

Abstract
Nickel compounds are carcinogenic to humans, possibly through induction of reactive oxygen species (ROS) that damage macromolecules including DNA and proteins. The aim of the present study is to elucidate the role of the ROS-mediated Akt/apoptosis-regulating signal kinase (ASK) 1/p38 pathway in nickel-induced apoptosis. Exposure of human bronchial epithelial cells (BEAS-2B) to nickel compounds induced the generation of ROS and activation of Akt that is associated with the activation of ASK1 and p38 mitogen-activated protein kinase. Immunoblotting suggested a down-regulation of several antiapoptotic proteins, including Bcl-2 and Bcl-xL in the nickel compound-treated cells. Indeed, a notable cell apoptosis following nickel compound treatment is evident as revealed by flow cytometry analysis. N-Acetyl-l-cysteine (NAC, a general antioxidant) and vitamin E or catalase (a specific H2O2 inhibitor) all decreased nickel-induced ROS generation. Scavenging of nickel-induced ROS by NAC or catalase attenuated Akt, ASK1, and p38 MAPK activation and apoptosis, which implies involvement of ROS in the Akt/ASK1/p38 pathway. In addition, nickel-induced activation of p38 MAPK was attenuated by a small interference of RNA specific to ASK1 (siRNA ASK1), implying that p38 MAPK was downstream of ASK1, while ASK1 activation was not reversely regulated by the inhibition of p38 MAPK by SB203580, a widely used p38 MAPK inhibitor. Silencing Akt by siRNA reduced the activation of ASK1 and p38 MAPK and cell apoptosis, whereas without nickel stimulation, siRNA Akt had no effect on the activation of ASK1 and p38 MAPK. Thus, these results suggest that the ROS-dependent Akt-ASK1-p38 axis is important for nickel-induced apoptosis.
Introduction
Nickel is a toxic transition metal and is widely used in many industries, including electroplating and the manufacture of steel, some special alloys, batteries, and electronic devices. Epidemiological studies have indicated that chronic occupational exposure to nickel (Ni) compounds increases the incidence of certain human cancers, such as lung and nasal cancers (1,2). Nickel exposure-induced generation of reactive oxygen species (ROS) has been considered a pivotal step in nickel-induced carcinogenesis (3). Recent studies also show that ROS production induced by nickel exposure is involved in nickel-induced apoptosis (4,5). Apoptosis is originally viewed as a normal physiologic process, removing cells carrying abnormal genetic information to maintain the functional integrity of the cell populations. In the case of metal-induced apoptosis, in contrast, it might allow the escape of cells with potentially carcinogenic ability from apoptosis under certain conditions, due to an abnormal apoptotic response (6). Therefore, investigation of nickel-induced apoptosis is necessary to understand the overall mechanism of nickel-induced carcinogenesis. One of the possible mechanisms of nickel-induced apoptosis and carcinogenesis is the induction of ROS by nickel compounds.
ROS, such as superoxide anion (O2•−), hydrogen peroxide (H2O2), and the hydroxyl radical (HO•), have numerous effects on essential biological processes, including normal cell growth, induction and maintenance of the transformed state, programmed cell death, and cellular senescence (7). An aberrant increase in the level of ROS may result in transient or permanent cellular alterations, such as irreversible oxidative damage on DNA, causing genomic instability and the consequent malignant transformation of the cells (8). Thus, ROS are thought to play multiple roles in tumor initiation, progression, and maintenance. Various studies have demonstrated that ROS induced by toxic metals are critical in metal-induced apoptosis and carcinogenesis (4,6,9).
ROS generation has been shown to be involved in the Akt (protein kinase B) signaling pathway (10,11). Akt serine/threonine kinases are one of the essential regulators of cell survival function in response to growth factor stimulations (12,13). It is generally believed that Akt kinases are antiapoptotic through phosphorylation and inhibition of a number of apoptosis regulatory proteins (14). Apoptosis signal-regulating kinase 1 (ASK1) has been reported to be phosphorylated by Akt at serine 83 (Ser83) and its activity reduced (15−17). ASK1 is a serine-threonine kinase that was initially discovered as a mitogen-activated protein kinase kinase kinase (MAPKKK) in the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 MAPK signaling cascades (18,19). A variety of stress-related stimuli activate ASK1. These stimuli include serum or trophic factor withdrawal, TNF-α, ROS, microtubule-interfering agents, and genotoxic stress. Some of these stress signals induce Thr838 (corresponding to Thr845 in mice) phosphorylation and activation of the ASK1 (20,21). Activated ASK1 will result in activation of the downstream kinases, leading to cell apoptosis (18,19,22).
To date, the majority of research on Akt has focused on its role in cell growth promotion. Little is known about its function in cell apoptosis. The present study demonstrates that nickel-induced generation of ROS activated Akt, which activated ASK1 through Thr838 phosphorylation, leading to downstream activation of p38 MAPK, eventually causing cell apoptosis.
Materials and Methods
Cell Culture and Other Reagents
Human bronchial epithelial cells cells (BEAS-2B) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Calbiochem) supplemented with 10% fetal bovine serum (FBS), 5% penicillin/streptomycin, and 2 mM l-glutamine (Invitrogen) at 37 °C in a humidified atmosphere with 5% CO2. Nickel subsulfide (Ni3S2), N-acetyl-l-cysteine (NAC), and vitamin E (α-tocopherol) were purchased from Sigma; catalase was from Roche Applied Science Co. Dihydroethidium (DHE) and 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) were from Invitrogen (Eugene, OR). Antibodies against Akt, phospho-JNK, JNK, and β-actin were purchased from Santa Cruz Biotechnology. Bcl-xL, phospho-Akt specific for Ser473 phosphorylation, phospho-ASK1 specific for Thr838 phosphorylation, phospho-ASK1 specific for Ser83 phosphorylation, ASK1, phospho-p38, and p38 were purchased from Cell Signaling. Bcl-2 was purchased from DAKO, anticatalase antibody was from Novus Biologicals, Inc., and anti-Cu/Zn SOD and anti-MnSOD antibody were from Upstate Biotechnology. All primary antibodies were diluted at 1:1000, except 1:2000 for actin and 1:200 for phospho-p38, and secondary antibodies were diluted at 1:4000.
Determination of ROS Production
ROS were detected by staining the cells with DHE or CM-H2DCFDA (Molecular Probes). DHE is oxidized to red fluorescent ethidium by O2•−, and CM-H2DCFDA is oxidized to green fluorescent DCF (dichlorofluorescein) by H2O2(23). Cells were loaded with 10 μM DHE and 5 μM CM-H2DCFDA for 30 min, respectively, at 37 °C, and 5% CO2 in PBS and then were washed with PBS and returned to media for a 30 min recovery period. The mean fluorescence intensity was determined as ROS generation by flow cytometry FACS Calibur (BD Bioscience, San Jose, CA). Quantitative assay of ROS generation was performed by normalization to the control group.
