

Abstract
Conjugated linoleic acid (CLA) inhibits tumorigenesis and tumor growth in most model systems, an effect mediated in part by its pro-apoptotic activity. We previously showed that trans-10,cis-12 CLA induced apoptosis of p53-mutant TM4t mouse mammary tumor cells through both mitochondrial and endoplasmic reticulum stress pathways. In the current study, we investigated the role of AMP-activated protein kinase (AMPK), a key player in fatty acid metabolism, in CLA-induced apoptosis in TM4t cells. We found that t10,c12-CLA increased phosphorylation of AMPK, and that CLA-induced apoptosis was enhanced by the AMPK agonist 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) and inhibited by the AMPK inhibitor compound C. The increased AMPK activity was not due to nutrient/energy depletion since ATP levels did not change in CLA-treated cells, and knockdown of the upstream kinase LKB1 did not affect its activity. Furthermore, our data do not demonstrate a role for the AMPK-modulated mTOR pathway in CLA-induced apoptosis. Although CLA decreased mTOR levels, activity was only modestly decreased. Moreover, rapamycin, which completely blocked the activity of mTORC1 and mTORC2, did not induce apoptosis, and attenuated rather than enhanced CLA-induced apoptosis. Instead, the data suggest that CLA-induced apoptosis is mediated by the AMPK-p38 MAPK-Bim pathway: CLA-induced phosphorylation of AMPK and p38 MAPK, and increased expression of Bim, occurred with a similar time course as apoptosis; phosphorylation of p38 MAPK was blocked by compound C; the increased Bim expression was blocked by p38 MAPK siRNA; CLA-induced apoptosis was attenuated by the p38 inhibitor SB-203580 and by siRNAs directed against p38 MAPK or Bim.
Keywords: conjugated linoleic acid, AMP kinase, p38 MAP kinase, mTOR, mammary tumor cells
1. Introduction
A number of studies have demonstrated that the polyunsaturated fatty acid conjugated linoleic acid (CLA)1 exerts chemopreventive and therapeutic activity in a number of rodent and human tumor models, including mammary, gastric, skin, prostate and colon cancers (reviewed in [1,2]). CLA is a collective term for a group of geometrically-related linoleic acid isomers with conjugated double bonds. The c9,t11 isomer is the major form of CLA found in dairy products and ruminant meats, while t10,c12-CLA is found with the c9,t11 isomer in commercial supplements. Both CLA isomers inhibit angiogenesis [3], as well as carcinogenesis and metastasis in several, although not all, in vivo mammary model systems [4-7]. Contributing to the in vivo efficacy of CLA is the incorporation of both isomers into the triglyceride fraction of mammary adipocytes, where they serve as a highly concentrated local CLA reservoir within the MG [4,8]. In vitro, however, our previous studies showed that t10,c12 is the effective form of CLA in inducing apoptosis of p53-mutant TM4t mouse mammary tumor cells, an event that occurs through both Bcl-2 mediated-mitochondrial and ER stress pathways [9,10].
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme, which is comprised of a catalytic α subunit and two regulatory subunits (β and γ). Each subunit has multiple isoforms (α1, α2, β1, β2, γ1, γ2, and γ3) with different subcellular and tissue distribution [11,12]. AMPK was first recognized as a pivotal sensor of cellular energy status, and dysregulation of the AMPK pathway has been implicated in several cardiovascular-related diseases and metabolic disorders [13-15]. When activated by intracellular ATP depletion, AMPK can orchestrate numerous metabolic processes to minimize ATP consumption and promote ATP generation, including reduced protein synthesis, inhibition of lipogenesis, enhanced glucose transport and increased fatty acid oxidation [16,17]. In addition to the energy stress-dependent mechanism, AMPK can also be activated by several stress-independent mechanisms, including osmotic and oxidative stress, calcium signaling, and fatty acids [18-24]. Activation of AMPK occurs through phosphorylation of Thr172 on the α subunit by several upstream kinases (AMPKKs) including LKB1 [25,26], calcium/calmodulin-dependent protein kinase kinase α (CaMKKα) [27], CaMKKβ [28], and TGFβ-activated kinase-1 (TAK1) [29]. In contrast, both AKT and cyclic AMP-dependent protein kinase (PKA) have been shown to inhibit the activation of AMPK by phosphorylating Ser485/491 on its α1 and α2 subunits respectively [30-32].
In addition to its role in the homeostatic regulation of ATP, AMPK was found to mediate apoptosis in response to energy stress and chemotherapeutic agents [33-36], and its activation was shown to inhibit cell proliferation and tumorigenesis [34,37-41]. The anti-tumor activity of AMPK has been attributed to its inhibitory action on the mTOR survival pathway, which occurs through AMPK-catalyzed phosphorylation and subsequent activation of tuberous sclerosis complex 2 (TSC2), a negative regulator of the mammalian target of rapamycin (mTOR) [42].
mTOR is a key regulator of cell growth and proliferation, and its dysregulation has been linked with many human diseases, including cancer, diabetes, and cardiovascular disease [43-46]. Recent studies revealed that mTOR is integrated within two structurally and functionally distinct multi-protein complexes, mTORC1 and mTORC2. mTORC1, which consists of mTOR, mLST8, Raptor, and proline-rich AKT substrate 40 (PRAS40, a negative regulator), is a nutrient/energy sensor [43,44,47,48]. On the other hand, mTORC2, which consists of mTOR, mLST8, mSIN1, Protor 1 and Rictor, responds to growth factors but is nutrient-insensitive. mTORC1 activates protein synthesis and cell growth through regulating p70S6 kinase (p70S6K) and eukaryotic initiation factor 4E-binding protein (4E-BP1) activity, while mTORC2 phosphorylates AKT on Ser473 [49], activating cell growth, proliferation, and survival [43,49]. A number of studies have suggested that the ATP-sensing capability of mTORC1 is regulated by the LKB1/AMPK/TSC1/2 pathway [44,48].
Because of the central role played by AMPK in modulating fatty acid metabolism and cell viability, the major objective of this study was to determine whether AMPK might be involved in mediating the apoptotic effect of t10,c12-CLA in TM4t mammary tumor cells. Our previous studies had shown that apoptosis induced by t10,c12-CLA is via the caspase 9 and 3 pathway [9]. In this study, we examined the upstream events responsible for mediating the caspase-dependent apoptosis. Here, we demonstrate that the AMPK-p38-Bim pathway is activated by t10,c12-CLA, but not by c9,t11-CLA. Blocking this pathway with an AMPK inhibitor or siRNAs against p38 or Bim reduced cleavage of PARP, caspase 9 and 3. Inhibiting mTOR activity by rapamycin was found to attenuate t10,c12-CLA-induced PARP cleavage, suggesting that the AMPK-mediated t10,c12-CLA apoptotic effect cannot be via the suppression of the AKT/mTOR/p70S6K survival pathway. Moreover, results from ATP assay and LKB1 knockdown suggest that the activation of AMPK is independent of the LKB1-dependent energy-depletion pathway.
