

Abstract
We provide a historical review and update on current thinking regarding the possibility of elimination of gastric cancer from Japan. Because Helicobacter pylori infection is the cause gastric cancer, its elimination forms the cornerstone of eradication of gastric cancer. However, simply eradicating H. pylori from the entire population will not immediately solve the problem because many patients with H. pylori infections have already developed the precursor lesion, atrophic gastritis. Cure of H. pylori in these high risk patients will only reduce the risk of subsequent cancer. In contrast, treatment of low risk patients will prevent cancer. Thus, to eliminate gastric cancer it is necessary to identify and treat all infected individuals. In addition, those at increased risk for gastric cancer (ie, atrophic gastritis irrespective a age) should be considered for endoscopic surveillance to identify those cancers that do develop at an early stage. We propose that selection be based on severity and extent of atrophy be used to separate those expected to benefit from endoscopy and annual surveillance from those with little or no potential benefit. We suggest an algorithm for eradicating gastric cancer that incorporates H. pylori and atrophic gastritis testing, H. pylori therapy, and surveillance. Institution of a program of surveillance restricted to those who could benefit most (ie, those with moderate or severe atrophy). This will also allow a much closer matching of surveillance capacity and surveillance need making surveillance more clinically and cost effective.
Introduction
Although Helicobacter pylori is recognized as the primary cause of gastric cancer, it is a “necessary but insufficient cause” meaning that while the infection is the primary cause, other factors are needed to produce gastric cancer. The underlying abnormality appears to be long standing acute and chronic inflammation that results in marked changes in gastric structure and function. The incidence of gastric cancer varies both between and among populations 1. The risk of developing gastric cancer correlates best with the severity and extent of atrophic gastritis/gastric atrophy.
Despite the requirement for H. pylori in the pathogenesis of gastric cancer, if H. pylori were to instantly disappear from the world, the incidence of gastric cancer would not perceptibly change at least for several years. Ultimately, however, eradication of H. pylori would result in elimination of gastric cancer as an important clinical problem. Eradication of the infection reliably results in healing of gastritis and elimination of the progression of the mucosal damage. However, its effect on subsequent risk of developing gastric cancer depends on the level of risk at the time of eradication (ie, the severity and extent of atrophic gastritis/gastric atrophy). Japan is a country with a high incidence of gastric cancer and is thus one in which there is a high prevalence of atrophic gastritis. This paper discusses an approach to the eradication of gastric cancer in Japan including how current resources and surveillance programs might be utilized more efficiently in cancer prevention.
History of gastric cancer in relation to gastric atrophy/atrophic gastritis
Gastric cancer was one of the most common cancers, if not the most common cancer, in Western countries until the second half of the 20th century (Figure 1) 2. The importance of gastric cancer as a clinical problem resulted in it being of major research interest of gastroenterologists, gastrointestinal physiologists, pathologists, and surgeons. In 1879, von den Velden reported that gastric cancer was linked to achlorhydria 3–6 (Figure 2). This observation suggested the availability of a possible simple diagnostic test and prompted many investigations into the relation between gastric cancer and acid secretion as well as studies regarding measurement of gastric secretion itself (reviewed in 7–9).
Figure 1. Gastric cancer mortality per 100,000 among representative countries from 1906 to 1910.

During that period atrophic gastritis/atrophy was common in Western countries and gastric cancer was generally the most common cancer (adapted from reference 2).
Figure 2.

Time line of events in the understanding of the relation between acid secretion, gastritis, and gastric cancer.
Before 1900, the study of gastric histology was largely restricted of the use of autopsy specimens and the results were plagued by artifacts produced by the rapid post mortem autolysis of the delicate mucosal surfaces. In the late 1800’s and early 1900’s Faber, following up on the observation of Damaschino, Chauffard, and Hayem 4, solved the autolysis problem by instilling formalin directly into the abdominal cavity and into the stomach immediately after death 4. Over a period of approximately 3 decades he used this approach to describe as well as to define normal vs. abnormal histology and the relationship of gastritis/atrophy to disease. He clearly recognized the association of different patterns of gastritis with different clinical diseases 10.