Cell Counting Assay
Cell counting was performed to assess the inhibitory effect of Ni3S2 on cell proliferation. BEAS-2B cells were seeded in each well of six well plates overnight and then treated without or with various doses of Ni3S2 for 72 h or with Ni3S2 2 μg/cm2 for various time points as indicated. After treatments, cells were washed by PBS and trypsinized, and then, cell counting was carried out using BECKMAN COULTER.
Annexin V/Propidium Iodide Assays for Apoptosis
For Annexin V/propidium iodide (PI) assays, BEAS-2B cells were stained with Annexin V-FITC and PI and then evaluated for apoptosis by flow cytometry according to the manufacturer’s protocol (BD Pharmingen). Briefly, 1 × 106 cells were washed twice with cold PBS and stained with 5 μL of Annexin V-FITC and 8 μL of PI (5 μg/mL) in 1 × binding buffer [10 mmol/L HEPES (pH 7.4), 140 mmol/L NaOH, and 2.5 mmol/L CaCl2] for 10 min at room temperature in the dark. The apoptotic cells were determined using a Becton Dickinson FACScan cytofluorometer. Both early apoptotic (Annexin V-positive and PI-negative) and late apoptotic (Annexin V-positive and PI-positive) cells were included in cell death determinations.
Western Blot Assay
Western blot analysis was performed using the NuPAGE Bis-Tris electrophoresis system (Invitrogen). The total cellular samples were washed once with ice cold PBS and lysed in 1 × RIPA buffer supplemented with 50 mmol/L DTT (Fisher Biotech) and then loaded with NuPAGE LDS sample buffer. The protein concentration was determined using Coomassie Protein Assay Reagent (Pierce). The total cellular protein extracts were separated by SDS-PAGE and transferred to nitrocellulose membrane in 20 mmol/L Tris-HCl (pH 8.0) containing 150 mmol/L glycine and 20% (v/v) methanol. Membranes were blocked with 5% fat-free dry milk in 1 × TBS containing 0.05% Tween 20 and incubated with antibodies. Protein bands were detected by incubation with horseradish peroxidase-conjugated antibodies (Kirkegaard and Perry Laboratories) and visualized with enhanced chemiluminescence reagent (Perkin-Elmer Life Sciences). Band densities in the Western blots were all analyzed with AlphaImager HP (Alpha Innotech).
Cell Transfection
The control and specific small interference RNA targeting ASK1 (siRNA ASK1, which inhibits expression of ASK1) was purchased from Santa Cruz Co. siRNA Akt (which inhibits expression of Akt1 and 2, not Akt3) and corresponding siRNA control were purchased from Cell Signaling Co. To block ASK1 or Akt signal, cells were transfected with the indicated siRNA, respectively, using Lipofectamine RNAiMAX from Invitrogen Co. The transfection procedure was followed by the protocol provided by the transfection reagent manufacturer. Briefly, control siRNA and siRNA ASK1 or Akt were incubated with Lipofectamine RNAiMAX in OPTI-MEM I for 30 min at room temperature and then added to cells in maintenance media without antibiotics (the final concentration of both control siRNA and ASK1 or Akt siRNA was 100 nM each). Media were replaced with maintenance media with antibiotics 24 h later after transfection, and then, nickel was added to the media. Experiments were performed approximately 72 h following transfection.
Statistical Analysis
For analysis of apoptosis and cell counting, values were presented as mean ± SEs. Statistical differences between controls and treated groups were determined by one-way ANOVA. Differences were considered statistically significant for P < 0.05.
Results
Nickel Exposure Induces Apoptosis in BEAS-2B Cells
BEAS-2B cells were treated with Ni3S2 for 48 h followed by cell apoptosis analyses using flow cytometry. Cell apoptosis was increased by 11.1, 14.7, and 29.5% at the concentrations of 1, 2, and 4 μg/cm2 of Ni3S2 treatment, respectively, whereas only 4.8% of the control cells were apoptotic (Figure 1A,B). Figure 1C shows that the cell number was also decreased with increased nickel concentration and treatment time, suggesting that cell growth arrest was also induced by nickel treatment. Other studies have shown the inhibitory effect of nickel on cell proliferation through interfering cell cycle progression. Ding et al. have demonstrated that up-regulation of cyclin B1 is responsible for nickel-induced M phase arrest and cell growth inhibition (24). Others revealed that soluble nickel compounds caused cell growth arrest and cyclin D1 degradation throught IKK α-dependent pathway (25). Figure 1D shows that nickel treatment, in addition to decreasing cell number, also induced concomitant morphological changes of the BEAS-2B cells. The majority of nickel-treated BEAS-2B cells that originally had an epithelial cell-like appearance became elongated and resembled fibroblasts (Figure 1D), as observed and reported by others (26). The elongation developed in the first 24 h of nickel exposure and persisted throughout the remaining 48 h of treatment.
Figure 1.

Nickel induces apoptosis. (A) BEAS-2B cells (4 × 105) were seeded in a 60 mm dish overnight, then treated without or with various concentrations of Ni3S2 for 48 h, and then stained with Annexin V/PI. Apoptosis was determined using flow cytometry as described in the Materials and Methods. Illustrated is a representative of at least three separate experiments. (B) Quantification of apoptotic cells. Data were obtained from Annexin V/PI assays and are represented as means ± SEs of three separate experiments. *P < 0.05 as determined by the treatment vs control. (C) BEAS-2B cells (1.5 × 105) were seeded in each well of six well plates overnight and then treated without or with various doses of Ni3S2 for 72 h or with 2 μg/cm2 Ni3S2 for various time points as indicated, and then, cell counting was carried out. Data represent means ± SEs of three individual experiments of cell counting. *P < 0.05 as determined by the treatment vs control. (D) Nickel induces morphological changes in BEAS-2B cells. BEAS-2B cells were seeded in 60 mm dishes. After they were cultured at 37 °C overnight, the cells were treated without or with 2 μg/cm2 Ni3S2 for 48 h. Pictures were taken using a phase contrast microscope. (E) Down-regulation of Bcl-2 and Bcl-xL is involved in nickel-induced apoptosis. BEAS-2B cells were seeded in each 100 mm dish and cultured in 10% FBS/DMEM at 37 °C. When the cell density reached 70−80%, the cells were exposed to different concentrations of Ni3S2 for 48 h. After treatments, total cellular extracts were prepared and subjected to Western blot assay using antibodies against Bcl-2, Bcl-xL, and β-actin. Each lane was loaded with 40 μg of protein. Blots were subsequently stripped and reprobed with antibody against β-actin to ensure equivalent loading and transfer. The results shown are representative of three separate experiments. Two additional studies yielded equivalent results. (F) Band densities in the Western blots were normalized against β-actin. Each bar represents the mean ± SE of the three independent experiments. All means marked with * (P < 0.05) are significantly different from the control.