2. Materials and methods
2.1. Cell culture
The TM4t mouse mammary tumor cell line was obtained from Dr. Dan Medina at Baylor College of Medicine. Cells were cultured in DMEM-F12 supplemented with 2% adult bovine serum (ABS), 10 μg/ml insulin, 5ng/ml EGF and 5 μg/ml gentamicin.
2.2. Materials
The CLA isomers, each ∼98% pure, were purchased from Larodan Fine Chemicals (Malmö, Sweden) and prepared as the sodium salt as described previously [50]. Anti-p38, anti-phospho AKT-Thr308, anti-phospho AKT-Ser473, anti-phospho ACC, anti-ACC, anti-phospho mTOR, anti-mTOR, anti-LKB1, anti-cleaved poly (ADP-ribose) polymerase (PARP), anti-cleaved caspase 9, anti-cleaved caspase 3, anti-phospho-AMPK-Thr172, anti-AMPK, anti-phospho p70S6K, anti-p70S6K, and AICAR were purchased from Cell Signaling Technology (Danvers, MA). Anti-Bim was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin and compound C were purchased from Calbiochem (La Jolla, CA). Rapamycin was purchased from LC laboratories. SRB (sulforhodamine B) and p38 inhibitor SB-203580 were purchased from Sigma (St. Louis, MO).
2.3. Western blot
One × 105/ml cells were plated in dishes and cultured overnight before CLA treatment. To generate the cell lysates for western blot, cells were collected and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM EDTA, 1% Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS), supplemented with phosphatase inhibitor cocktail 1 and 2 (Sigma), and 1% (v/v) protease inhibitor cocktail (Sigma). Protein concentration was determined by Bio-Rad protein assay. Lysates were separated by SDS-PAGE and proteins detected by chemiluminescence. All data reported are representative of at least two independent experiments.
2.4. Cell number determination
The SRB assay modified from the original protocol [51] was used to estimate cell number. Briefly, cells were plated in a 24-well plate and cultured overnight before CLA treatment. After 72h CLA treatment with or without drug, culture media were aspirated followed by the addition of 400 μl PBS plus 200 μl ice cold 50% (w/v) TCA and incubation for 1h at 4°C to fix the viable cell monolayer. After fixation, cells were washed 5 times with distilled water and the plates inverted to drain. The cells were then stained with 300 μl of 0.4% SRB solution for 5 min at room temperature. The excess dye was removed by washing 5 times with 1% (v/v) acetic acid. The protein-bound dye was dissolved in 1 ml of 10 mM Tris base solution for OD determination at 570 nm using a microplate reader.
2.5. ATP assay
Cellular ATP concentration was determined by the luminescence ATP detection assay system from PerkinElmer (Boston, MA), using a modification of the company's protocol. Briefly, at each time point, cells were harvested and lysed in 100 μl lysis buffer for 10 min. at room temperature. After incubation, mixtures were passed through a 21G syringe 5 times. Supernates were collected by centrifugation at 16,000 × g for 10 min at 4°. Lysates were used directly for luciferase and protein assays. Protein concentrations were determined by the DC protein assay kit from Bio-Rad (Hercules, CA) and used to normalize luciferase activity.
2.6. siRNA transfection
Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) was used for siRNA transfection. Negative control siRNA (4611) was purchased from Ambion (Austin, TX). LKB1, p38α and p38β siRNAs were purchased from Santa Cruz. Before transfection, TM4t cells were plated in 60 mm dishes, cultured overnight, then transfected with 1 μg of siRNA. Cells were transfected for 6 h, followed by replacement of fresh medium with or without 40 μM CLA for various times as indicated.
2.7. Statistical analysis
ANOVA followed by the Holm-Sidak procedure for all pairwise multiple comparison was used to evaluate statistical differences in the SRB assay ratios. The t-test was used to compare the experimental versus control groups in the ATP assay. P<0.05 was considered statistically significant.
3. Results
3.1. t10,c12-CLA activates AMPK and induces apoptosis
We previously demonstrated that t10,c12-CLA induced apoptosis of TM4t cells, as measured by flow cytometric evaluation of annexin V-PI double staining, as well as cleavage of caspase 9, caspase 3 and PARP [9]. To construct the signaling network underlying the induction of apoptosis by CLA, we first treated TM4t cells with t10,c12- or c9,t11-CLA for 1-3 days. Using PARP cleavage as an index of apoptosis, we found that t10,c12-CLA, but not c9,t11-CLA, induced PARP cleavage as early as the 48 hr time point (Fig. 1A). t10,c12-CLA also activated AMPK activity at 48 hr, and more extensively (10-fold) at 72 hr, by enhancing its phosphorylation at Thr172 (Fig. 1B). Activation of AMPK was further confirmed by the t10,c12-CLA-induced increase in phosphorylation of acetyl coenzyme A carboxylase (ACC), an AMPK substrate (Fig. 1B).
Figure 1.
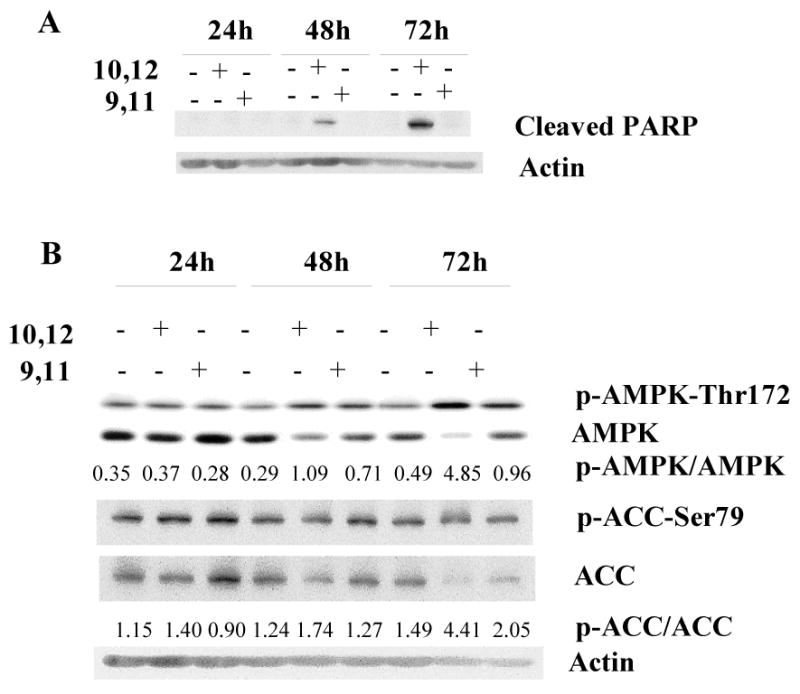
t10,c12-CLA induces PARP cleavage, concurrent with phosphorylation of AMPK and acetyl CoA carboxylase (ACC). TM4t cells were treated with 40 μM t10,c12- or c9,t11-CLA for various times and proteins evaluated by western blot. This figure shows that CLA-induced PARP cleavage is isomer-specific, and that phosphorylation of AMPK and ACC occurs to a much greater extent with t10,c12-CLA than with c9,t11-CLA. A, PARP cleavage. B, Phospho-Thr172 AMPK, total AMPK, phospho-Ser79 ACC and total ACC. Actin was used as a loading control. The numbers indicate the ratio of the phospho-enzyme to the total enzyme.