The early 20th century saw many breakthroughs in the analysis of gastric secretion, gastric histology, barium contrast radiology, and gastrointestinal surgery. This resulted in numerous studies and new insights regarding the relationships between gastric cancer, gastric acid secretion, and gastritis. This was a period in which gastric atrophy and gastric cancer were still common in Western countries (Figure 3) 4 such that investigators had a wealth of clinical material to examine. For example, in 1934 Comfort and Vanzant reported studies on 619 men and 186 women with gastric cancer seen at the Mayo clinic between 1927 and 1931 7. They compared acid secretion of patients with cancer to those of normal individuals of similar age and sex and showed that the proportion with achlorhydria in both gastric cancer and normal individuals increased with age. Importantly, they showed that the prevalence of achlorhydria was higher than controls at each age interval among those with gastric cancer. They also noted that although the mean acidity decreased with age, at each age interval the mean acidity of those with cancer was always lower than normal individuals confirming that the abnormalities in gastric secretion preceded development of gastric cancer. By the late 1930’s the importance of gastritis in peptic ulcer and gastric cancer was widely recognized. For example, in 1938 G.E. Konjetzny prophesied “Ulcer and gastric cancer will be developed through silent inflammation of gastric mucosa. We are not able to distinguish between gastritis, which forms benign ulcer and that developing gastric cancer. When we were able to prevent gastritis or treat it, we would be able to prevent ulcer and gastric cancer: Prophylaxis of gastritis means prophylaxis against ulcer and gastric cancer”) (as quoted by 11).
Figure 3. Prevalence of atrophic gastritis/atrophy in the early 20th century.

Example of studies in the United States and Europe showing the high prevalence of atrophic gastritis/atrophy as evidenced by the age-related prevalence of achlorhydria in the first part of the 20th century (adapted from reference 4).
At mid-century, Comfort summarized the research relating acid secretion, gastritis, and gastric cancer that had been done in the first half of the 20th century (Table 2) 5. These data showed that 1) gastric cancer was associated with loss of secretory activity, 2) the loss of gastric secretory was progressive, 3) gastric secretory activity was subnormal before cancer develops, 4) was subnormal in each decade of life among patients destined to develop gastric cancer, and 5) that these facts were true no matter how many years before cancer develop that the secretion was tested. He concluded that atrophy of the acid secreting cells was the most likely the cause of abnormal gastric acidity in the precancerous stomach and that the soil in which a majority of gastric cancers develop appeared was characterized by markedly subnormal acid secretion and was the seat of chronic atrophic gastritis. He listed five possible causes of loss of gastric secretory activity (Table 1) and, from the available data, was able to exclude two possibilities (loss of secretory activity was developmental and that it was due to the presence of the tumor). The second half of the 20th century produced explanations for the remaining 3 possibilities with the “degenerative process attacking the acid cells” being explained by the autoimmune reaction that produced Addisonian pernicious anemia, “destruction of acid cells by atrophic gastritis” being explained as an effect of H. pylori infection, and “inhibitory substances capable of depressing gastric secretion” being identified (eg, the acid inhibitory cytokine, IL-1β produced) as part of the inflammatory response to H. pylori infection.
Table 2.
Identification of the extent and severity of gastric atrophy
| Invasive tests |
| Endoscopy |
| Location of gastric border |
| Chromoendoscopy |
| Targeted Biopsy protocols (eg, OLGA staging) |
| Non-invasive |
| Pepsinogen levels |
| Gastrin levels |
Table 1.
Possible causes of loss of acid secretory activity in gastric cancer
|
The next 30 years saw the development of fiberoptic endoscopy, advances in understanding of gastric secretion, and development of increasingly powerful drugs to suppress acid secretion. There were also many epidemiology of gastritis, largely prompted by attempts to define the relation between gastric diseases and gastritis (eg, 12. In 1975, the entire process starting with the first year of life combined into what is now known as the Correa hypothesis 13.