Bcl-2-family proteins are evolutionarily conserved regulators of apoptosis (27,28). Within this family, Bc1-2 and Bcl-xL proteins are potent antiapoptotic proteins that inhibit a mitochondria-operated pathway of apoptosis in many types of cells. Both Bcl-2 and Bcl-xL were down-regulated by nickel treatment (Figure 1E,F).
Generation of ROS Stimulated by Nickel Is Required for Nickel-Induced Apoptosis
It has been reported that nickel may induce ROS generation of the cells under some circumstances (29−31). To study the relationship between ROS generation and apoptosis, nickel-induced ROS production was determined by staining the cells with CM-H2DCFDA and DHE, fluorescent dyes for H2O2 and O2•−, respectively. Figure 2A shows that cells treated with Ni3S2 stimulated generation of H2O2, whereas there was no apparent alteration in O2•− generation (Figure 2B). Pretreatment of the cells with antioxidant NAC decreased H2O2 production (Figure 2C). The addition of catalase, a scavenger of H2O2, also inhibited ROS generation (Figure 2D). Vitamin E, another well-established antioxidant, was also used to evaluate effect on ROS generation stimulated by nickel. As shown in Figure 2E, pretreatment of BEAS-2B cells with vitamin E reduced nickel-induced ROS generation.
Figure 2.

Oxidative stress is involved in nickel-induced apoptosis. (A, B) Nickel mainly induces H2O2 generation, not O2•−. BEAS-2B (4 × 105) cells were seeded in a 60 mm dish overnight and then treated without or with different concentrations of Ni3S2 for 48 h. The cells were labeled with CM-H2DCFDA and DHE, respectively, followed by flow cytometry as described in the Materials and Methods. The rightward shift of the overlay reflected the ROS generation. Illustrated overlays are representatives of at least three separate experiments. Quantifications of both H2O2 and O2•− generation are displayed on the right panel. Each bar represents the mean ± SE of the three independent experiments. *P < 0.05 as determined by the treatment vs control. (C, D) NAC and catalase inhibited nickel-induced ROS generation. BEAS-2B cells (4 × 105) were seeded in a 60 mm dish overnight and then pretreated with or without NAC (10 mM) or catalase (2000 units) for 2 h, respectively, and then treated without or with 2 μg/cm2 Ni3S2 for 48 h. The cells were stained with CM-H2DCFDA and measured by flow cytometry. Overlays shown here are representatives of at least three separate experiments. Quantifications of H2O2 generation are displayed on the right panel. Each bar represents the mean ± SE of the three independent experiments. *P < 0.05 between the indicated two groups. (E) Vitamin E ameliorated nickel-induced ROS generation. BEAS-2B cells (4 × 105) were seeded in a 60 mm dish overnight and then pretreated with or without vitamin E (20 μM) for 2 h and then treated without or with 2 μg/cm2 Ni3S2 for 48 h. The cells were stained with CM-H2DCFDA and measured by flow cytometry. The overlay shown here is representative of at least three separate experiments. Quantifications of H2O2 generation are displayed on the right panel. Each bar represents the mean ± SE of the three independent experiments. *P < 0.05 between the indicated two groups. (F) NAC attenuated nickel-induced apoptosis. BEAS-2B cells (4 × 105) were seeded in a 60 mm dish overnight, then pretreated with or without NAC (10 mM) for 2 h, and then treated without or with 4 μg/cm2 Ni3S2 for 48 h. The cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry as described in the Materials and Methods. The results shown are the means of triplicate determinations ± SE. *P < 0.05 as determined by the indicated two groups. (G) Nickel decreased the protein expression of catalase, not Cu/Zn SOD or Mn SOD. BEAS-2B cells were treated without or with various concentrations of Ni3S2 for 48 h as indicated and then measured by Western blot assay. Four more additional experiments achieved equivalent results. Band densities of catalase in the Western blots were normalized against β-actin (shown on right panel). Each bar represents the mean ± SE of the three independent experiments. All means marked with * (P < 0.05) are significantly different from the control.
To investigate the possible role of ROS in nickel-induced apoptosis, the effects of specific modifiers of ROS on apoptosis were determined. The results show that pretreatment of the cells with NAC attenuated nickel-induced apoptosis (p < 0.05) (Figure 2F). We also pretreated BEAS-2B cells with antioxidant vitamin E, and our result shows that apoptosis induced by nickel was also ameliorated by vitamin E treatment (data not shown). In addition, the protein level of catalase was decreased with the stimulation of Ni3S2, while the protein level of Cu/Zn SOD (SOD1) and Mn SOD (SOD2) remained the same (Figure 2G). Accordingly, H2O2 is likely the main ROS induced by nickel treatment.
Signaling Pathway of ASK1/p38 MAPK Is Involved in Nickel-Induced Apoptosis
Since its discovery by Ichijo et al. in 1997 (19), ASK1 has drawn much attention in cell apoptosis, especially in oxidative stress-induced cell apoptosis through Thr838 (corresponding to Thr845 in mice) phosphorylation (20,21). Since nickel induced ROS generation, we speculated that ASK1 could be involved in nickel-induced apoptosis. By performing immunoblotting analysis, our results showed that ASK1 phosphorylation at Thr838, which is correlated with its activity, was increased with the nickel treatment (Figure 3A), whereas phosphorylation at Ser83, which attenuates its activity and promotes cell survival (15), remained unchangeable (Figure 3A). Since ASK1 is located upstream of the SEK1/MKK4-JNK/SAPK and MKK3/MKK6-p38 pathways (19), we examined the activation of the multiple downstream protein kinases by Western blot using phospho-specific antibodies (JNK and p38). Figure 3B shows that treatment with nickel resulted in the activation of p38 MAPK but not JNK.
Figure 3.

