ly (h−1), (e) interstitial-to-cell infection, hy (genomes ml−1 h−1), (f) cell-to-cell infection, hf (cell−1), (g) reduced infection, m (genomes ml−1), (h) rate infected cells die, lf (h−1), (i) virus released cell−1 (proportion of total), b (genomes ml−1 cell−1), (j) initial dose, X(0) (genomes ml−1) and (k) key of scaling factors.
3.2.1. Flow between interstitial and blood compartments, kxy, kyx (h−1) and r.
If the flow of virus is decreased from the blood (kxy) to the interstitial space then few viruses are able to infect the cells creating a delay in the virus production. As more viruses are retained in the blood, an increase in the virus in the blood is also observed (figure 7a). Increasing the virus flow in the other direction (kyx) increases the virus in the blood since few viruses are cleared via the interstitial space. This then increases the infection rate of the cells as the virus re-enters the interstitial space (figure 7b).
The model may be simplified since the variables for the blood, X, and the interstitial space, Y, are in equilibrium with one another, related by the equation 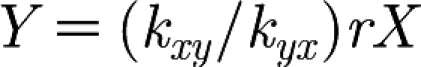 , although the results presented here refer to the full model (see appendix A). The sensitivity of changing the ratio between the blood and the interstitial space, given by constant r, is shown in figure 7c. Parameters kxy and kyx, which are fitted, can account for any variability in r. Therefore, we are not too concerned with the individual values of kxy, kyx and r.
, although the results presented here refer to the full model (see appendix A). The sensitivity of changing the ratio between the blood and the interstitial space, given by constant r, is shown in figure 7c. Parameters kxy and kyx, which are fitted, can account for any variability in r. Therefore, we are not too concerned with the individual values of kxy, kyx and r.
3.2.2. Exit rate of FMDV genomes, ly (h−1).
The parameter ly controls the exit of virus from the interstitial space and therefore also controls the exit of virus from the blood. This parameter has two main roles: first to create the initial dip in the viral load, and second to create a decrease in the viral load after the peak.
As the value of ly is increased, the virus in the interstitial space is cleared quicker delaying the production of virus within the proportion of cells. The magnitude of the peak of the viral load also decreases since the rate at which virus is produced within the proportion of cells is met sooner by the clearance rate (figure 7d). For all of the individual fits, except UQ11 and UQ19, the upper bound of the 95% CI is unbounded. This is due to the exit rate to the blood, kyx, also being a variable and can be set as high as required, so that all of the viruses are not lost through clearance. Unfortunately, this means that only a lower bound is found for ly for these animals, showing that an almost instant clearance of virus from the interstitial space cannot be ruled out. The exit rate for the model fit of UQ11 is virtually zero, shown by the flat peak. The model fit of UQ19 relies less on virus production via the interstitial space and is therefore limited in magnitude.
3.2.3. Interstitial space infection rate, hy (genomes−1 ml h−1).
The parameter hy controls the rate at which the proportion of cells becomes infected by the free virion population in the interstitial space. As the value of hy is decreased the production of virus is delayed (figure 7e), and if it were set to zero the virus would quickly disappear. Since the virus produced from the proportion of cells is returned to the interstitial space, this creates a positive feedback loop and the virus increases exponentially in the proportion of cells and the interstitial space. Lower values of hy create a larger initial dip and delay the onset of virus production (as mentioned above for the animal UQ14).
We can calculate typical values for the interstitial space infection rate of the proportion of available epithelial cells for infection, given by the expression hy(Y2/(m2 + Y2))YU, using the common fit model 24 hours before the peak in the viral load, at the peak and 24 hours after the peak, as shown in table 3 (animal UQ10 was used with a peak time of 138.64 hours). Since these values are for the proportion of available epithelial cells for infection, these values must be multiplied by the total number of available epithelial cells for infection to obtain values of the number of individual cells infected per hour.
3.2.4. Cell-to-cell infection, hf (cell−1).
The parameter hf controls the rate at which the proportion of infected cells infects the proportion of available uninfected cells. Increasing hf causes the proportion of available uninfected cells to become infected quicker and so exhausting the supply of cells to infect sooner (figure 3f). If this parameter is set to zero, then cell-to-cell infection cannot occur. Therefore, the rate of infection of the proportion of cells by free virions in the interstitial space (hy) must be sufficiently high to ensure that enough of the cells become infected to increase virus in the system. If hy is too low, then the virus exits the system via the interstitial space quicker than it can be produced and so decreases to zero. The proportion of infected cells, F, increases exponentially provided that cells first become infected from the interstitial space, and, second, the proportion of available cells for infection does not decrease too low.