The H. pylori era
In 1983, Warren and Marshall reported culture of H. pylori and recognized that, if it were the cause of gastritis, that it would likely also be the cause of peptic ulcer and gastric cancer (Table 2) 6. Many new investigators became involved in research related to H. pylori infection, gastritis, and gastric diseases. Unfortunately, the many advances and insights of the prior 80 years went either unread or unheeded such that what was considered well know and accepted by our fathers had to be redone and relearned. For example, many epidemiologists who may have been unaware of the rich history of the relation between atrophic gastritis and gastric cancer, focused on results of serology of H. pylori and underestimated the importance of H. pylori in the pathogenesis of gastric cancer. The fact that estimates of attributable risk based on H. pylori serology were inconsistent with the results of 80 years of carefully done studies were unrecognized or remained explored. Only recently has it become generally recognized that standard IgG anti-H. pylori serology is a relatively blunt instrument in part because advanced atrophy results in loss of the infection leading to false negative serologic results 14. This can be partially overcome by examining for the presence of anti-CagA antibody 15, 16.
Nonetheless, the critical experiments proving that H. pylori was the major cause of gastritis, peptic ulcer, and gastric cancer were completed and confirmed Warren and Marshall’s suggestion that H. pylori is a bacterial pathogen that caused gastroduodenal inflammation and resulted in alterations in gastroduodenal structure and function resulting in duodenal ulcer disease, gastric ulcer disease, and atrophic gastritis 17. H. pylori-induced atrophic gastritis in turn could result in iron and/or vitamin B12 deficiency, gastric adenocarcinoma and/or primary B-cell gastric lymphoma.18–21. Although the road was possibly longer than necessary, now H. pylori is now generally accepted as the cause of gastric cancer.
Eradication of gastric cancer
H. pylori is typically acquired in childhood. Transmission of the infection is enhanced by poor sanitation, poor household hygiene, and poor standards of living. Improvements in the standards of living, sanitation and clear water supplies in regions where the infection was common resulted in disruption of the chain of transmission such that the disease began to disappear. Naturally this process requires many generations. The prevalence of the infection at about age 20 for any birth cohort is the prevalence for that birth cohort throughout life. The fall in prevalence in any population was therefore dependent on a succession of birth cohorts of increasingly lower prevalence replacing those born in eras when the acquisition of the infection was common. This happened long ago in Europe and the United States and is currently in progress in Japan and some other Asian countries 22.
The prevalence of different clinical outcomes varies both within and between populations 1. However, the likelihood of a particular clinical outcome of an infection can be predicted based on the pattern of and severity of gastritis (atrophic vs. non-atrophic and antral predominant vs. pangastritis or corpus predominant) with duodenal ulcer disease being associated with antral predominant (corpus sparing) gastritis) and gastric cancer with atrophic gastritis. The predominant pattern of gastritis depends upon interactions between the host, the bacteria, and the environment. Environmental factors appear to be the dominant factors as evidenced by the rapid change in outcome experienced by migrants (ie, Japanese to Hawaii) or associated with changes in food preservation (eg, use of refrigeration instead of salt) and year around availability of fresh fruits and vegetables instead of seasons diets with long periods without fresh fruits and vegetables 1, 23.
Within any population there are subpopulations at increased risk of developing atrophic gastritis and gastric cancer 24. Host factors are known to increase this risk 23. The best studied host factors relate to those associated with an enhanced inflammatory response to the infection (ie, polymorphisms in genes controlling the inflammatory response) 25. However, such polymorphisms are actually uncommon and despite there importance in enhancing our understanding of disease pathogenesis, they have little or no effect on the overall risk to the population. Similarly, some H. pylori strains are associated with enhanced inflammation (eg, cag pathogenicity island-containing H. pylori) and also increase the risk of disease. However, strains lacking these putative virulence factors also cause gastric inflammation, peptic ulcer and gastric cancer leading one to conclude that when one considers elimination of H. pylori-related diseases from a population, there is scant evidence to support a strategy of targeting “high risk groups” in preference to targeting the entire population for H. pylori eradication.