In order to determine whether AMPK activation could trigger apoptosis, cells were treated with t10,c12-CLA in combination with the AMPK activator, AICAR, or the AMPK inhibitor, compound C. Fig. 2A shows that AICAR itself induced PARP cleavage and potentiated t10,c12-CLA-induced PARP cleavage at 24 and 48 hr. In contrast, inhibition of AMPK activity with compound C reduced CLA-induced cleavage of PARP at 48 and 72 hr (Fig. 2B). Cleavage of caspases 9 and 3 essentially followed the same pattern. Concurrent with its effects on PARP cleavage, compound C also increased the number of viable CLA-treated cells in a concentration-dependent manner, as indicated in the microscope images (Fig. 2C, compare iii, vi and viii with iv), and the SRB assay on the attached viable cells (Fig. 2D). These findings strongly suggest that AMPK activation by CLA plays an important role in mediating t10,c12-CLA-induced apoptosis.
Figure 2.



The AMPK activator AICAR enhances and the AMPK inhibitor Compound C (Comp C or CC) attenuates CLA-induced cleavage of PARP, caspase 9 and caspase 3. A and B, TM4t cells were treated with or without 40 μM of t10,c12-CLA and/or 500 μM AICAR (A) or 20 μM Compound C (B) for various times and cleavage of PARP, caspase-9 and caspase-3 was evaluated by western blot. C and D, TM4t cells were treated for 72 hr with or without 40 μM of t10,c12-CLA and/or various concentrations of Compound C, photographed under the 10× microscope objective (C), then viable cell number estimated by the SRB assay on the attached (viable) cell monolayer (D). Compound C alone at a concentration of 20 μM modestly decreased cell number (P<0.05, left panel). The data in the right panel of D show the ratio of the ODs from the SRB assay from CLA-treated to untreated cells. The asterisk denotes a statistically significant difference of the 20 μM compound C group from each of the other groups.
3.2. Activation of AMPK by t10,c12-CLA does not result from energy depletion
We next tested the hypothesis that the activation of AMPK by t10,c12-CLA was a result of energy depletion. To do this, ATP levels were measured in whole cell lysates from cells treated with or without t10,c12-CLA for various times. As shown in Fig. 3A, cellular ATP levels were not significantly reduced in cells treated with t10,c12-CLA at any of the time points tested. Moreover, t10,c12-CLA actually maintained ATP levels up to 72 hr. It is unlikely, then, that activation of AMPK results from energy depletion. To further confirm this notion, we used siRNA to knock down the expression of LKB1, since LKB1 has been identified as one of the upstream effectors of the AMPK pathway in response to cellular energy crisis [52]. Fig. 3B shows that reducing LKB1 protein levels did not affect the phosphorylation pattern of AMPK in the presence of t10,c12-CLA.
Figure 3.


Decreased cellular energy levels are not responsible for activation of AMPK. A. Intracellular ATP levels, measured using a luminescence assay, are maintained in cells treated with t10,c12-CLA. TM4t cells were treated without or with 40 μM t10,c12-CLA for 24, 48, or 72 hr and cells harvested for measurement of ATP levels. Each bar represents the mean ± SEM of triplicate wells from a representative experiment. *, indicates a statistically significant difference compared to the control at the same time point. B. Attenuation of LKB1 expression by siRNA does not affect CLA-induced activation of AMPK. TM4t cells were transfected with LKB1 siRNA or control siRNA for 6 hr followed by treatment without or with 40 μM t10,c12-CLA for 24, 48 or 72 hr. Cells were harvested, and proteins evaluated by western blot.
3.3. mTOR does not play a significant role in mediating t10,c12-CLA – induced apoptosis
We next focused on the downstream route used by CLA-activated AMPK to initiate apoptosis. In this regard, mTOR has been recognized as one of the major downstream targets of the AMPK pathway [44,48]. AMPK can exert its inhibitory action on mTOR indirectly via phosphorylation and activation of TSC2 [42], or by phosphorylation of the mTOR binding partner raptor [53]. To determine if mTOR might play a role in mediating the apoptotic effect of t10,c12-CLA in the TM4t mammary tumor cell model, we initially measured the phosphorylation status of one of its targets, p70S6K, as an index of mTOR activity. Inhibition of AMPK by compound C showed the expected increase in phosphorylation of p70S6K at all time points (Fig. 4A), demonstrating the presence of the AMPK-mTOR pathway in TM4t cells. Treatment with t10,c12-CLA modestly reduced the phosphorylation status of p70S6K at 24 and 48 hr, but increased it slightly at 72 hr. The initial decrease was blocked by the AMPK inhibitor compound C at 24 hr, although not at 48 hr (Fig. 4A). Moreover, at the 72 hr time point when the CLA-treated cells were undergoing apoptosis (Fig. 2), the combination of compound C with CLA unexpectedly significantly increased the phosphorylation status of 70S6K.
Figure 4.
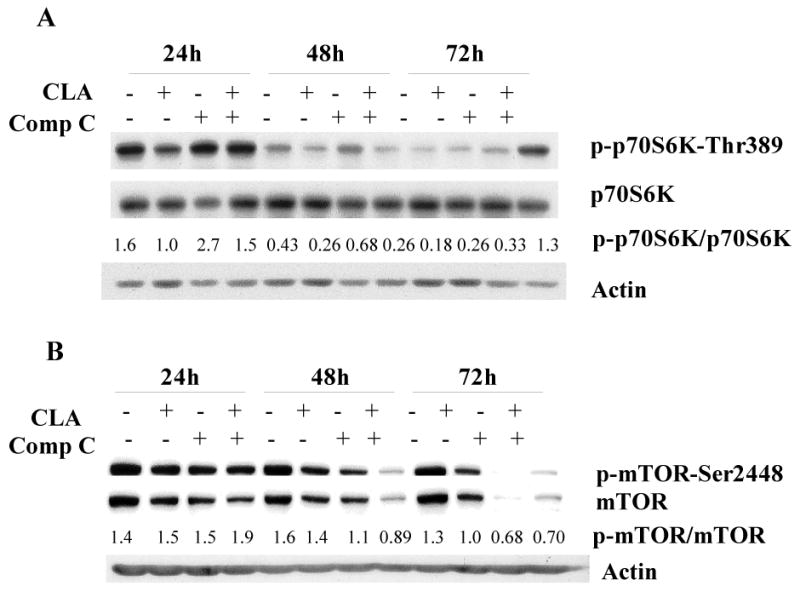
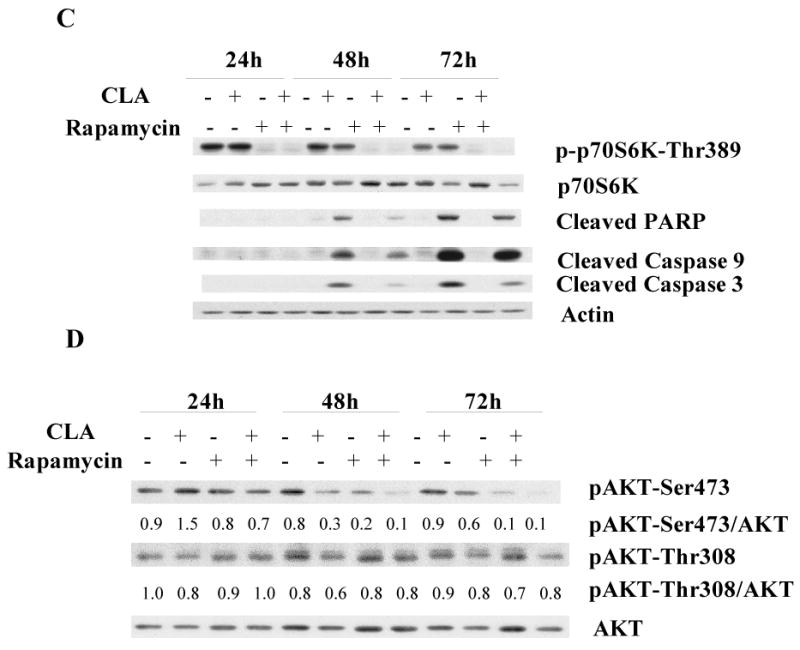
The effect of CLA on mTOR pathway signaling. TM4t cells were treated with or without 40 μM t10,c12-CLA, in the presence or absence of 20 μM compound C or 200 nM rapamycin for 24, 48 or 72 hr, and phosphorylation of p70S6K, mTOR and AKT, as well as cleavage of PARP, caspase 9 and caspase 3 examined by western blot. A. Effect of CLA and compound C on phosphorylation of p70S6K-Thr389. B. Effect of CLA and compound C on phosphorylation of mTOR1-Ser2448. C. Effect of CLA and rapamycin on apoptosis and on phosphorylation of p70S6K-Thr389. D. Effect of CLA and rapamycin on phosphorylation of AKT at Ser473 and Thr308. The numbers indicate the ratio of the phospho-enzyme to the total enzyme.
