

Abstract
Over 160 rare genetic variants in presenilin 1 (PSEN1) are known to cause Alzheimer’s disease (AD). In this study we screened a family with early-onset AD for mutations in PSEN1 using direct DNA sequencing. We identified a novel PSEN1 genetic variant which results in the substitution of a Proline with an Alanine at codon 117 (P117A). The P117A variant was present in all demented individuals and fifty percent of at risk individuals. This variant occurs at a site where three other disease-causing variants have been previously observed. In vitro functional studies demonstrate that the P117A variant results in an altered Aβ42/total Aβ ratio consistent with an AD causing mutation. The P117A variant is a novel mutation in PSEN1, which causes early-onset AD in an autosomal dominant manner.
Keywords: Alzheimer’s disease, presenilin 1, mutation, amyloid
The presenilins form the catalytic core of the γ-secretase complex, which is required to produce amyloid-beta (Aβ) from full length amyloid-beta precursor protein (APP)[9]. Over 160 rare genetic variants in PSEN1 are known to cause Alzheimer’s disease (AD)[1] (http://www.molgen.ua.ac.be/ADMutations/). With few exceptions[3] these variants result in onset of dementia before age 55yrs, or early-onset AD (EOAD). Most of these variants occur in transmembrane regions of PSEN1 and appear to alter the production and deposition of 42 amino acid Aβ fragments (Aβ42)[10]. Thirteen variants, including P117L, P117R and P117S occur in the first hydrophilic loop of PSEN1[2, 6, 8, 11 ]. In this study we sequenced PSEN1 in a family with EOAD and identified a novel variant which segregates with AD.
The pedigree is shown in figure 1. Eight individuals over four generations of this family have developed dementia during the fourth decade of life. The proband (IV-3) was identified in the local hospital of a small town in the state of Valle del Cauca, Colombia at 35 years at age. The clinical evaluation included a comprehensive interview with the subject as well as collection of information from medical records and family members. The clinical diagnoses were made using the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable Alzheimer’s disease along with a mini-mental state exam (MMSE). The routine evaluation of a patient with dementia included computed tomography (CT) head scan, and the following laboratory tests: complete blood count (CBC), standard chemistry, thyroid stimulant hormone (TSH), Venereal Disease Research Laboratory test (VDRL), Folic acid and Vitamin B12 levels. Information regarding the affected deceased family members was obtained through family members and review of hospital records.
Figure 1.
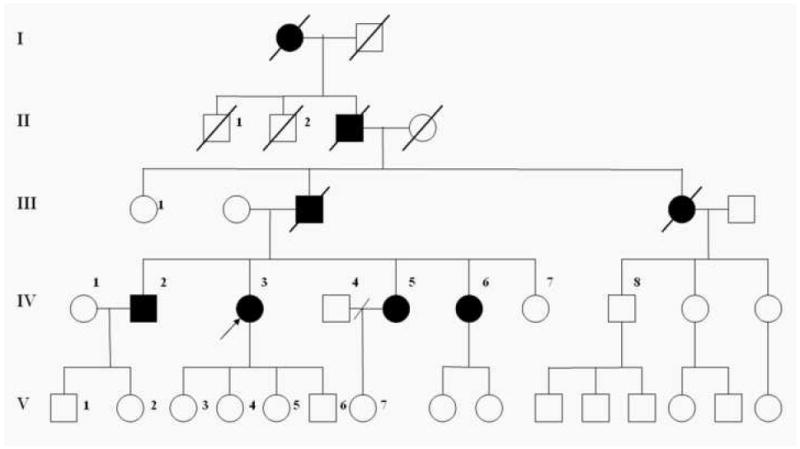
Pedigree. Open symbols denote unaffected individuals; closed symbols denote affected individuals, diagonal lines denote deceased individuals. The index case is denoted with an arrow.