Risk stratification as part of population wide H. pylori eradication
Population-wide H. pylori eradication will eliminate H. pylori-induced disease 26, 27. Eradication of H. pylori results in healing of the gastritis, prevention of progression of gastritis, elimination of the ongoing inflammatory response, recovery of the normal feedback loops controlling acid secretion, and removal of cytokine-associated suppression of parietal cells. However, there are few data supporting recovery of lost cellular components or reversal of metaplastic epithelia to normal 28. The greatest yield comes from H. pylori eradication of those with non-atrophic gastritis as there risk of subsequent development of gastric cancer is minimal such that no follow-up is needed or indeed indicated. In contrast, those with atrophic gastritis have an elevated risk and while one can expect their risk to no longer increase, it will be unlikely to completely disappear, at least over the near future. Risk stratification is therefore a necessary part of any population-based eradication program 29.
Risk stratification can be based on the extent and severity of atrophy and both invasive and non-invasive methods are available to determine the extent and severity of atrophy (Table 2). The most accurate methods are those in which targeted gastric biopsies are used to stage cancer risk. The most recent system is the OLGA staging system 30 (Figure 4). However, this approach requires close cooperation between the endoscopist and the pathologist, accurate specimen collection and identification, as well as an experienced pathologist able to identify pseudopyloric metaplasia 31. The most practical approach for large studies is assessment of serum pepsinogen levels as this method is both non-invasive and widely available24, 32, 33. Pepsinogen testing is predicated on the fact that pepsinogen I is found almost exclusively in the gastric corpus such that corpus atrophy leads to a fall in the pepsinogen I levels and of the ratio of pepsinogen I/II. The method is not useful for identifying those with severe antral atrophy associated with only mild corpus damage 34. Nonetheless, pepsinogen testing has proven its value for identification of moderate to severe atrophic gastritis and is currently being used to enhance the yield of endoscopic screening programs designed to identify incident gastric cancers at an early stage 32, 33. Those at moderate to high risk of developing gastric cancer post H. pylori eradication should be considered for post treatment surveillance programs.
Figure 4. OLGA staging system of gastric cancer risk.
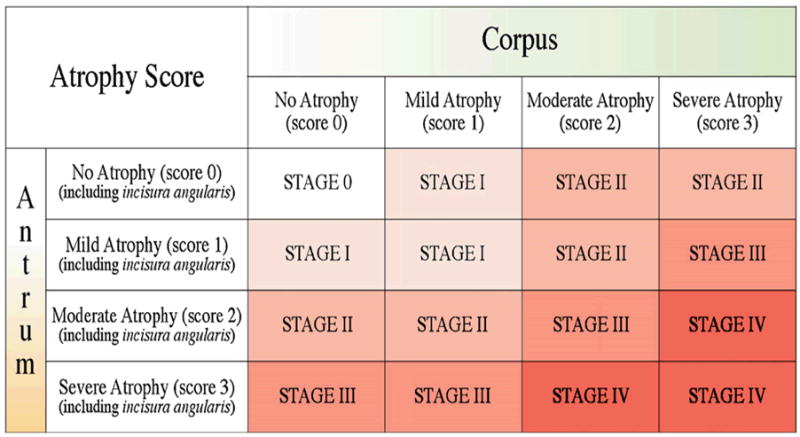
The OLGA staging system 30 is based on the severity and topography of gastric atrophy. Such a system may allow fine tuning of risk stratification and individualization of surveillance programs to be more efficient.
Gastric cancer surveillance programs: today and tomorrow
Surveillance programs were currently designed to identify gastric cancers at an early stage where they were still curable (ie, to discover incident cancer). When these programs were introduced they were state-of-the-art and have provided an important service. However, now that we have evidence that we can both identify and modify an individual’s risk of developing gastric cancer, and also we can prevent progression to cancer, there is a need to rethink how surveillance can best be used. The weaknesses of the current surveillance programs include: 1) they can only examine a small proportion of the population at risk (eg, 20%), 2) they includes those that benefit as well as those who can not benefit, 3) they not prevent progression of risk, and 4) they have limited or no “preventive” role 29.