These data did not provide strong support for an AMPK-mTOR link in contributing to the apoptotic effect of CLA, thus we used a different approach to investigate this concept further. In response to short-term nutrient deprivation, AMPK has been shown to phosphorylate mTOR at Thr2446; moreover, phosphorylation at this site hinders the phosphorylation of mTOR at Ser2448, which is required for mTOR activation [54]. To determine whether AMPK activation by t10,c12-CLA can directly reduce mTOR activity by suppressing phosphorylation at its Ser2448 site, cells were treated with t10,c12-CLA in the absence or presence of the AMPK inhibitor compound C. CLA slightly decreased both phospho-Ser2448-mTOR and mTOR at 48 and 72 hr, resulting in a modest decrease in its phosphorylation status (Fig. 4B). With compound C, our expectation was that by inhibiting AMPK-catalyzed phosphorylation of mTOR at Thr2446, phosphorylation of mTOR at Ser2448 would be increased. This was not the case, however, and in fact a decrease was observed at both the 48 and 72 hr time points (Fig. 4B); moreover, compound C markedly reduced the level of mTOR protein. In combination, CLA and compound C showed an additive inhibition of phosphorylation at 48 hr, but the combination was similar to compound C alone at 72 hr.
Our observation that compound C decreased mTOR protein levels suggested that this agent was not a good tool to address the relationship between AMPK and mTOR in these cells. Thus, as an additional clarification of a potential role of the mTORC1-p70S6K pathway in t10,c12-CLA-induced apoptosis of TM4t cells, we utilized the mTOR inhibitor rapamycin. As expected, rapamycin markedly suppressed mTORC1 activity as indicated by the significant reduction in the phosphorylation of p70S6K (Fig 4C). This inhibition of mTORC1 signaling, however, did not appear to alter cell viability, since rapamycin did not induce apoptosis. Moreover, rapamycin actually decreased t10,c12-CLA-induced cleavage of PARP, caspase 9 and caspase 3 (Fig. 4C), suggesting that the t10,c12-CLA-induced apoptosis in TM4t cells does not result from the inhibition of the mTORC1-p70S6K pathway.
Although mTORC2 was thought to be rapamycin-insensitive, a study has demonstrated that mTORC2 activity can be inhibited by long-term treatment with rapamycin [55]. A key substrate of mTORC2 is AKT, which is phosphorylated at its Ser473 site by mTORC2. AKT is also phosphorylated by PI3K/PDK1 signaling at Thr308, and phosphorylation at both sites is believed to be required for the full activity of AKT [43,56]. Since mTORC2-AKT is a well-known survival pathway, we next investigated whether t10,c12-CLA might exert its effects via this route. As shown in Fig. 4D, t10,c12-CLA alone reduced AKT phosphorylation at Ser473 at the 48 and 72 hr time points, while having no effect on Thr308 phosphorylation. This suggests that t10,c12-CLA could suppress AKT activity through inhibition of mTORC2 activity, rather than through the PI3K/PDK1 pathway. Treating cells with rapamycin alone reduced AKT phosphorylation at Ser473, but not at Thr308, demonstrating the specificity of rapamycin in inhibiting mTORC2 activity. Concurrent treatment of t10,c12-CLA and rapamycin further reduced AKT phosphorylation at Ser473 (Fig. 4D). This additive effect on AKT phosphorylation is not consistent with a role for mTORC2 activity in mediating t10,c12-CLA-induced apoptosis, since rapamycin was able to block the apoptotic effect of CLA, at least in part (Fig. 4C).
3.4. p38 MAPK mediates the apoptotic effect of t10,c12-CLA
Since the above experiments ruled out a role for the mTOR pathway in mediating the apoptotic effect of t10,c12-CLA, we next investigated the potential role of p38 MAPK. Previous studies had shown that p38 plays an important role in mediating apoptosis in response to different extracellular stimuli, including myocardial injury [57], chemotherapeutic agents [58,59], environmental toxicants [60,61], death receptor stimulation [62], serum deprivation [63], and free fatty acid exposure [64,65]. Moreover, the p38 MAPK cascade has also been identified as part of the AMPK signaling network [66-68]. To investigate whether the AMPK-p38 pathway might be involved in the apoptotic effect of CLA, we first asked whether p38 could be activated by t10,c12-CLA. In a preliminary experiment, we demonstrated that the AMPK agonist AICAR stimulated p38 phosphorylation in this cell line (data not shown). Data in Fig. 5A show that p38 was robustly phosphorylated by t10,c12-CLA treatment, and that this effect was isomer specific. Blocking AMPK activity with compound C abrogated the t10,c12-CLA-induced p38 phosphorylation (Fig. 5B), demonstrating that AMPK was acting upstream of p38. In addition, when added to CLA-treated cultures, the p38 inhibitor SB-203580 increased the percentage of viable cells in a concentration-dependent manner as shown in the cell micrographs (Fig. 5C, compare iii, vi and viii with iv) and in the SRB assay on the attached viable cell monolayer (Fig. 5D). Moreover, SB-203580 attenuated t10,c12-CLA-induced cleavage of PARP and caspase 9 at 48h, and of caspase 3 at 48 and 72h (Fig. 5E). To further validate the involvement of p38 in mediating the t10,c12-CLA apoptotic effect, we utilized siRNAs targeting either p38α or p38β. As seen in Fig. 4F, reduction in p38 protein levels resulted in a reduced level of cleaved PARP, caspase 9 and caspase 3 in CLA-treated cells. Since partial knockdown of p38 with either siRNA showed similar efficacy in inhibiting PARP cleavage, these data suggest that both p38 isoforms may be involved in signaling t10,c12-CLA-induced apoptosis.