We sequenced PSEN1 in three AD cases and two at risk individuals. Primers were designed to target each exon of PSEN1 and at least 50bp of 3’ and 5’ flanking intronic sequence. Primers for sequencing were designed using consensus sequence from Ensembl (http://www.ensembl.org/index.html) and the software PRIMER3[5] (primer sequences will be provided on request). Sequencing was performed using ABI Big Dye version 3.1 (Applied Biosystems, Foster City, CA). Sequence analysis was performed using Sequencher software (Gene Codes, Ann Arbor, MI).
To test the effects of the P117A mutation, we transfected P117A mutant PSEN1 cDNA into HEK cells and measured secreted Aβ40 and Aβ42. The QuickChange II site-directed mutagenesis kit (Stratagene, Cedar Creek, TX) was used to introduce the P117A point mutation into wtPSEN1 cDNA. The construct was confirmed by direct sequencing. HEK cells were transfected with APPΔNL and wtPSEN1, P117A or ΔE9 (a PSEN1 familial AD variant which is known to have a large effect on APP processing[4]). The cDNA constructs for wtPSEN1, APPΔNL and ΔE9 and methods for 40 amino acid Aβ (Aβ40) and Aβ42 measurement have been described previously[7]. Total Aβ (Aβ42 + Aβ40) and Aβ42/total Aβ ratio were compared between the wtPSEN1 and P117A variant cells using a t-test.
We have identified a pedigree with a four-generation history of early onset dementia of the Alzheimer type. Eight individuals in this pedigree have been diagnosed with a dementing disorder. The transmission of AD in this family is consistent with autosomal dominant inheritance (Figure 1). The proband (IV-3) was visited the local hospital and was evaluated by Dr. Henao-Martinez.
Subject IV-3
The proband (IV-3) developed progressive memory impairment with gradual onset at 32 years of age. She visited the hospital repeatedly (even several times a day), often forgetting the information she had provided previously. She is a single mother and experienced difficulties in caring for her children; preparing baby bottles several times and misplacing documents or personal objects frequently. This difficulty led to the need for a neighbor’s assistance. Finally local government authorities took custody of her children because she was unable to provide them with appropriate care. Her speech was characterized by confabulation and repetitive phrases. At age 35 she exhibited significant cognitive impairment affecting more than two cognitive areas, with an MMSE score of 10/30, showing severe memory dysfunction, acalculia, agraphia, impairment in visuospatial tasks and some degree of ideational apraxia. The CT of her head showed diffuse cerebral atrophy with all laboratory measurements in the dementia workup within normal limits.
Subject IV-2
The proband’s brother developed symptoms at 33 years of age. He was married with two children and worked as an auto mechanic. He started to forget and misplace objects frequently. He became unable to perform his duties satisfactorily and was released from his job. Later, he attempted to assist another auto mechanic without success. He required his wife’s assistance in several daily activities and wandered frequently. At the time of the evaluation he presented with marked cognitive deficits, including disorientation, memory dysfunction, acalculia, agraphia and ideational apraxia, with an MMSE score of 5/30. The physical exam and laboratory workup was unremarkable but the CT showed diffuse cerebral atrophy.
Subject IV-5
Subject IV-5 was also a single mother with one child. She started to develop symptoms at the age of 33 years. She required her daughter’s assistance for common tasks at home. At 36 years old she exhibited repetitive speech with marked forgetfulness. She worked as a housekeeper, but gradually became incapable of performing her job satisfactorily. When examined by a physician the patient presented with acalculia, agraphia and memory impairment (MMSE of 15/30), with an otherwise normal dementia workup.
A transversion from C to G, resulting in a change from Proline (CCA) to Alanine (GCA) at codon 117 (P117A) was found in each of the demented subjects and fifty percent of at risk individuals (Figure 1, 2). Codon 117 is located in the first hydrophilic loop of the PSEN1 protein[6] and is the site of three other disease-causing variants[8, 11]. No variants that segregate with disease were found elsewhere in the PSEN1 gene in members of this family.
Figure 2.