The natural history of gastritis is to progress toward increasing atrophy 35–38. This tendency is reflected in the well-described age-related increase in the incidence in atrophy and in gastric cancer. Annual surveillance does not affect the “natural history” of the disease such that the cancer risk steadily increases each year irrespective of whether the patient is or is not participating in a surveillance program. The advantage of participation is that, if cancer develops, it would likely be detected at an early stage. However, any population-based program will include those with and without H. pylori infection (ie, include those with some risk as well as those with no risk). Among those with gastritis, it will include those at very low risk (non-atrophic gastritis, duodenal ulcer, etc) and those at high risk (severe atrophic gastritis/atrophy). The surveillance capacity (number that can be examined in a year) has always been limited and non-selectivity has resulted in most of the at risk population being excluded because of their number greatly exceeds the capacity of any program. The new knowledge regarding the ability to prevent progression focus of low risk patients to becoming high risk patients and the ability to focus on those at significant cancer risk (risk stratification) allows the possibility that, by excluding those who have little or no benefit, surveillance capacity can be brought into line with need.
An example of using risk stratification to qualify patients for surveillance programs is the cohort of asymptomatic Japanese followed by Ohata et al. 24. The presence of chronic atrophic gastritis was determined using pepsinogen values. Among 4655 asymptomatic Japanese, average age approximately 49, were followed for an average of 7.7 years. Chronic atrophic gastritis was identified using pepsinogen testing and H. pylori infection by ELISA; 967 (21%) were H. pylori negative without chronic atrophic gastritis, 2341 (52%) had H. pylori infections but had non-atrophic gastritis, 1316 (28%) had H. pylori and atrophic gastritis, and 31 (0.7%) had severe atrophic gastritis. The rates of development of gastric cancer per 100,000/year were none, 107, 238, and 871, respectively. Thus the number of endoscopies per year needed to find one cancer by annual surveillance ranged from no cancers/1000 endoscopies, to 1/1000, 1/410, and 1/114, respectively. The true high risk group was only about 0.8% of the total group. In one included those with mild o moderate atrophic gastritis the proportion would increase to 29% allowing at least 70% of the population to be excluded as they would not benefit from annual surveillance following H. pylori eradication (Figure 5). Similar data are available form the study of Watabe et al. 39 of 6985 patients (approximately one-third women) average age approximately 50. In that group 47.6% were uninfected and an additional 30.5% had non-atrophic H. pylori gastritis; thus, only approximately 22% qualified for surveillance following H. pylori eradication (6% of the total were in the highest risk group of atrophy with loss of the H. pylori infection). If one examined a group with a higher average age (eg, 70) one would expect the proportion with severe atrophic gastritis and with moderate to severe atrophic gastritis to increase. However, the proportion not benefiting from surveillance would likely remain above 50%.
Figure 5. Proportion of an asymptomatic Japanese cohort in each risk group for gastric cancer.

The illustration shows th risk groups identified by a surveillance programs of a cohort of 4655 asymptomatic Japanese followed by Ohata et al. 24. H. pylori status was determined by ELISA and chronic atrophic gastritis (CAG) by pepsinogen values. The pie chart is superimposed on the age-specific gastric cancer incidence among Japanese men from 1986 41. If H. pylori eradication had been done at the outset, only the minority of subjects would have been candidates for annual surveillance (ie, those with mild [28.3%] and severe CAG [0.7%].
The relatively large group with H. pylori and pepsinogen levels below the range of normal but above the cut-off for severe atrophic gastritis (ie, atrophic gastritis but not severe atrophic gastritis) contains patients with varying degrees of cancer risk. It would be desirable to further stratify those patients into those who would benefit from surveillance from those for whom surveillance would be optional. Because pepsinogen testing has only a modest sensitivity and specificity other markers for risk will probably be required. Since the group is manageable in terms of size, we suggest research using targeted gastric biopsy and a validated histology staging system. This should allow better stratification and improved identification of the population with those likely to obtain the greatest benefit from surveillance. For example, those with OLGA stage IV might need annual surveillance, OLGA III every two years, OLGA II every 5 years, and OLGA I, no surveillance. Clearly, further work is needed to refine risk markers as well as surveillance methods and intervals and duration. Other areas of study include the role of adjuvants such as anti-inflammatory agents or gastroprotectives in terms of further risk reduction.