Figure 5.




Activation of p38 MAPK signaling plays an important role in CLA-induced apoptosis. TMT4t cells were treated with or without 40 μM CLA in the absence or presence of the indicated inhibitors for 24, 48 or 72 hr, and the samples prepared for western blot analysis. A. t10,c12-CLA, but not c9,t11-CLA stimulates phosphorylation of p38. B. The AMPK inhibitor compound C (20 μM) attenuates the t10,c12-CLA-induced phosphorylation of p38. C and D. TM4t cells were treated for 72 hr with or without 40 μM of t10,c12-CLA and/or various concentrations of SB-203580 (SB), photographed under the 10× microscope objective (C), then viable cell number estimated by the SRB assay on the attached (viable) cell monolayer (D). The data in the right panel of D show the ratio of the ODs from the SRB assay from CLA-treated to control cells. The asterisks denote a statistically significant difference from the control group. E. The p38 inhibitor SB-203580 (10 μM) attenuates t10,c12-CLA-induced cleavage of PARP, caspase 3 and caspase 9. F. p38 siRNAs attenuate t10,c12-CLA-induced cleavage of PARP, caspase 3 and caspase 9, as well as the t10,c12-CLA induction of Bim. TM4t cells were transfected with p38α, p38β or control siRNA for 6 hr followed by treatment with t10,c12-CLA for 72 hr.
Bim, a pro-apoptotic BH3-only member of the Bcl-2 family, has been implicated in executing the apoptotic signal from p38 MAPK [69-71]. We therefore hypothesized that p38 activation by t10,c12-CLA might stimulate Bim activity and induce apoptosis. In support of this notion, we found that t10,c12-CLA increased both BimEL and BimL, and that transfection of the cells with siRNA against either p38α or p38β partially abrogated this increase (Fig. 5F). Furthermore, since t10,c12-CLA but not c9,t11-CLA induced apoptosis, we predicted that only the former isomer would induce Bim. At the 72 hr time point, this indeed was found to be the case (Fig. 6A). Finally, knockdown of Bim using siRNA reduced the t10,c12-CLA-induced cleavage of PARP, caspase 9, and caspase 3 (Fig. 6B). Concurrently, Bim siRNA increased the +CLA/-CLA viable cell ratio from 0.38 ± 0.01 to 0.58 ± 0.02 (SRB assay on the viable attached monolayer; data not shown). These results demonstrate that the p38-Bim pathway can be specifically activated by t10,c12-CLA and mediate its apoptotic effect.
Figure 6.
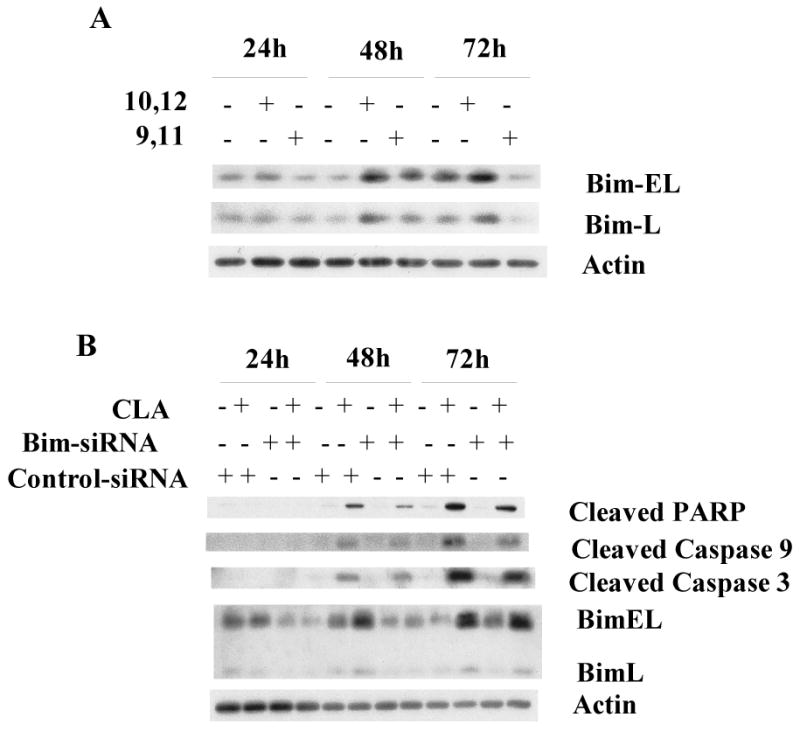
t10,c12-CLA induces Bim, and knockdown of Bim attenuates CLA-induced apoptosis. TMt4t cells were treated with 40 μM t10,c12-CLA (A,B) or c9,t11-CLA (A) for 24, 48, or 72 hr, without (A) or with (B) Bim siRNA or control siRNA, and samples prepared for western blot analysis. A. t10,c12-CLA, but not c9,t11-CLA increases expression of Bim-EL and Bim-L. B. Knockdown of Bim attenuates CLA-induced apoptosis.
4. Discussion
The current study provides strong support for a central role of AMPK in mediating the apoptotic response of TM4t mammary tumor cells to the t10,c12 isomer of CLA. Activation of AMPK, monitored by phosphorylation at its Thr172 site, was increased 4-fold at 48 hr and 10-fold at 72 hr, concurrent with the increase in apoptosis. Activation of AMPK was further confirmed by phosphorylation of its downstream target acetyl CoA carboxylase. The physiological relevance of CLA-induced AMPK activation was demonstrated using the AMPK inhibitor compound C, which reduced CLA-induced apoptosis. Moreover, the AMPK activator AICAR potentiated CLA-induced apoptosis and increased apoptosis on its own, demonstrating a critical role of this enzyme in the life and death balance of the cells.