ASK1/p38 MAPK pathway is activated in nickel-induced apoptosis. (A) Nickel treatment increased protein phosphorylation of ASK1 at Thr838, not Ser83. BEAS-2B cells were treated without or with different concentrations of Ni3S2 for 48 h. After treatments, a Western blot assay was performed using antibodies against ASK1 at Thr838 or Ser83, respectively. Each lane was loaded with 40 μg of protein. Blots were subsequently stripped and reprobed with antibody against ΑSK1 to ensure equivalent loading and transfer. The results shown are representative of three separate experiments. Two additional studies yielded equivalent results. Band densities of ASK1 phosphorylation at Thr838 in the Western blots were normalized against ASK1 (shown on lower panel). Each bar represents the mean ± SE of the three independent experiments. All means marked with * (P < 0.05) are significantly different from the control. (B) Nickel treatment increased protein phosphorylation of p38, not JNK. BEAS-2B cells were treated without or with different concentrations of Ni3S2 for 48 h. After treatments, a Western blot assay was performed using antibodies against phosphor-p38 or phosphor-JNK, respectively. Each lane was loaded with 40 μg of protein. Blots were subsequently stripped and reprobed with antibody against p38 or JNK to ensure equivalent loading and transfer. The results shown are representative of three separate experiments. Two additional studies yielded equivalent results. Band densities of p38 phosphorylation in the Western blots were normalized against p38 (shown on lower panel). Each bar represents the mean ± SE of the three independent experiments. All means marked with * (P < 0.05) are significantly different from the control. (C) siRNA ASK1 decreased nickel-induced p38 activation. BEAS-2B cells were transfected with 100 nM ASK1 siRNA and control siRNA, and 24 h later, cells were treated with 2 μg/cm2 Ni3S2 for 48 h. The protein expression was measured by Western Blotting. ASK1 expression and p38 phosphorylatin were decreased by siRNA ASK1. All results demonstrated here are representatives of three separate experiments. Band densities of ASK1 and p38 phosphorylation in the Western blots were normalized against actin and p38, respectively (shown on right panel). Each bar represents the mean ± SE of the three independent experiments. All means marked with * (P < 0.05) are significantly different from the control. (D) SB203580, a p38 MAPK inhibitor, has no effect on ASK1 phosphorylation at Thr838. BEAS-2B cells were pretreated with or without SB203580 (10 μM) for 2 h and then coincubated with 2 μg/cm2 Ni3S2 for 48 h. The protein levels were measured by Western blot assays and represented from three separate experiments. Blots were late stripped and reprobed with antibody against ASK1 to ensure equivalent loading and transfer.
To investigate the role of ASK1 in regulating p38 MAPK, we used siRNA that specifically silences ASK1, an approach of loss-of-function analysis utilizing RNA interference. Expression of siRNAs is able to silence gene expression and allows the functional inactivation of the targeted gene (32,33). Both siRNA control and siRNA ASK1 products that we used here are from Santa Cruz Co. and have been tested to reduce protein expression in human cells. By Western blotting analysis, our results show that ASK1 was down-regulated after transfection with siRNA specific to ASK1 (Figure 3C). We then examined the effect of siRNA ASK1 on p38 MAPK. Figure 3C shows that siRNA ASK1 decreased nickel-induced activation of p38 MAPK, suggesting that ASK1 mediated nickel-induced p38 MAPK activation. Utilizing a pharmacological inhibition method, we checked the effect of p38 MAPK inhibition through SB203580, a widely used p38 inhibitor, on ASK1 activation, to see whether ASK1 activation is inversely regulated by p38 MAPK. Immunoblot results show that activation of ASK1 induced by nickel was not altered by SB203580 (Figure 3D).
Akt Kinase Is Activated and Involved in ASK1/p38 MAPK Signaling Pathway in Nickel-Induced Apoptosis
Akt has been revealed by many researches to play an essential role in promoting cell survival, inhibiting apoptosis, and so on. By immunoblotting, we observed a pronounced activation of Akt by nickel treatment (Figure 4A). It has been reported that Akt can phosphorylate ASK1 on Ser83 and inactivates the apoptotic function of ASK1, leading to the enhancement of cell survival (15). It is unknown whether activation of ASK1/p38 pathway by nickel is mediated by Akt.
Figure 4.

Akt is activated and involved in ASK1/p38 MAPK signaling pathway in nickel-induced apoptosis. (A) Nickel induced Akt phosphorylation at Ser473. BEAS-2B cells were treated with different concentrations of Ni3S2 for 48 h and followed by immunoblot analysis of phospho-Akt at Ser473. Blots were late stripped and reprobed with antibody against total Akt. The results shown are representative of three separate experiments. All studies yielded equivalent results. The quantification analyses of Akt phosphorylation are shown on lower panel. * (P < 0.05) represents significantly different from the control. (B) Small interfering RNAs designed to target Akt mRNA (Akt siRNA) decreased both Akt phosphorylation at Ser473 and Akt expression. BEAS-2B cells were transfected with 100 nM Akt siRNA and control siRNA for 24 h and then treated with 2 μg/cm2 Ni3S2 for 48 h. The protein expression was measured by Western Blotting. All results demonstrated here are representatives of three separate experiments. (C) Akt siRNAs attenuated nickel-induced protein phosphorylation of ASK1 and p38 (experimental conditions are the same as in panel B described above). Blots were late stripped and reprobed with antibody against ASK1 and p38, respectively. Quantification of phospho-ASK1 at Thr 838 and phospho-p38 was normalized to ASK1 and p38, respectively. * (P < 0.05) means significantly different from the control, or # (P < 0.05) means significantly different between the indicated two groups. (D) Without nickel stimulation, siRNA Akt has no effect on ASK1 phosphorylation and p38 phosphorylation. BEAS-2B cells were transfected with 100 nM Akt siRNA and control siRNA, and 72 h later, the protein expression was measured by Western blotting. All results demonstrated here are representatives of three separate experiments. (E) siRNA Akt attenuates nickel-induced apoptosis. BEAS-2B cells were transfected with 100 nM Akt siRNA and control siRNA, and 24 h later, cells were treated with 2 μg/cm2 Ni3S2 for 48 h. Apoptosis was measured by flow cytometry as described in the Materials and Methods. Data displayed here are representatives of three separate experiments. Results shown are the means of triplicate determinations ± SE. * (P < 0.05) means significantly different as compared with the control. # (P < 0.05) represents a significant difference between the two indicated groups.
To obtain direct evidence for the involvement of Akt in mediating the ASK1/p38 pathway in nickel-induced apoptosis, we used siRNA that specifically silences Akt. Both siRNA control and siRNA Akt products that we used here are from Cell Signaling Co. and have been tested in-house and shown to reduce protein expression of Akt. As shown in Figure 4B, both expression of Akt and phosphorylatd Akt at Ser473 were all decreased by siRNA specific to Akt but not the control siRNA. As compared with siRNA control, protein levels of phosphorylated Akt and Akt after siRNA Akt were decreased by almost 70 and 60%, respectively, through quantitative analysis (data not shown here).
As shown in Figure 4C, activation of ASK1 and downstream kinase p38 was attenuated by siRNA Akt. In a control experiment, we transfected BEAS-2B cells with siRNA control and siRNA Akt. All conditions and procedures are exactly the same as before except omitting nickel stimulation. Our results showed that, in the absence of nickel stimulation, siRNA Akt had no effect on ASK1 phosphorylation at both Thr838 and Ser83 and p38 MAPK phosphoryltion demonstrated by Western Blot analysis (Figure 4D). Flow cytometric analysis further indicated that apoptosis induced by nickel was decreased by Akt-specific siRNA (Figure 4E). Accordingly, these observations demonstrate that Akt plays a role in mediating ASK1/p38 pathway and apoptosis induced by nickel.