As above, we calculate typical values for the cell-to-cell infection rate of the proportion of available epithelial cells for infection, given by the expression, hf lf FU, as shown in table 3. The overall infection rate given by the first two terms of dF/dt, (hy(Y2/(m2 + Y2))YU + hflfFU), is given in table 4. This shows that infection from the interstitial space contributes 50–60 per cent.
Table 4.
Infection rate of the proportion of available epithelial cells for infection evaluated from the common fit model using animal UQ10 with a peak time of 138.64 hours. (The table shows the infection rate 24 hours before the peak, at peak and 24 hours after the peak.)
| time since inoculation | infection rate (cell h−1) | 95% CI | percentage of total cell infection |
|---|---|---|---|
| interstitial space infection rates | |||
| 24 hours before peak | 6.78×10−4 | (9.04×10−5, 6.35×10−3) | 50.0 |
| at peak | 3.36×10−2 | (4.39×10−3, 3.51×10−2) | 58.9 |
| 24 hours after peak | 1.03×10−3 | (1.35×10−4, 3.51×10−2) | 59.2 |
| cell-to-cell infection rates | |||
| 24 hours before peak | 6.79×10−4 | (1.14×10−4, 6.66×10−3) | 50.0 |
| at peak | 2.34×10−2 | (5.71×10−3, 2.58×10−2) | 41.1 |
| 24 hours after peak | 7.09×10−4 | (1.60×10−4, 3.69×10−3) | 40.8 |
| overall infection rates of the proportion of cells available for infection | |||
| 24 hours before peak | 1.36×10−3 | (2.18×10−4, 1.27×10−2) | 100.0 |
| at peak | 5.70×10−2 | (1.06×10−2, 6.00×10−2) | 100.0 |
| 24 hours after peak | 1.74×10−3 | (3.15×10−4, 6.95×10−3) | 100.0 |
3.2.5. Reduced infectious ability, m (genomes ml−1).
The parameter m represents a virus level below which there is a much reduced infectious ability, delaying the production of virus in the cell system (figure 7g). The maximum value of the 95% CIs of m for each of the high-dose animals is similar to one another. This is due to all of these datasets having few or no values below the detection threshold of 102 genomes ml−1 (figure 6). Since virus levels in the high-dose animals are high, the reduced infectious term, Y2/(m2 + Y2), has less of an effect than it does for lower dose animals with lower virus levels. This is why higher doses have a larger confidence interval for m.
The common fit model shows that m is a necessary parameter, and hence justifies the inclusion of the term Y2/(m2 + Y2), having a lower 95% CI greater than zero. The value of m for the common fit model enables the correct dynamics to be displayed for all animals by only varying the initial condition. The parameter, m (genomes ml−1), is estimated by the 95% CI for the common fit model: (2.11×10−1, 1.68×102).
3.2.6. Rate infected cells die, lf (h−1).
The sensitivity to the constant lf apparent in figure 7h is considerable, but these graphs correspond to cell attachment and intracellular infection times of 24.24, 12.12, 6.06, 1.52, 0.76 and 0.38 hours for (1/8)lf, (1/4)lf, (1/2)lf, 2lf, 4lf and 8lf, respectively. These times are outside the calculated approximate time of between 2.5 and 3.5 hours.
3.2.7. Released FMDV genomes, b (genomes ml−1cell−1).
When the amount of virus released per proportion of cells is increased, the infection rate of the proportion of cells also increases, consequently, exhausting the available cells for infection more quickly (figure 7i).
3.2.8. Initial dose, X(0) (genomes ml−1).
Decreasing the initial dose delays the production of virus, causing a viral load peak of the same magnitude later (figure 7j).
4. Discussion
The parameter X(0) controls the initial quantity of virus in the system. A higher initial concentration of FMDV in the blood results in an earlier peak of virus in both the data and in the model. The model peak always has the same dynamics for any value of X(0), which can be seen clearly from the common fit graphs in figures 4 and 7d. From the 95% CI plots of X(0) from the individual fits, as shown in figure 5a, we can see that this fitted parameter is in good agreement with the initial dose given to each animal. In particular, it reflects that animals UQ10–UQ13 were given a low dose, animals UQ14–UQ17 a medium dose and animals UQ18–UQ21 a high dose. This is the only parameter that seems to reflect the fact that different animals were given different doses. From the 95% CI plots of X(0) from the common fit model, as shown in figure 5b, we also see the relationship with the initial dose. Although there is no clear difference between the low- and medium-dose animals, the timing of the peaks is similar.