Risk reduction and surveillance prevention programs
Risk can be stratified into risk groups (eg, Table 3). Importantly, because H. pylori produces progressive inflammation, risk stratification to a low risk group without H. pylori eradication is likely to be temporary as infected individuals are expected to progress to higher risk groups over time 26, 27, 40. Thus, without H. pylori eradication the groups are: never infected (no need for surveillance), non-atrophic or mild atrophic gastritis (no current need for surveillance but expected them to become candidates later), and moderate of severe atrophic gastritis (current surveillance candidates). Because H. pylori eradication stops that natural progression of atrophic gastritis and thus stabilizes or reduces risk H. pylori eradication is the factor that allows for a major change it practice (ie, because it changes the natural history of the process). It is therefore critical to identify and eradicate H. pylori infections irrespective of current cancer risk as this is the basis for any successful cancer prevention/eradication program. H. pylori eradication reduces the number of risk groups to two post eradication groups: those with moderate or severe atrophic gastritis who are candidates for surveillance and everyone else (ie, those with no risk and those with low risk not needing surveillance). A potentially ideal schema for accomplishing this goal is show in Figure 6.
Table 3.
Cancer risk groups
|
Figure 6. Ideal schema for eradication of gastric cancer.

Adults receive noninvasive testing for H. pylori infection and atrophic gastritis. All H. pylori infected have confirmed H. pylori eradication. Those with atrophic gastritis are candidates for further evaluation for possible surveillance. The type and frequency of surveillance needed would depend on the relative cancer risk.
Summary
The natural history of H. pylori infection in Japan has been to fall, such that at any age, the proportion not requiring surveillance will steadily fall although without an H. pylori eradication program, it will require approximately 70 more years before gastric cancer becomes a rare disease 26. Because Helicobacter pylori infection is the cause gastric cancer, its elimination forms the cornerstone of eradication of gastric cancer. However, simply eradicating H. pylori from the entire population will not immediately solve the problem because many patients with H. pylori infections have already developed the precursor lesion, atrophic gastritis. Cure of H. pylori in these high risk patients will only reduce the risk of subsequent cancer. In contrast, treatment of low risk patients will prevent cancer. Thus, to eliminate gastric cancer it is necessary to identify and treat all infected individuals. In addition, those at increased risk for gastric cancer (ie, atrophic gastritis irrespective a age) should be considered for endoscopic surveillance to identify those cancers that do develop at an early stage. We propose that selection be based on severity and extent of atrophy be used to separate those expected to benefit from endoscopy and annual surveillance from those with little or no potential benefit. Institution of a program of surveillance restricted to those who could benefit most (ie, those with moderate or severe atrophy). This will also allow a much closer matching of surveillance capacity and surveillance need making surveillance more clinically and cost effective. We look forward to the day when Japan declares a population wide program to eradicate H. pylori while restricting cancer surveillance to those who will benefit most (Figure 2)
Acknowledgments
This material is based upon work supported in part by the Office of Research and Development Medical Research Service Department of Veterans Affairs and by Public Health Service grant DK56338 which funds the Texas Gulf Coast Digestive Diseases Center.