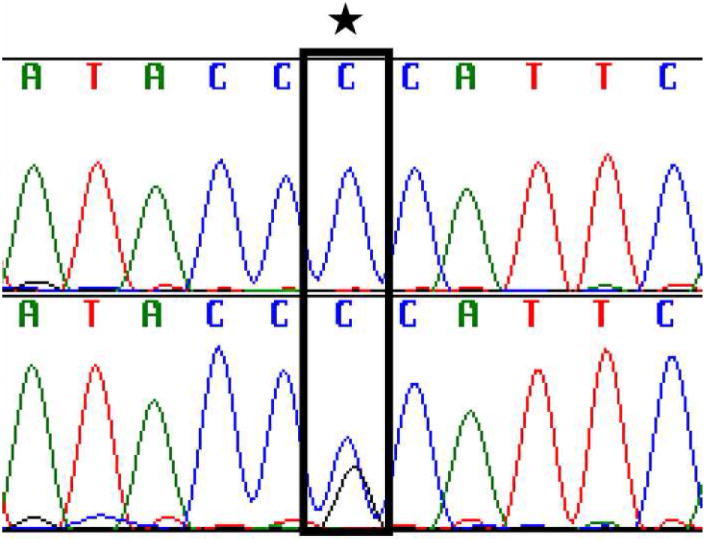
Chromatogram of a portion of PSEN1 exon 5 sequence. This displays a base pair change (P117A; bottom) compared to the family control (top).
The Aβ42/total Aβ ratio in media from P117A cells was significantly higher than that of wtPSEN1 cells (p=2.79×10-7; Figure 3). Total Aβ levels from P117A were not significantly different from wtPSEN1.
Figure 3.
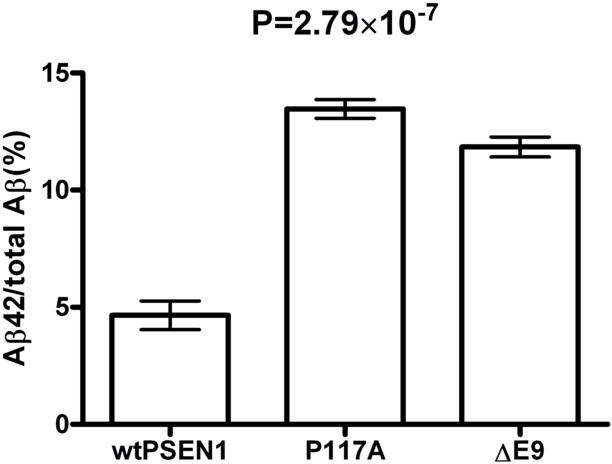
Ratio of Aβ42 to total Aβ (Aβ42 + Aβ40) in the media of wild type PSEN1 versus P117A HEK cells. The y-axis represents Aβ42 as a percentage of total Aβ levels. The x-axis shows the levels for cells with wild type PSEN1 (wtPSEN1), cells with the PSEN1 P117A variant (P117A), and cells with the PSEN1 mutation delta E9 (ΔE9). P-value is for a t-test contrasting wtPSEN1 and P117A.
The P117A variant segregates with disease in this Columbian kindred with an autosomal dominant pattern of early-onset AD. Three disease-causing variants at codon 117 of PSEN1 have been described previously. The P117L mutation was described in a single family with onset between 24 and 34 years[8]. The P117R variant was identified in a female patient, who has had complaints of memory loss beginning at 36 years of age[11]. The P117S mutation was observed in a family with AAO ranging from 29 to 33 years[2].
AAO for the P117A variant is similar to that of the previously reported variants at codon 117, with mean AAO at 32.7 years. Clinical features of individuals with these four variants are quite similar; cognitive impairment in memory, learning and visuospatial tasks and rapid disease progression were observed with all four variants. Variants at this codon consistently cause a very severe early onset form of the disease even for PSEN1 mutations.