From the graphs in figure 4, it appears that the model predicted peak values of X are identical for each animal. When the peak value is calculated for each animal, we see that all the peak values are equal at 1.79×108 FMDV genomes ml−1. Unfortunately, it is not possible to write down an analytical solution for the peak value of X in terms of the given parameter set, since we must solve the system of differential equations numerically. However, in all the analyses performed in producing the best-fit common parameters, the peak value has always been the same across all animals.
In conclusion, although there is some variation in the dynamics of virus spread between animals, there is a general pattern that can be explained in terms of the initial dose of FMDV in the blood. The model predicts that, when the initial dose is varied, only the timing of the virus spreading is varied, with the dynamics remaining the same: the higher the initial dose the earlier the development of detectable viral load. This was shown by fitting the model to all 12 animal datasets only allowing the initial dose to vary between animals, as shown in figure 4.
The use of a model was important in this exercise providing estimates for missing data and providing a mechanistic explanation for the observed data. Furthermore, estimates for the parameters governing the within-host dynamics of FMDV were obtained. Some of the authors are currently involved in the ongoing experimental work to estimate the infectious and incubation periods of animals with FMDV, together with the implications for vaccines, an important component of this is the study of within-host dynamics.
Acknowledgements
Animal experimentation was approved by the Institute for Animal Health (IAH) ethical review board under the authority of a Home Office project licence in accordance to the Home Office Guidance on the Operation of the Animals (Scientific Procedures) Act of 1986 and associated guidelines.We would like to thank the BBRSC for funding this research.
Appendix A. Central compartment and interstitial space
In the model, the blood, X, and interstitial space, Y, are connected by a constant flow from X to Y with rate kxy. Similarly, there is a constant flow from Y to X with rate kyx, where r acts as a conversion factor for the concentration of FMDV between the different-sized spaces. Considering only the interactions between X and Y we have the following:
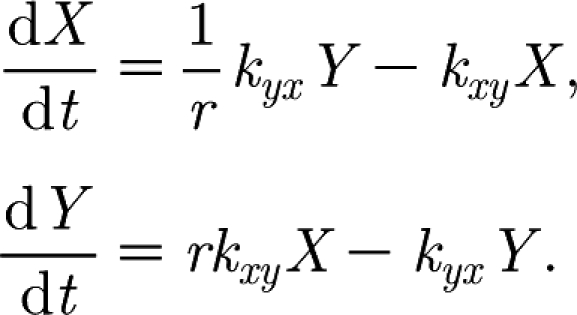 |
These equations have a set of steady states (i.e. X and Y remain constant when dx/dt and dy/dt equals zero) given by the line 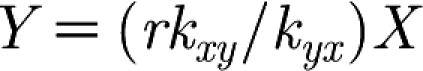 . It is easily shown that this set of steady states is stable. The equations have the following solution:
. It is easily shown that this set of steady states is stable. The equations have the following solution:
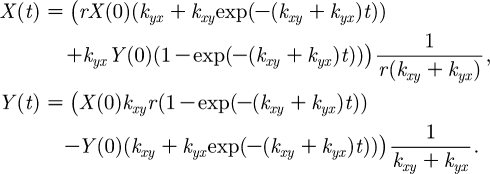 |
Therefore, as t→∞,  and
and 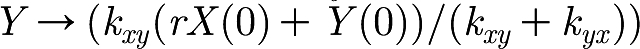 . Furthermore, since
. Furthermore, since 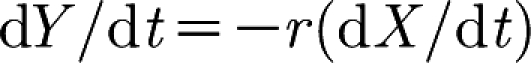 , we know that X and Y converge exponentially fast towards the stationary point (which is on the line kyxY=rkxyX) along the line Y=r(X(0)−X)+Y(0).
, we know that X and Y converge exponentially fast towards the stationary point (which is on the line kyxY=rkxyX) along the line Y=r(X(0)−X)+Y(0).
When we consider X and Y in the model, we observe that Y has extra interactions that increase and decrease the value of Y. However, the interactions of Y with X remain the same; therefore, when Y changes, X converges exponentially fast to the corresponding value on the line 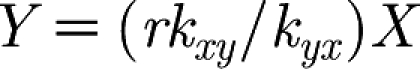 . This can be observed in figure 2, where the values of X and Y track one another.