References
- 1.Graham DY, Lu H, Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: facts or medical myths. J Dig Dis. 2009;10:77–84. doi: 10.1111/j.1751-2980.2009.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman FL. The mortality from cancer throughout the world. Newark: Prudential Press; 1915. [Google Scholar]
- 3.von den Velden R. Ueber vorkommen und mandgel der freien salzsaure in magensaft bei gastrektasie. Deutsches Arch F Klin Med. 1879;23:369–399. [Google Scholar]
- 4.Faber K. Gastritis and its consequences. Paris: Oxford University Press; 1935. [Google Scholar]
- 5.Comfort MW. Gastric acidity before and after development of gastric cancer: its etiologic, diagnostic and prognostic significance. Ann Intern Med. 1951;36:1331–1348. doi: 10.7326/0003-4819-34-6-1331. [DOI] [PubMed] [Google Scholar]
- 6.Warren RJ, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1:1273–1275. [PubMed] [Google Scholar]
- 7.Comfort MW, Vanzant FR. Gastric acidity in carcinoma of the stomach. Am J Surg. 1934;26:447–456. [Google Scholar]
- 8.Shay H, Gershon-Cohen J, Fels SS. Gastric anacidity: Its physiologic and clinical significance and its management. Am J Dig Dis. 1941;8:115–123. [Google Scholar]
- 9.Berkson J, Butt HR, Comfort MW. Occurrence of gastric cancer in persons with achlorhydria and with pernicious anemia. Proc Staff Meet Mayo Clin. 1956;31:583–596. [PubMed] [Google Scholar]
- 10.Faber K. Chronic gastritis: its relation to achylia and ulcer. Lancet. 1927;2:902–907. [Google Scholar]
- 11.Massarrat S. Konjetzny: a German surgeon of the past century and his pioneering hypothesis of a bacterial aetiology for gastritis, peptic ulcer and gastric cancer. Z Gastroenterol. 2005;43:411–413. doi: 10.1055/s-2005-858023. [DOI] [PubMed] [Google Scholar]
- 12.Cheli R, Perasso A, Giacosa A. Gastritis a clinical review. Berlin: Springer-Verlag; 1987. [Google Scholar]
- 13.Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975;2:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 14.Kokkola A, Kosunen TU, Puolakkainen P, Sipponen P, Harkonen M, Laxen F, et al. Spontaneous disappearance of Helicobacter pylori antibodies in patients with advanced atrophic corpus gastritis. APMIS. 2003;111:619–624. doi: 10.1034/j.1600-0463.2003.1110604.x. [DOI] [PubMed] [Google Scholar]
- 15.Weck MN, Gao L, Brenner H. Helicobacter pylori infection and chronic atrophic gastritis: associations according to severity of disease. Epidemiology. 2009;20:569–574. doi: 10.1097/EDE.0b013e3181a3d5f4. [DOI] [PubMed] [Google Scholar]
- 16.Ekstrom AM, Held M, Hansson LE, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology. 2001;121:784–791. doi: 10.1053/gast.2001.27999. [DOI] [PubMed] [Google Scholar]
- 17.Graham DY, Sung JY. Helicobacter pylori. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger & Fordtran’s Gastrointestinal and liver disease. Pathophysiology, diagnosis, management. 7. Philadelphia: WB Saunders Co; 2006. pp. 1049–1066. [Google Scholar]
- 18.Cardenas VM, Mulla ZD, Ortiz M, Graham DY. Iron deficiency and Helicobacter pylori infection in the United States. Am J Epidemiol. 2006;163:127–134. doi: 10.1093/aje/kwj018. [DOI] [PubMed] [Google Scholar]
- 19.Dholakia KR, Dharmarajan TS, Yadav D, Oiseth S, Norkus EP, Pitchumoni CS. Vitamin B12 deficiency and gastric histopathology in older patients. World J Gastroenterol. 2005;11:7078–7083. doi: 10.3748/wjg.v11.i45.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DuBois S, Kearney DJ. Iron-deficiency anemia and Helicobacter pylori infection: a review of the evidence. Am J Gastroenterol. 2005;100:453–459. doi: 10.1111/j.1572-0241.2005.30252.x. [DOI] [PubMed] [Google Scholar]
- 21.Hershko C, Lahad A, Kereth D. Gastropathic sideropenia. Best Pract Res Clin Haematol. 2005;18:363–380. doi: 10.1016/j.beha.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Tan HJ, Goh KL. Changing epidemiology of Helicobacter pylori in Asia. J Dig Dis. 2008;9:186–189. doi: 10.1111/j.1751-2980.2008.00344.x. [DOI] [PubMed] [Google Scholar]