4.1. AMPK-modulated mTOR is unlikely to mediate CLA-induced cell death
The serine-threonine kinase mTOR plays a critical role in cell survival, and is regulated by AMPK at several levels. These include phosphorylation and activation of TSC2, a negative regulator of mTOR [42], phosphorylation of raptor, a member of the activated mTORC1 binding complex, resulting in raptor dissociation and thus inactivation of mTORC1 [53], and direct phosphorylation of mTOR at Thr2446, consequently interfering with phosphorylation at nearby Ser2448, a site required for mTOR activation [54]. We therefore predicted that CLA-activated AMPK would inhibit the mTOR survival pathway, thus permitting the cells to undergo apoptosis. Results from our current studies, however, do not support a role for the mTOR pathway in mediating CLA-induced apoptosis. First, although CLA decreased mTOR protein levels, it had only modest effects on mTOR activity, decreasing the phosphorylation of one of its downstream effectors, p70S6K, at 24 and 48 hr, as the hypothesis would predict, but modestly increasing phosphorylation at 72 hr (Fig. 4A,4C), a time when there was very significant apoptosis (Fig. 1 and [9]) and when we would expect an even greater decrease in phosphorylation of p70S6K. Second, although CLA decreased the activating phosphorylation of mTOR at Ser2448, this effect was very modest. Third, reduction of mTOR protein levels by compound C decreased CLA-induced apoptosis. Fourth, and perhaps most compelling, was the observation that rapamycin, which completely inhibited the activity of both mTORC1 and mTORC2, as measured by the almost complete loss of phosphorylation of p70S6K and of AKT-Ser473, respectively, did not enhance CLA-induced apoptosis as we had predicted, but instead, attenuated it. A similar observation was made using a lower dose of rapamycin (10 ng/ml; data not shown). Thus, in spite of the multiple mechanisms by which AMPK can mediate mTOR, our data do not support a role for either mTOR complex in mediating the effect of CLA. Interestingly, rapamycin by itself did not induce apoptosis, demonstrating that the TM4t mammary tumor cell line does not require, nor is “addicted” to, mTOR, as has been suggested for some cancer cells [43]. Whether the mutant p53 status of this cell line contributes to this finding will require further study. Importantly, however, our data with rapamycin add caution to the generalization that rapamycin analogues will be useful clinically.
The mechanism by which rapamycin attenuates CLA-induced apoptosis remains unknown, but may be independent of mTOR. For example, it is of interest that rapamycin was also found to be protective against apoptosis induced by some cytotoxic drugs [72], hydrogen peroxide [73], and hypoxia [74], and in at least one cell line, this protection against drugs was associated with an increase in Bcl-2 [72]. We have recently shown that t10,c12-CLA induces apoptosis through both ER stress and mitochondrial pathways [9,10], and that Bcl-2 levels are reduced concurrently. Furthermore, overexpression of Bcl-2 attenuated CLA-induced apoptosis [9]. This observation suggests that rapamycin may induce partial resistance to the apoptotic effect of CLA by attenuating the loss of Bcl-2. Protection against hydrogen peroxide may be mediated in the same way, since this chemical has also been shown to utilize both the ER stress and mitochondrial pathways to induce apoptosis [75]. Finally, it is important to recognize that rapamycin can alter the expression of numerous genes [76], therefore providing cells with a number of opportunities to bypass an apoptotic signal.
4.2. AMPK mediates the CLA apoptotic signal through activation of the p38 MAPK-Bim pathway
In contrast to our initial expectation that CLA-activated AMPK would block the mTOR pathway and therefore induce apoptosis, our data clearly point to an important role for p38 MAPK in mediating the apoptotic effect of CLA. First, CLA induction of p38 phosphorylation was robust, and followed the same time course as activation of AMPK and induction of apoptosis. Second, a link between AMPK, p38 activation and apoptosis was further confirmed by the observation that the AMPK inhibitor compound C significantly blocked p38 phosphorylation as well as apoptosis. Third, CLA-induced apoptosis was blocked with SB-203580, a p38 inhibitor, and with both p38α and β siRNAs. Inhibition with SB-203580 was most effective at 48 hr, suggesting that other pathways may contribute to the strengthening apoptotic signal at later times, and/or that negative feedback from another AMPK target limits the apoptotic signaling along this particular pathway.
Phosphorylation by p38 MAPK activates a number of proteins, inducing a wide variety of biological effects in a cell type and cell context specific manner. Among the p38 targets are transcription factors, several of which, including CREB, FOXOA3, FOXO1, CHOP and NFκB stimulate expression of the pro-apoptotic protein Bim, and have been identified as direct substrates of p38 [77-88]. Unpublished studies in our lab have shown that CREB phosphorylation is specifically enhanced by t10,c12-CLA, but not by c9,t11-CLA (Hsu and Ip, data not shown), thus linking the t10,c12-CLA-induced activation of p38 to Bim via CREB, at least in part. We cannot be absolutely certain that the p38 pathway is the sole route by which Bim is activated, since it was not possible to completely knock down p38. However, our data that a reduction in p38 protein levels is associated with a reduction in CLA-induced Bim-EL and Bim-L is consistent with that theory. Another mechanism by which Bim levels can be increased is through protein stabilization via protein phosphatase 2A-mediated dephosphorylation. This pathway was shown by Puthalakath et al to be triggered by ER stress [84]. Concurrently, CHOP was elevated, and the authors demonstrated the importance of this transcription factor in the transcriptional induction of Bim. Based on our previous studies which demonstrated a role for ER stress-induced CHOP in t10,c12-CLA-induced apoptosis [10], we used CHOP siRNA to examine the importance of CHOP in the CLA induction of Bim. This treatment did not alter CLA-induced Bim upregulation (Hsu and Ip, data not shown), suggesting that CHOP is not required for the increased expression of Bim in response to CLA.
The robust activation of p38 MAPK by CLA raises the possibility that the signaling pathway may branch at this node. In addition to phosphorylation of transcription factors which may increase Bim transcription, p38 has been shown to phosphorylate Bim and Bcl-2, resulting in an increase and decrease, respectively, in their pro-apoptotic (Bim) or anti-apoptotic (Bcl-2) activities [70,89]. Together with the CLA-induced decrease in Bcl-2 protein [9], a loss of function of the remaining Bcl-2, concurrent with an increase in expression and activity of Bim, would dramatically tip the balance towards the apoptotic cell death that we observed upon treatment with t10,c12-CLA. Our observation that Bim siRNA rendered cells resistant to the apoptotic effect of CLA, together with our previous studies demonstrating that CLA induces ER stress [10], is consistent with the report of Morishima et al [90] who found that apoptosis resulting from tunicamycin-induced ER stress was partially blocked by Bim siRNA.
4.3. Activation of AMPK is not via an energy-sensing mechanism
The observation that CLA does not deplete ATP suggests that AMPK activation does not occur as a result of nutrient or energy depletion. This conclusion is supported by the observation that ATP levels were actually maintained in CLA-treated cells which were fed only at the start of the 72 hr treatment period, in comparison to control cultures in which ATP levels declined over this time probably as a result of nutrient consumption. In this regard, TM4t cells may use CLA as an energy source. Additionally, or alternatively, AMPK activation, with subsequent phosphorylation and inactivation of ACC, may preserve ATP, since the ACC-catalyzed conversion of acetyl coA to malonyl CoA requires ATP.