. This can be observed in figure 2, where the values of X and Y track one another.
From these observations of X and Y, we can conclude that it would be sufficient to model X and Y with only one variable. For example, we can drop the variable X from the model and achieve the same behaviour. The initial values of Y are then given by the original model initial values of X as  .
.
References
- Åberg A.-M., Hultin M., Abrahamsson P., Larsson J.E. 2004. Circulatory effects and kinetics following acute administration of carbon monoxide in a porcine model. Life Sci. 75, 1029–1039. 10.1016/j.lfs.2003.12.030. [DOI] [PubMed] [Google Scholar]
- Alexandersen S., Donaldson A.I. 2002. Further studies to quantify the dose of natural aerosols of foot-and-mouth disease virus for pigs. Epidemiol. Infect. 128, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandersen S., Oleksiewicz M.B., Donaldson A.I. 2001. The early pathogenesis of foot-and-mouth disease in pigs infected by contact: a quantitative time-course study using TaqMan RT-PCR. J. Gen. Virol. 82, 747–755. [DOI] [PubMed] [Google Scholar]
- Alexandersen S., Brotherhood I., Donaldson A.I.Natural aerosol transmission of foot-and-mouth disease virus to pigs: minimal infectious dose for strain O1 lausanne. Epidemiol. Infect. 128, 2002a301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandersen S., Zhang Z., Donaldson A.I.Aspects of the persistence of foot-and-mouth disease virus in animals—the carrier problem. Microbes Infect. 4, 2002b1099–1110. 10.1016/S1286-4579(02)01634-9. [DOI] [PubMed] [Google Scholar]
- Alexandersen S., Zhang Z., Donaldson A.I., Garland A.J.The pathogenesis and diagnosis of foot-and-mouth disease. J. Comp. Pathol. 129, 2003a1–36. 10.1016/S0021-9975(03)00041-0. [DOI] [PubMed] [Google Scholar]
- Alexandersen S., Zhang Z., Reid S.M., Hutchings G.H., Donaldson A.I.Studies of quantitative parameters of virus excretion and transmission in pigs and cattle experimentally infected with foot-and-mouth disease virus. J. Comp. Pathol. 129, 2003b268–282. 10.1016/S0021-9975(03)00045-8. [DOI] [PubMed] [Google Scholar]
- Alexandersen S., Kitching R.P., Mansley L.M., Donaldson A.I.Clinical and laboratory investigations of five outbreaks of foot-and-mouth disease during the 2001 epidemic in the United Kingdom. Vet. Rec. 152, 2003c489–496. [DOI] [PubMed] [Google Scholar]
- Baccam P., Beauchemin C., Macken C.A., Hayden F.G., Perelson A.S. 2006. Kinetics of influenza A virus infection in humans. J. Virol. 80, 7590–7599. 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright S.F., Pay T.W.F., Henderson W.M. 1957. Multiplication of the virus of foot and mouth disease virus in culture. J. Gen. Mircobiol. 16, 730–748. [DOI] [PubMed] [Google Scholar]
- Chang K., et al. 2002. Effective generation of transgenic pigs and mice by linker based sperm-mediated gene transfer. BMC Biotechnol. 2, 5. 10.1186/1472-6750-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciupe S.M., Ribeiro R.M., Nelson P.W., Dusheiko G., Perelson A.S. 2007. The role of cells refractory to productive infection in acute hepatitis B viral dynamics. Proc. Natl Acad. Sci. USA. 104, 5050–5055. 10.1073/pnas.0603626104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corfec E.L., Rouzioux C., Costagliola D. 2000. HIV-1 quantitative dynamics in vivo: a review of mathematical models. Revue d'Epidemiologie et de Sante Publique. 48, 168–181. [PubMed] [Google Scholar]
- Cottral G. E., Bachrach H. L. 1968. Foot-and-mouth disease viraemia. In Proc. Annual Meeting of the United States Animal Health Association, vol. 72, pp. 383–399. [PubMed] [Google Scholar]
- Gilks W., Richardson S., Spiegelhalter D.J. 1995New York, NY:Chapman and Hall [Google Scholar]
- Herz V.A., Bonhoeffer S., Anderson R.M., May R.M., Nowak M.A. 1996. Viral dynamics in vivo: limitations on estimates of intracellar delay and virus decay. Proc. Natl Acad. Sci. USA. 93, 7247–7251. 10.1073/pnas.93.14.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurvich C.M., Chih-Ling T. 1989. Regression and time series model selection in small samples. Biometrika. 76, 297–307. 10.1093/biomet/76.2.297. [DOI] [Google Scholar]