- 23.Graham DY. Helicobacter pylori infection in the pathogenesis of duodenal ulcer and gastric cancer: a model. Gastroenterology. 1997;113:1983–1991. doi: 10.1016/s0016-5085(97)70019-2. [DOI] [PubMed] [Google Scholar]
- 24.Ohata H, Kitauchi S, Yoshimura N, Mugitani K, Iwane M, Nakamura H, et al. Progression of chronic atrophic gastritis associated with Helicobacter pylori infection increases risk of gastric cancer. Int J Cancer. 2004;109:138–43. doi: 10.1002/ijc.11680. [DOI] [PubMed] [Google Scholar]
- 25.Snaith A, el-Omar EM. Helicobacter pylori: host genetics and disease outcomes. Expert Rev Gastroenterol Hepatol. 2008;2:577–585. doi: 10.1586/17474124.2.4.577. [DOI] [PubMed] [Google Scholar]
- 26.Graham DY, Uemura N. Natural history of gastric cancer after Helicobacter pylori eradication in Japan: after endoscopic resection, after treatment of the general population, and naturally. Helicobacter. 2006;11:139–143. doi: 10.1111/j.1523-5378.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- 27.Graham DY, Shiotani A. The time to eradicate gastric cancer is now. Gut. 2005;54:735–738. doi: 10.1136/gut.2004.056549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham DY, Shiotani A, El-Zimaity HM. Chromoendoscopy points the way to understanding recovery of gastric function after Helicobacter pylori eradication. Gastrointest Endosc. 2006;64:686–690. doi: 10.1016/j.gie.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Graham DY. Gastric cancer surveillance or prevention plus targeted surveillance. Jpn J Helicobacter Res. 2009;10:9–14. [PMC free article] [PubMed] [Google Scholar]
- 30.Rugge M, Correa P, Di Mario F, el-Omar E, Fiocca R, Geboes K, et al. OLGA staging for gastritis: a tutorial. Dig Liver Dis. 2008;40:650–658. doi: 10.1016/j.dld.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 31.Graham DY, Kato M, Asaka M. Gastric endoscopy in the 21st century: appropriate use of an invasive procedure in the era of non-invasive testing. Dig Liver Dis. 2008;40:497–503. doi: 10.1016/j.dld.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miki K, Morita M, Sasajima M, Hoshina R, Kanda E, Urita Y. Usefulness of gastric cancer screening using the serum pepsinogen test method. Am J Gastroenterol. 2003;98:735–739. doi: 10.1111/j.1572-0241.2003.07410.x. [DOI] [PubMed] [Google Scholar]
- 33.Urita Y, Hike K, Torii N, Kikuchi Y, Kanda E, Sasajima M, Miki K. Serum pepsinogens as a predicator of the topography of intestinal metaplasia in patients with atrophic gastritis. Dig Dis Sci. 2004;49:795–801. doi: 10.1023/b:ddas.0000030091.92379.91. [DOI] [PubMed] [Google Scholar]
- 34.El-Zimaity HM, Ota H, Graham DY, Akamatsu T, Katsuyama T. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer. 2002;94:1428–1436. doi: 10.1002/cncr.10375. [DOI] [PubMed] [Google Scholar]
- 35.Oi M, Oshida K, Sugimura S. The location of gastric ulcer. Gastroenterology. 1959;36:45–56. [PubMed] [Google Scholar]
- 36.Kimura K, Takemoto T. An endoscopic recognition of the atrophic border and its significance in chronic gastritis. Endoscopy. 1969;1:87–97. [Google Scholar]
- 37.Kimura K. Chronological transition of the fundic-pyloric border determined by stepwise biopsy of the lesser and greater curvatures of the stomach. Gastroenterology. 1972;63:584–592. [PubMed] [Google Scholar]
- 38.Hebbel R. The topography of chronic gastritis in otherwise normal stomachs. Am J Pathol. 1949;25:125–141. [PMC free article] [PubMed] [Google Scholar]
- 39.Watabe H, Mitsushima T, Yamaji Y, Okamoto M, Wada R, Kokubo T, et al. Predicting the development of gastric cancer from combining Helicobacter pylori antibodies and serum pepsinogen status: a prospective endoscopic cohort study. Gut. 2005;54:764–768. doi: 10.1136/gut.2004.055400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–397. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 41.Cancer incidence and incidence rates in Japan in 1986: estimates based on data from nine population-based cancer registries. The Research Group for Population-based Cancer Registration in Japan. Jpn J Clin Oncol. 1991;21:318–323. [PubMed] [Google Scholar]