LKB1 is an upstream kinase which is known to activate AMPK [25,26]; however the inability of LKB1 siRNA to block CLA-induced AMPK activation rules out this pathway. Additional possibilities remain to be fully explored. For example, it has been shown that AKT can phosphorylate Ser485/Ser491 in AMPK, and as a result, antagonize the activating Thr172 phosphorylation by LKB1 or CaMKK [30]. Since t10,c12-CLA inhibited activation of AKT (Fig. 4D), it is possible that this relieved a restraint on AMPK-Thr172 phosphorylation, thus contributing to AMPK activation. CLA has been shown to bind to and activate PPARγ [91-93], and the observation that rosiglitazone, a PPARγ activator, can activate AMPK [94] suggests that CLA might be acting through this transcription factor. Experiments in the TM4t model utilized in the current studies, however, do not support a role for PPARs in CLA-induced apoptosis (Hsu and Ip, (unpublished), probably ruling out this pathway. The most likely mechanism by which t10,c12-CLA activates AMPK in TM4t cells is through the calcium/calmodulin-dependent protein kinase kinase (CaMKK). As noted above, t10,c12-CLA induces ER stress [10], which generally results in Ca+2 release from the ER [95-97], thus activating CaMKK, which is an upstream kinase for AMPK. This concept is currently being tested in our laboratory, and preliminary data showing that BAPTA-AM, a Ca+2 chelator, and STO-609, a selective inhibitor of CaMKK can each reduce CLA-induced cleavage of PARP, caspase-3 and caspase-9, is strongly supportive of this pathway.
In summary, the t10,c12 isomer of CLA induces apoptosis of TM4t mammary tumor cells through activation of the AMPK-p38 MAPK-Bim signaling pathway, resulting in an increased ratio of Bim to Bcl-2, culminating in activation of the intrinsic pathway that we have previously demonstrated [9]. Cell survival is also compromised by a CLA-induced decrease in AKT activity (Fig. 4D). Initiation of the apoptotic cascade occurs as a result of CLA-induced ER stress [10], which our preliminary data suggests results in activation of AMPK.
Acknowledgments
These studies were supported by National Institutes of Health Grant CA61763 and by the shared resources of the National Cancer Institute Cancer Center Support Grant CA16056. XM was supported by an Avon-AACR Scholar Award.
Footnotes
Abbreviations The abbreviations used are as follows: ACC, acetyl coenzyme A carboxylase; AICAR, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (activator of AMPK); AMPK, AMP-activated protein kinase; CaMKKα, calcium/calmodulin-dependent protein kinase kinase α; CC or Comp C, compound C (inhibitor of AMPK); CLA, conjugated linoleic acid; t10,c12-CLA, trans-10, cis-12 CLA; c9,t11-CLA, cis-9, trans-11 CLA; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; p70S6K, p70S6 kinase; PPAR, peroxisome-proliferator activated protein; PKA, cyclic AMP-dependent protein kinase; PRAS40, proline-rich AKT substrate 40; SB, SB-203580 (inhibitor of p38 MAPK); SRB, sulforhodamine B; TAK1, TGFβ-activated kinase; TSC2, tuberous sclerosis complex 2; 4EBP1, eukaryotic initiation factor 4E-binding protein.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ip MM, Masso-Welch PA, Ip C. J Mammary Gland Biol Neoplasia. 2003;8:101–116. doi: 10.1023/a:1025739506536. [DOI] [PubMed] [Google Scholar]
- 2.Kelley NS, Hubbard NE, Erickson KL. J Nutr. 2007;137:2599–2607. doi: 10.1093/jn/137.12.2599. [DOI] [PubMed] [Google Scholar]
- 3.Masso-Welch PA, Zangani D, Ip C, Vaughan MM, Shoemaker SF, McGee SO, Ip MM. J Nutr. 2004;134:299–307. doi: 10.1093/jn/134.2.299. [DOI] [PubMed] [Google Scholar]
- 4.Ip C, Dong Y, Ip MM, Banni S, Carta G, Angioni E, Murru E, Spada S, Melis MP, Saebo A. Nutr Cancer. 2002;43:52–58. doi: 10.1207/S15327914NC431_6. [DOI] [PubMed] [Google Scholar]
- 5.Hubbard NE, Lim D, Erickson KL. Cancer Lett. 2003;190:13–19. doi: 10.1016/s0304-3835(02)00515-3. [DOI] [PubMed] [Google Scholar]
- 6.Ip MM, McGee SO, Masso-Welch PA, Ip C, Meng X, Ou L, Shoemaker S. Carcinogenesis. 2007;28:1269–1276. doi: 10.1093/carcin/bgm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng X, Shoemaker SF, McGee SO, Ip MM. Carcinogenesis. 2008;29:1013–1021. doi: 10.1093/carcin/bgn035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banni S, Angioni E, Casu V, Melis MP, Carta G, Corongiu FP, Thompson H, Ip C. Carcinogenesis. 1999;20:1019–1024. doi: 10.1093/carcin/20.6.1019. [DOI] [PubMed] [Google Scholar]
- 9.Ou L, Ip C, Lisafeld B, Ip MM. Biochem Biophys Res Commun. 2007;356:1044–1049. doi: 10.1016/j.bbrc.2007.03.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou L, Wu Y, Ip C, Meng X, Hsu YC, Ip MM. J Lipid Res. 2008;49:985–994. doi: 10.1194/jlr.M700465-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie DG, Carling D, Carlson M. Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 12.Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Trends Biochem Sci. 1999;24:22–25. doi: 10.1016/s0968-0004(98)01340-1. [DOI] [PubMed] [Google Scholar]
- 13.Dyck JR, Lopaschuk GD. J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo Z, Saha AK, Xiang X, Ruderman NB. Trends Pharmacol Sci. 2005;26:69–76. doi: 10.1016/j.tips.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Viollet B, Mounier R, Leclerc J, Yazigi A, Foretz M, Andreelli F. Diabetes Metab. 2007;33:395–402. doi: 10.1016/j.diabet.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Osler ME, Zierath JR. Endocrinology. 2008;149:935–941. doi: 10.1210/en.2007-1441. [DOI] [PubMed] [Google Scholar]
- 17.Kola B, Grossman AB, Korbonits M. Front Horm Res. 2008;36:198–211. doi: 10.1159/000115366. [DOI] [PubMed] [Google Scholar]
- 18.Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D. Biochem J. 2002;363:167–174. doi: 10.1042/0264-6021:3630167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo KS, Ha J. Biochem Biophys Res Commun. 2001;287:92–97. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 20.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 21.Clark H, Carling D, Saggerson D. Eur J Biochem. 2004;271:2215–2224. doi: 10.1111/j.1432-1033.2004.04151.x. [DOI] [PubMed] [Google Scholar]
- 22.Hickson-Bick DL, Buja LM, McMillin JB. J Mol Cell Cardiol. 2000;32:511–519. doi: 10.1006/jmcc.1999.1098. [DOI] [PubMed] [Google Scholar]
- 23.Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA. J Physiol. 2006;574:139–147. doi: 10.1113/jphysiol.2006.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang S, Wang Z, Riethoven JJ, Xia Y, Miner J, Fromm M. J Nutr. 2009;139:2244–2251. doi: 10.3945/jn.109.112417. [DOI] [PubMed] [Google Scholar]