- Hurvich C.M., Chih-Ling T. 1995. Model selection for extended quasi-likelihood models in small samples. Biometrics. 51, 1077–1084. 10.2307/2533006. [DOI] [PubMed] [Google Scholar]
- Lewin S.R., Ribeiro R.M., Walters T., Lau G.K., Bowden S., Locarnini S., Perelson A.S. 2001. Analysis of hepatitis B viral load decline under potent therapy: complex decay profiles observed. Hepatology. 34, 1012–1020. 10.1053/jhep.2001.28509. [DOI] [PubMed] [Google Scholar]
- Nowak M.A., May R.M.Virus dynamics: mathematical principles of immunology and virology. In Oxford University Press 2000Oxford, UK:Oxford University Press [Google Scholar]
- Nowak M.A., et al. 1997. Viral dynamics of primary viraemia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71, 7518–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmann T., Rau M., Morley S.J., Pain V.M. 1995. Proteolytic cleavage of initiation factor eIF-4 gamma in the reticulocyte lysate inhibits translation of capped mRNAs but enhances that of uncapped mRNAs. Nucleic Acids Res. 23, 334–340. 10.1093/nar/23.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelson A.S. 2002. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2, 28–36. 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- Perelson A.S., Neumann A.U., Markowitz M., Leonard J.M., Ho D.D. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 271, 1582–1586. 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Perelson A.S., Essunger P., Cao Y., Vesanen M., Hurley A., Saksela K., Markowitz M., Ho D.D. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 387, 188–191. 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- Perelson A.S., Herrmann E., Micol F., Zeuzem S. 2005. New kinetic models for the hepatitis C virus. Hepatology. 42, 749–754. 10.1002/hep.20882. [DOI] [PubMed] [Google Scholar]
- Pond W.G., Hought K.A.The biology of the pig. In Cornell University Press 1978Ithaca, NY:Cornell University Press [Google Scholar]
- Press W.H., Flannery B.P., Teukolsky S.A., Vetterling W.T.Numerical recipes: the art of scientific computing. In Cambridge University Press 1986Cambridge, UK:Cambridge University Press [Google Scholar]
- Quan M. 2005 Quantitative dynamics of foot-and-mouth disease virus infection in pigs. PhD thesis, University of Edinburgh.
- Quan M., Murphy C.M., Zhang Z., Alexandersen S. 2004. Determinants of early foot-and-mouth disease virus dynamics in pigs. J. Comp. Pathol. 131, 294–307. 10.1016/j.jcpa.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Riviere J.E.Comparative pharmacokinetics: principles, techniques, and applications. In Iowa State University Press 1999Ames, IA:Iowa State University Press [Google Scholar]
- Stewart W.E., Shon Y., Box G.E.P. 1998. Discrimination and goodness of fit of multiresponse mechanistic models. AIChE J. 44, 1404–1412. 10.1002/aic.690440618. [DOI] [Google Scholar]
- Sugiura N. 1978. Further analysis of the data by Akaike's information criterion and the finite corrections. Commun. Stat. Theory Methods A. 7, 13–26. [Google Scholar]
- Sutmoller P., McVicar J.W. 1976. Pathogensis of foot-and-mouth disease: clearance of the virus from the circulation of cattle and goats during experimental viraemia. J. Hyg. 77, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D., Muriel P., Russel D., Osbourne P., Bromley A., Rowland M., Creigh-Tyte M., Brown S. 2002. Economic costs of the foot and mouth disease outbreak in the United Kingdom in 2001. Revue Scientifique et Technique, Office International des Epizooties. 21, 675–687. [DOI] [PubMed] [Google Scholar]
- Wodarz D., Nowak M.A., Bangham C.R. 1999. The dynamics of HTLV-I and the CTL response. Immunol. Today. 20, 220–227. 10.1016/S0167-5699(99)01446-2. [DOI] [PubMed] [Google Scholar]
- Woolhouse M.E.Foot and mouth disease: current perspectives. In Mathematical models of the epidemiology and control of foot-and-mouth disease Sobrino F., Domingo E. 2004. pp. 355–381. Eds. Wymondham, UK:Horizon Bioscience; ch. 13. [Google Scholar]