Oxidative Stress Involved in the Akt/ASK1/p38 MAPK Pathway in Nickel-Induced Apoptosis
As signal molecules, ROS have been implicated in a wide array of apoptotic processes by mediating signal transduction. Our results have already demonstrated that nickel could induce ROS generation (Figure 2). Here, to dissect the role of ROS in mediating signal transduction pathways in nickel-induced apoptosis, BEAS-2B cells were preincubated with NAC and catalase for 2 h, and then, the cells were used to study the alteration of signaling pathway in response to nickel. As shown in Figure 5A−C, treatment of NAC and catalase attenuated nickel-induced phosphorylation of Akt, ASK1, and downstream p38 MAPK. The effects of these ROS modifiers on signaling changes are in agreement with their effects on nickel-induced apoptosis (Figure 2F). Thus, the results show that the generation of ROS stimulated by nickel is involved in nickel-induced apoptotic signaling pathway.
Figure 5.

Oxidative stress involved in Akt/ASK1/p38 MAPK pathway in nickel-induced apoptosis. (A) Effects of NAC and catalase on the Akt phosphorylation at Ser473 and ASK1 phosphorylation at Thr838. BEAS-2B cells were pretreated with or without NAC (10 mM) and catalase (2000 units/ml) for 2 h and then coincubated with 2 μg/cm2 Ni3S2 for 48 h. The protein levels were measured by Western blot assays and were from three separate experiments. Blots were stripped and reprobed with antibody against Akt and ASK1. All conditions are the same as described in Figure 1E. (B) Quantitative analyses of the blots are normalized to Akt and ASK1, respectively. * (P < 0.05) is significantly different between the indicated two groups. (C) Effects of NAC and catalase on p38 phosphorylation. BEAS-2B cells were similarly treated with the agents indicated above, and p38 phosphorylation was determined by Western blotting. Representatives here are from three separate experiments. Blots were stripped and reprobed with antibody against p38. Quantitative analyses of the blots are normalized to p38 and displayed on lower panel. * (P < 0.05) represents a significant difference between the two indicated groups.
Discussion
The present study addressed the importance of ROS in mediating Akt/ASK1/p38 signal cascades in nickel-induced apoptosis. Nickel is known to induce genotoxic stress. However, very limited information is available with regard to the mechanisms of nickel-induced apoptosis and related signaling pathways. The nickel-induced apoptosis was first reported in Chinese hamster ovary cells (34). The phenomenon of apoptosis was also observed in other cell lines, for example, T cell hybridoma cells, Jurkat cells, and mouse epidermal JB6 cells (35−37). Fas ligand (FasL) expression, cell cycle alteration, and activation of c-Myc through the ERK pathway are reportedly involved in nickel-induced apoptosis (35,36).
Our study demonstrates that nickel could induce apoptosis in BEAS-2B cells. Down-regulation of bcl-2 and bcl-xL, two of the central players of the Bcl-2 family members, was involved in nickel-induced apoptosis. The Bcl-2 family proteins are the principal regulators of apoptosis, which consist of two groups, antiapoptotic members, including Bcl-2, Bcl-xL, Bcl-w, and Mcl-1, and pro-apoptotic members, including Bax and the BH3-only families (38). Bcl-2 down-regulation is reportedly involved in arsenic-induced apoptosis (39). Our study shows that nickel could down-regulate expression of both bcl-2 and bcl-xL proteins. Accompanied by apoptosis under stimulation of nickel is the cell morphological alteration from epithelial cell-like appearance to becoming elongated and fibroblast-like. This is in agreement with the observation reported by others (26).
ROS are generated in various biological systems and are well-known to be important determinants in regulation of cell signaling pathways involved in proliferation, apoptosis, and senescence (40,41). The generation of ROS in response to certain metals has been implicated in the triggering of apoptosis (42−44). Nickel compounds have been reported to induce oxidative damage resulting from an increase of ROS production (45). Results from the present study demonstrate that nickel exposure caused ROS production and cell apoptosis. By using molecular probes CM-H2DCFDA and DHE, we found that nickel mainly induced H2O2 generation, for no apparent increase of O2•− was observed, which is in agreement with the data of protein expression of catalase and SOD after nickel treatment. Among antioxidant defense mechanisms in mammalian cells, antioxidant enzymes, such as catalase, SOD1, and SOD2, play key roles in the detoxification of H2O2 and O2•−. Our study showed that nickel treatment decreased protein expression of catalase, whereas SOD1 and SOD2 remained unchanged. These results indicate that H2O2 is likely the main ROS involved in nickel-induced apoptosis. Moreover, pretreatment of BEAS-2B cells with NAC, vitamin E, or catalase all reduced nickel-induced ROS generation, respectively. Even though the exact mechanism of ROS generation stimulated by nickel is unknown, several sources of ROS generation could exist in cells, including cyclooxygenase, lipoxygenase, mitochondrial electron transfer system, and cytochrome P450. The major source of ROS has been suggested to be the NADPH oxidase (NOX) (46).
ROS induced by nickel may act as upstream-mediating molecules of the Akt/ASK1/p38 signaling pathway in nickel-induced apoptosis in BEAS-2B cells. The Akt kinases play critical roles in regulating growth, proliferation, survival, metabolism, and other cellular activities. Akt can phosphorylate a number of pro-apoptotic proteins, including glycogen synthase kinase 3 (GSK-3), BAD, caspase 9, and Forkhead transcription factors, to suppress apoptosis (47−51). A study by others shows that nickel compounds can activate the Akt pathway to induce hypoxia-inducible factor transactivation and cap43 expression (52).
However, in contrast to its well-established survival-promoting role, we found here that Akt also plays a pro-apoptotic role in nickel-induced apoptosis. The same concentration of nickel treatment leading to cell apoptosis also induced activation of Akt. In agreement with our study, recent researches performed by others also indicate that Akt is not just a single-function kinase. Under certain conditions, activation of Akt may be beneficial to cell death. Nogueira et al. showed that Akt activation increases oxidative stress, which in turn further increases Akt phosphorylation and renders cells susceptible to ROS-triggered cell senescence or death (53). Additionally, anticancer drugs, such as methotrexate, docetaxel, and doxorubicin, can also activate the Akt/CDK2 (cyclin dependent kinase) pathway to promote, rather than suppress, cell death (54). Apoptin, a viral protein, has also been reported to selectively kill cancer cell death through Akt activation followed by Cdk activation (55). Furthermore, in the case of the death receptor pathway, activation of Akt by Fas ligand stimulation leads to apoptosis in epidermal C141 cells (56). Activation of Akt in nickel-induced apoptosis has also been observed in JB6 cells by another group (37).