HEK cells carrying the P117A variant showed significantly higher Aβ42/total Aβ ratio than wild type cells. This finding is clearly consistent with the observation of increased Aβ42/total Aβ ratio in several other known PSEN1 AD mutations, including P117L[8] and P177S[2] and suggests that this mutation causes AD via a similar mechanism. These findings reiterate the central role of PSEN1 in AD and add to the list of known disease causing variants in PSEN1.
Acknowledgments
We gratefully acknowledge the subjects who participated in this study. We also acknowledge Sarah Jacquart, Brittny Davis and David Grotsky for their assistance in data collection, Luis Felipe Diago, Hector Henao, and Gladys Martinez for their cooperation in the collection and shipment of the samples and Dr. Francisco Lopera for his invaluable guidance and support through the entire project. This study was supported by grant P50 AG05681 from the National Institutes of Health/National Institute of Aging, Bethesda, MD (Washington University Alzheimer’s Disease Research Center, St Louis, MO) and by Barnes Jewish Foundation. JSKK was a Ford Foundation predoctoral fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dillen K, Annaert W. A two decade contribution of molecular cell biology to the centennial of Alzheimer’s disease: are we progressing toward therapy? Int Rev Cytol. 2006;254:215–300. doi: 10.1016/S0074-7696(06)54005-7. [DOI] [PubMed] [Google Scholar]
- 2.Dowjat WK, Kuchna I, Wisniewski T, Wegiel J. A novel highly pathogenic Alzheimer presenilin-1 mutation in codon 117 (Pro117Ser): Comparison of clinical, neuropathological and cell culture phenotypes of Pro117Leu and Pro117Ser mutations. J Alzheimers Dis. 2004;6:31–43. doi: 10.3233/jad-2004-6105. [DOI] [PubMed] [Google Scholar]
- 3.Kauwe JS, Jacquart S, Chakraverty S, Wang J, Mayo K, Fagan AM, Holtzman DM, Morris JC, Goate AM. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann Neurol. 2007;61:446–53. doi: 10.1002/ana.21099. [DOI] [PubMed] [Google Scholar]
- 4.Mehta ND, Refolo LM, Eckman C, Sanders S, Yager D, Perez-Tur J, Younkin S, Duff K, Hardy J, Hutton M. Increased Abeta42(43) from cell lines expressing presenilin 1 mutations. Ann Neurol. 1998;43:256–8. doi: 10.1002/ana.410430217. [DOI] [PubMed] [Google Scholar]
- 5.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 6.Spasic D, Tolia A, Dillen K, Baert V, De Strooper B, Vrijens S, Annaert W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem. 2006;281:26569–77. doi: 10.1074/jbc.M600592200. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Beher D, Nyborg AC, Shearman MS, Golde TE, Goate A. C-terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation. J Neurochem. 2006;96:218–27. doi: 10.1111/j.1471-4159.2005.03548.x. [DOI] [PubMed] [Google Scholar]
- 8.Wisniewski T, Dowjat WK, Buxbaum JD, Khorkova O, Efthimiopoulos S, Kulczycki J, Lojkowska W, Wegiel J, Wisniewski HM, Frangione B. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport. 1998;9:217–21. doi: 10.1097/00001756-199801260-00008. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe MS, De Los Angeles J, Miller DD, Xia W, Selkoe DJ. Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry. 1999;38:11223–30. doi: 10.1021/bi991080q. [DOI] [PubMed] [Google Scholar]
- 10.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–82. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 11.Zekanowski C, Styczynska M, Peplonska B, Gabryelewicz T, Religa D, Ilkowski J, Kijanowska-Haladyna B, Kotapka-Minc S, Mikkelsen S, Pfeffer A, Barczak A, Luczywek E, Wasiak B, Chodakowska-Zebrowska M, Gustaw K, Laczkowski J, Sobow T, Kuznicki J, Barcikowska M. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol. 2003;184:991–6. doi: 10.1016/S0014-4886(03)00384-4. [DOI] [PubMed] [Google Scholar]