Our study demonstrates that nickel induced BEAS-2B cell apoptosis through the Akt-mediated ASK1/p38 pathway. ASK1 is one of the MAP3K that activates p38 and JNK via activating the MAP2Ks, MKK4/MKK7 and MKK3/MKK6 (18,19). ASK1 is activated by a variety of stresses including calcium influx, endoplasmic reticulum (ER) stress, lipopolysaccharide (LPS), ROS, and tumor necrosis factor (57−59). These stresses induce activation of ASK1 through Thr838 (Thr845 in mice) phosphorylation (20,21). In recent years, researches have revealed that activation of ASK1 plays pivotal roles in a wide variety of cellular responses, such as cell differentiation, apoptosis, and immune response, with special focus on oxidative stress-induced apoptosis (60). Our results show that nickel treatment induced ASK1 phosphorylation at Thr838, not Ser83, which decreases ASK1 activity. The results suggest that nickel induced apoptosis mainly through ASK1 phosphorylation at Thr838. Activation of ASK1 can selectively activate JNK and p38 MAP kinases, leading to apoptosis. Here, we found that nickel treatment caused p38 MAPK phosphorylation, not JNK. To obtain evidence that ASK1 regulates p38 MAPK, we employed siRNA method to specifically down-regulate ASK1 expression. We found that phosphorylation of p38 by nickel stimulation was attenuated by siRNA ASK1. However, inhibition of p38 MAPK with pharmacological inhibitor, SB203580, had no effect on phosphorylation of ASK1 at Thr838, probably implicating that ASK1 was not reversely regulated by p38 MAPK.
It has been shown that ASK1 activity can be regulated by a number of ASK1-interacting proteins. Among them, thioredoxin (Trx) and 14-3-3 can directly bind to ASK1 leading to inhibition of ASK1 activity (59,61). Protein phosphatase 5 (PP5) is also capable of binding to and dephosphorylating Thr838 (Thr845 in mice) to inactivate ASK1 in response to oxidative stress (62). Previous studies show Akt that can also act as an upstream kinase of ASK1 to phosphorylate and negatively regulate ASK1 at the site of Ser83 (15−17). Nevertheless, in nickel-induced apoptosis in BEAS-2B cells, both Akt and ASK1 were all activated. To elucidate the direct regulation of Akt on ASK1, we down-regulated Akt by utilizing siRNA specific to Akt. Our study shows that under the stimulation of nickel, phosphorylation of ASK1 at Thr838 and p38 MAPK, downstream of ASK1, were all attenuated by siRNA Akt. In addition, siRNA Akt also partially ameliorated nickel-induced apoptosis. On the contrary, in the absence of nickel stimulation, siRNA Akt showed no effect on ASK1 phosphorylation at Thr838 and Ser83 as well as p38 phosphorylation. These observations suggest that Akt acted upstream of ASK1/p38 MAPK pathway in the nickel-induced BEAS-2B cell apoptosis. Given the results that we obtained in siRNA ASK1 (Figure 3C) that p38 phosphorylation was almost completely inhibited by siRNA ASK1, we propose that Akt may phosphorylate ASK1 at Thr838 first, followed by p38 phosphorylation. Regulation of p38 activity by Akt through ASK1 was also demonstrated by others (17,63). In addition, a number of substrates have been shown to be downstream of p38 MAPK activation that are involved in regulating diverse cellular functions. Among them, the p38/p53 pathway is reportedly involved in G1-S arrest induced by loss of centrosome integrity (64). Activation of STAT1 by p38 MAPK has been shown to be important for LPS-induced death of macrophages (65). Whether those substrates of p38 MAPK mentioned above are involved in nickel-induced apoptosis remains to be investigated.
There is increasing evidence within the literature that ROS contribute to apoptosis stimulated by diverse stimuli. Our study also showed that nickel-induced apoptosis was attenuated by ROS scavenger, indicating that ROS are probably involved in nickel-induced apoptosis in BEAS-2B cells. Furthermore, activation of signaling kinases including Akt, ASK1, and downstream p38 MAPK were all ameliorated by scavenging ROS, implying that ROS mediated the Akt/ASK1/p38 pathway in nickel-induced cell apoptosis. Several lines of evidence have indicated the involvement of ROS in these pathways (66). For instance, ROS mediate PI3K/Akt signaling and apoptosis induced by FasL(56). ROS are also required for lipopolysaccharide (LPS)-induced activation of ASK1/p38 pathway and so on (57). However, we found that neither NAC nor siRNA Akt could completely block nickel-induced apoptosis pathway. It suggests that there are some other factors or pathways contributing to nickel-induced apoptosis. Apoptosis is a complicated process. In mammalian cells, there are two major apoptotic pathways, the death-receptor pathway and the mitochondrial pathway. In the case of nickel-induced apoptosis, increased FasL expression, cell cycle alteration, activation of c-Myc through ERK pathway, caspase-8/AIF-mediated pathways, and so on have been reportedly involved (24,35−37). Also of note is the fact that some other unknown pathways are probably also involved in nickel-induced apoptosis.
In summary, the present study has demonstrated that ROS induced by nickel probably play a role in nickel-induced apoptosis. ROS mediates nickel-induced apoptosis through the Akt-ASK1-p38 axis (Figure 6). Considering the important role of the Akt signaling pathway in cell transformation and cancer, understanding the mechanism of nickel-induced apoptosis through the Akt signaling pathway will be important in understanding the mechanism of nickel-induced carcinogenesis.
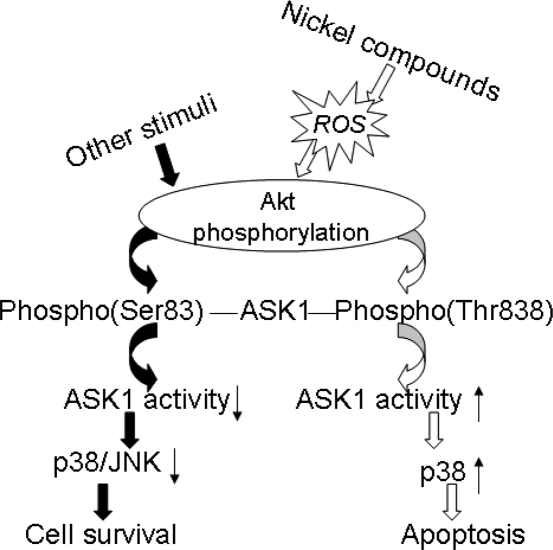
Figure 6.

Schematic model for the role of ROS/Akt/ASK1/p38 signaling pathway in nickel-induced apoptosis. Nickel-generated ROS stimulate the phosphorylation of Akt, which in turn activates the ASK1 by phosphorylating ASK1 on Thr838, then followed by p38 activation, leading to an apoptotic effect.
Acknowledgments
X.S. is grateful for research support from NIH Grants 5R01CA119028-05, R01CA116697, R01ES015518, and R01ES015375.
Funding Statement
National Institutes of Health, United States
References
- Costa M. (1991) Molecular mechanisms of nickel carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 31, 321–337. [DOI] [PubMed] [Google Scholar]
- Doll R.; Morgan L. G.; Speizer F. E. (1970) Cancers of the lung and nasal sinuses in nickel workers. Br. J. Cancer 24, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K.; Costa M. (2000) Epigenetic mechanisms of nickel carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 19, 307–318. [PubMed] [Google Scholar]
- Pulido M. D.; Parrish A. R. (2003) Metal-induced apoptosis: Mechanisms. Mutat. Res. 533, 227–241. [DOI] [PubMed] [Google Scholar]
- Trombetta D.; Mondello M. R.; Cimino F.; Cristani M.; Pergolizzi S.; Saija A. (2005) Toxic effect of nickel in an in vitro model of human oral epithelium. Toxicol. Lett. 159, 219–225. [DOI] [PubMed] [Google Scholar]
- Rana S. V. (2008) Metals and apoptosis: recent developments. J. Trace Elem. Med. Biol. 22, 262–284. [DOI] [PubMed] [Google Scholar]
- Finkel T. (2003) Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 15, 247–254. [DOI] [PubMed] [Google Scholar]
- Benhar M.; Engelberg D.; Levitzki A. (2002) ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 3, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K.; Zhitkovich A. (2008) Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol. 21, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; McCullough K. D.; Franke T. F.; Holbrook N. J. (2000) Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 275, 14624–14631. [DOI] [PubMed] [Google Scholar]
- Huang C.; Li J.; Ding M.; Leonard S. S.; Wang L.; Castranova V.; Vallyathan V.; Shi X. (2001) UV Induces phosphorylation of protein kinase B (Akt) at Ser-473 and Thr-308 in mouse epidermal Cl 41 cells through hydrogen peroxide. J. Biol. Chem. 276, 40234–40240. [DOI] [PubMed] [Google Scholar]
- Dudek H.; Datta S. R.; Franke T. F.; Birnbaum M. J.; Yao R.; Cooper G. M.; Segal R. A.; Kaplan D. R.; Greenberg M. E. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665. [DOI] [PubMed] [Google Scholar]
- Franke T. F.; Kaplan D. R.; Cantley L. C. (1997) PI3K: Downstream AKTion blocks apoptosis. Cell 88, 435–437. [DOI] [PubMed] [Google Scholar]
- Lawlor M. A.; Alessi D. R. (2001) PKB/Akt: A key mediator of cell proliferation, survival and insulin responses. J. Cell Sci. 114, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Kim A. H.; Khursigara G.; Sun X.; Franke T. F.; Chao M. V. (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 21, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T.; Furuta S.; Mitsushita J.; Shang W. H.; Ito M.; Yokoo Y.; Yamaura M.; Ishizone S.; Nakayama J.; Konagai A.; Hirose K.; Kiyosawa K.; Kamata T. (2006) Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 25, 3699–3707. [DOI] [PubMed] [Google Scholar]
- Yuan Z. Q.; Feldman R. I.; Sussman G. E.; Coppola D.; Nicosia S. V.; Cheng J. Q. (2003) AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: Implication of AKT2 in chemoresistance. J. Biol. Chem. 278, 23432–23440. [DOI] [PubMed] [Google Scholar]
- Ichijo H. (1999) From receptors to stress-activated MAP kinases. Oncogene 18, 6087–6093. [DOI] [PubMed] [Google Scholar]
- Ichijo H.; Nishida E.; Irie K.; ten Dijke P.; Saitoh M.; Moriguchi T.; Takagi M.; Matsumoto K.; Miyazono K.; Gotoh Y. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94. [DOI] [PubMed] [Google Scholar]
- Tobiume K.; Saitoh M.; Ichijo H. (2002) Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J. Cell Physiol. 191, 95–104. [DOI] [PubMed] [Google Scholar]
- Saito J.; Toriumi S.; Awano K.; Ichijo H.; Sasaki K.; Kobayashi T.; Tamura S. (2007) Regulation of apoptosis signal-regulating kinase 1 by protein phosphatase 2Cepsilon. Biochem. J. 405, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. Y.; Nishitoh H.; Yang X.; Ichijo H.; Baltimore D. (1998) Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281, 1860–1863. [DOI] [PubMed] [Google Scholar]
- Gao N.; Rahmani M.; Dent P.; Grant S. (2005) 2-Methoxyestradiol-induced apoptosis in human leukemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene 24, 3797–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J.; He G.; Gong W.; Wen W.; Sun W.; Ning B.; Huang S.; Wu K.; Huang C.; Wu M.; Xie W.; Wang H. (2009) Effects of nickel on cyclin expression, cell cycle progression and cell proliferation in human pulmonary cells. Cancer Epidemiol. Biomarkers Prev. 18, 1720–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W.; Zhang D.; Li J.; Verma U. N.; Costa M.; Huang C. (2009) Soluble and insoluble nickel compounds exert a differential inhibitory effect on cell growth through IKKalpha-dependent cyclin D1 down-regulation. J. Cell Physiol. 218, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M. (1978) Alteration in morphology of chinese hamster ovary cells by Ni3S2 and dibutyryl cAMP. Toxicol. Appl. Pharmacol. 44, 555–566. [DOI] [PubMed] [Google Scholar]
- Gross A.; McDonnell J. M.; Korsmeyer S. J. (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Thompson C. B. (1999) Bcl-2 proteins: Regulators of apoptosis or of mitochondrial homeostasis?. Nat. Cell Biol. 1, E209–E216. [DOI] [PubMed] [Google Scholar]
- Huang C.; Li J.; Costa M.; Zhang Z.; Leonard S. S.; Castranova V.; Vallyathan V.; Ju G.; Shi X. (2001) Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel subsulfide. Cancer Res. 61, 8051–8057. [PubMed] [Google Scholar]
- Huang X.; Klein C. B.; Costa M. (1994) Crystalline Ni3S2 specifically enhances the formation of oxidants in the nuclei of CHO cells as detected by dichlorofluorescein. Carcinogenesis 15, 545–548. [DOI] [PubMed] [Google Scholar]
- Huang X.; Frenkel K.; Klein C. B.; Costa M. (1993) Nickel induces increased oxidants in intact cultured mammalian cells as detected by dichlorofluorescein fluorescence. Toxicol. Appl. Pharmacol. 120, 29–36. [DOI] [PubMed] [Google Scholar]
- Elbashir S. M.; Harborth J.; Lendeckel W.; Yalcin A.; Weber K.; Tuschl T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Brummelkamp T. R.; Bernards R.; Agami R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Shiao Y. H.; Lee S. H.; Kasprzak K. S. (1998) Cell cycle arrest, apoptosis and p53 expression in nickel(II) acetate-treated Chinese hamster ovary cells. Carcinogenesis 19, 1203–1207. [DOI] [PubMed] [Google Scholar]
- Kim K.; Lee S. H.; Seo Y. R.; Perkins S. N.; Kasprzak K. S. (2002) Nickel(II)-induced apoptosis in murine T cell hybridoma cells is associated with increased fas ligand expression. Toxicol. Appl. Pharmacol. 185, 41–47. [DOI] [PubMed] [Google Scholar]
- Li Q.; Suen T. C.; Sun H.; Arita A.; Costa M. (2009) Nickel compounds induce apoptosis in human bronchial epithelial Beas-2B cells by activation of c-Myc through ERK pathway. Toxicol. Appl. Pharmacol. 235, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Bowman L.; Zhang X.; Shi X.; Jiang B.; Castranova V.; Ding M. (2009) Metallic nickel nano- and fine particles induce JB6 cell apoptosis through a caspase-8/AIF mediated cytochrome c-independent pathway. J. Nanobiotechnol. 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S.; Adams J. M. (2002) The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656. [DOI] [PubMed] [Google Scholar]
- Ai Z.; Lu W.; Qin X. (2006) Arsenic trioxide induces gallbladder carcinoma cell apoptosis via downregulation of Bcl-2. Biochem. Biophys. Res. Commun. 348, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Gamaley I. A.; Klyubin I. V. (1999) Roles of reactive oxygen species: Signaling and regulation of cellular functions. Int. Rev. Cytol. 188, 203–255. [DOI] [PubMed] [Google Scholar]
- Powis G.; Briehl M.; Oblong J. (1995) Redox signalling and the control of cell growth and death. Pharmacol. Ther. 68, 149–173. [DOI] [PubMed] [Google Scholar]
- Jing Y.; Dai J.; Chalmers-Redman R. M.; Tatton W. G.; Waxman S. (1999) Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 94, 2102–2111. [PubMed] [Google Scholar]
- Shen H. M.; Zhang Z.; Zhang Q. F.; Ong C. N. (2001) Reactive oxygen species and caspase activation mediate silica-induced apoptosis in alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L10–L17. [DOI] [PubMed] [Google Scholar]
- Ye J.; Wang S.; Leonard S. S.; Sun Y.; Butterworth L.; Antonini J.; Ding M.; Rojanasakul Y.; Vallyathan V.; Castranova V.; Shi X. (1999) Role of reactive oxygen species and p53 in chromium(VI)-induced apoptosis. J. Biol. Chem. 274, 34974–34980. [DOI] [PubMed] [Google Scholar]
- Dally H.; Hartwig A. (1997) Induction and repair inhibition of oxidative DNA damage by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis 18, 1021–1026. [DOI] [PubMed] [Google Scholar]
- Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189. [DOI] [PubMed] [Google Scholar]
- Cross D. A.; Alessi D. R.; Cohen P.; Andjelkovich M.; Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Pap M.; Cooper G. M. (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932. [DOI] [PubMed] [Google Scholar]
- Datta S. R.; Dudek H.; Tao X.; Masters S.; Fu H.; Gotoh Y.; Greenberg M. E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241. [DOI] [PubMed] [Google Scholar]
- Cardone M. H.; Roy N.; Stennicke H. R.; Salvesen G. S.; Franke T. F.; Stanbridge E.; Frisch S.; Reed J. C. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282, 1318–1321. [DOI] [PubMed] [Google Scholar]
- Brunet A.; Bonni A.; Zigmond M. J.; Lin M. Z.; Juo P.; Hu L. S.; Anderson M. J.; Arden K. C.; Blenis J.; Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Li J.; Davidson G.; Huang Y.; Jiang B. H.; Shi X.; Costa M.; Huang C. (2004) Nickel compounds act through phosphatidylinositol-3-kinase/Akt-dependent, p70(S6k)-independent pathway to induce hypoxia inducible factor transactivation and Cap43 expression in mouse epidermal Cl41 cells. Cancer Res. 64, 94–101. [DOI] [PubMed] [Google Scholar]
- Nogueira V.; Park Y.; Chen C. C.; Xu P. Z.; Chen M. L.; Tonic I.; Unterman T.; Hay N. (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S.; Ande S. R.; Wiechec E.; Hansen L. L.; Wesselborg S.; Los M. (2008) Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J. Cell Sci. 121, 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S.; Panigrahi S.; Wiechec E.; Wesselborg S.; Fischer U.; Schulze-Osthoff K.; Los M. (2009) Unscheduled Akt-triggered activation of cyclin-dependent kinase 2 as a key effector mechanism of apoptin’s anticancer toxicity. Mol. Cell. Biol. 29, 1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B.; Wang L.; Stehlik C.; Medan D.; Huang C.; Hu S.; Chen F.; Shi X.; Rojanasakul Y. (2006) Phosphatidylinositol 3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in epidermal Cl41 cells. J. Immunol. 176, 6785–6793. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A.; Saegusa K.; Noguchi T.; Sadamitsu C.; Nishitoh H.; Nagai S.; Koyasu S.; Matsumoto K.; Takeda K.; Ichijo H. (2005) ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587–592. [DOI] [PubMed] [Google Scholar]
- Nishitoh H.; Saitoh M.; Mochida Y.; Takeda K.; Nakano H.; Rothe M.; Miyazono K.; Ichijo H. (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 2, 389–395. [DOI] [PubMed] [Google Scholar]
- Saitoh M.; Nishitoh H.; Fujii M.; Takeda K.; Tobiume K.; Sawada Y.; Kawabata M.; Miyazono K.; Ichijo H. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa A.; Ichijo H. (2008) Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 1780, 1325–1336. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Chen J.; Fu H. (1999) Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 8511–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K.; Saitoh M.; Tobiume K.; Matsuura H.; Enomoto S.; Nishitoh H.; Ichijo H. (2001) Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 20, 6028–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.; Hung M. C. (2003) Regulation of the activity of p38 mitogen-activated protein kinase by Akt in cancer and adenoviral protein E1A-mediated sensitization to apoptosis. Mol. Cell. Biol. 23, 6836–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikule K.; Delaval B.; Kaldis P.; Jurcyzk A.; Hergert P.; Doxsey S. (2007) Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 9, 160–170. [DOI] [PubMed] [Google Scholar]
- Kim H. S.; Lee M. S. (2005) Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol. Cell. Biol. 25, 6821–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Noguchi T.; Naguro I.; Ichijo H. (2008) Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 48, 199–225. [DOI] [PubMed] [Google Scholar]