- 25.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 26.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 28.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Momcilovic M, Hong SP, Carlson M. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 30.Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. J Biol Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- 31.Soltys CL, Kovacic S, Dyck JR. Am J Physiol Heart Circ Physiol. 2006;290:H2472–H2479. doi: 10.1152/ajpheart.01206.2005. [DOI] [PubMed] [Google Scholar]
- 32.Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 33.Hwang JT, Park IJ, Shin JI, Lee YK, Lee SK, Baik HW, Ha J, Park OJ. Biochem Biophys Res Commun. 2005;338:694–699. doi: 10.1016/j.bbrc.2005.09.195. [DOI] [PubMed] [Google Scholar]
- 34.Hwang JT, Kim YM, Surh YJ, Baik HW, Lee SK, Ha J, Park OJ. Cancer Res. 2006;66:10057–10063. doi: 10.1158/0008-5472.CAN-06-1814. [DOI] [PubMed] [Google Scholar]
- 35.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee P, Mulrooney TJ, Marsh J, Blair D, Chiles TC, Seyfried TN. Mol Cancer. 2008;7:37. doi: 10.1186/1476-4598-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D, Matsumoto K, Toyonaga T, Asano T, Nishikawa T, Araki E. Circ Res. 2005;97:837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 38.Saha AK, Persons K, Safer JD, Luo Z, Holick MF, Ruderman NB. Biochem Biophys Res Commun. 2006;349:519–524. doi: 10.1016/j.bbrc.2006.08.107. [DOI] [PubMed] [Google Scholar]
- 39.Motoshima H, Goldstein BJ, Igata M, Araki E. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Huang W, Tao R, Ibaragi S, Lan F, Ido Y, Wu X, Alekseyev YO, Lenburg ME, Hu GF, Luo Z. Oncogene. 2009;28:1993–2002. doi: 10.1038/onc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu SY, Chan DW, Liu VW, Ngan HY. Tumour Biol. 2009;30:80–85. doi: 10.1159/000216843. [DOI] [PubMed] [Google Scholar]
- 42.Inoki K, Zhu T, Guan KL. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 43.Guertin DA, Sabatini DM. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Kopelovich L, Fay JR, Sigman CC, Crowell JA. Cancer Epidemiol Biomarkers Prev. 2007;16:1330–1340. doi: 10.1158/1055-9965.EPI-07-0045. [DOI] [PubMed] [Google Scholar]
- 45.Dann SG, Selvaraj A, Thomas G. Trends Mol Med. 2007;13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Tsang CK, Qi H, Liu LF, Zheng XF. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Hay N, Sonenberg N. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 48.Meric-Bernstam F, Gonzalez-Angulo AM. J Clin Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 50.Ip MM, Masso-Welch PA, Shoemaker SF, Shea-Eaton WK, Ip C. Experimental Cell Research. 1999;250:22–34. doi: 10.1006/excr.1999.4499. [DOI] [PubMed] [Google Scholar]
- 51.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 52.Carling D, Sanders MJ, Woods A. Int J Obes (Lond) 2008;32 4:S55–S59. doi: 10.1038/ijo.2008.124. [DOI] [PubMed] [Google Scholar]
- 53.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng SW, Fryer LG, Carling D, Shepherd PR. J Biol Chem. 2004;279:15719–15722. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 55.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 56.Brazil DP, Hemmings BA. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 57.Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- 58.Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. Mol Biol Cell. 2003;14:2071–2087. doi: 10.1091/mbc.E02-10-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Losa JH, Parada CC, Viniegra JG, Sanchez-Arevalo L, Cajal VS, Sanchez-Prieto R. Oncogene. 2003;22:3998–4006. doi: 10.1038/sj.onc.1206608. [DOI] [PubMed] [Google Scholar]
- 60.Cai B, Xia Z. Apoptosis. 2008;13:803–810. doi: 10.1007/s10495-008-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung YS, Jeong EM, Park EK, Kim YM, Sohn S, Lee SH, Baik EJ, Moon CH. Eur J Pharmacol. 2008;578:11–18. doi: 10.1016/j.ejphar.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 62.Brenner B, Koppenhoefer U, Weinstock C, Linderkamp O, Lang F, Gulbins E. J Biol Chem. 1997;272:22173–22181. doi: 10.1074/jbc.272.35.22173. [DOI] [PubMed] [Google Scholar]
- 63.Kummer JL, Rao PK, Heidenreich KA. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 64.Chai W, Liu Z. Endocrinology. 2007;148:1622–1628. doi: 10.1210/en.2006-1068. [DOI] [PubMed] [Google Scholar]
- 65.Dersch K, Ichijo H, Bhakdi S, Husmann M. Cell Death Differ. 2005;12:1107–1114. doi: 10.1038/sj.cdd.4401633. [DOI] [PubMed] [Google Scholar]
- 66.Xi X, Han J, Zhang JZ. J Biol Chem. 2001;276:41029–41034. doi: 10.1074/jbc.M102824200. [DOI] [PubMed] [Google Scholar]
- 67.Cheng Z, Pang T, Gu M, Gao AH, Xie CM, Li JY, Nan FJ, Li J. Biochim Biophys Acta. 2006;1760:1682–1689. doi: 10.1016/j.bbagen.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 68.Pelletier A, Joly E, Prentki M, Coderre L. Endocrinology. 2005;146:2285–2294. doi: 10.1210/en.2004-1565. [DOI] [PubMed] [Google Scholar]
- 69.Kirschnek S, Ying S, Fischer SF, Hacker H, Villunger A, Hochrein H, Hacker G. J Immunol. 2005;174:671–679. doi: 10.4049/jimmunol.174.2.671. [DOI] [PubMed] [Google Scholar]
- 70.Cai B, Chang SH, Becker EB, Bonni A, Xia Z. J Biol Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- 71.Lu J, Quearry B, Harada H. FEBS Lett. 2006;580:3539–3544. doi: 10.1016/j.febslet.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 72.Calastretti A, Rancati F, Ceriani MC, Asnaghi L, Canti G, Nicolin A. Eur J Cancer. 2001;37:2121–2128. doi: 10.1016/s0959-8049(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 73.Zohlnhofer D, Nuhrenberg TG, Neumann FJ, Richter T, May AE, Schmidt R, Denker K, Clauss MA, Schomig A, Baeuerle PA. Mol Pharmacol. 2004;65:880–889. doi: 10.1124/mol.65.4.880. [DOI] [PubMed] [Google Scholar]
- 74.Ronellenfitsch MW, Brucker DP, Burger MC, Wolking S, Tritschler F, Rieger J, Wick W, Weller M, Steinbach JP. Brain. 2009;132:1509–1522. doi: 10.1093/brain/awp093. [DOI] [PubMed] [Google Scholar]
- 75.Min SK, Lee SK, Park JS, Lee J, Paeng JY, Lee SI, Lee HJ, Kim Y, Pae HO, Lee SK, Kim EC. J Oral Pathol Med. 2008;37:490–498. doi: 10.1111/j.1600-0714.2008.00679.x. [DOI] [PubMed] [Google Scholar]
- 76.Peng T, Golub TR, Sabatini DM. Mol Cell Biol. 2002;22:5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thuerauf DJ, Arnold ND, Zechner D, Hanford DS, DeMartin KM, McDonough PM, Prywes R, Glembotski CC. J Biol Chem. 1998;273:20636–20643. doi: 10.1074/jbc.273.32.20636. [DOI] [PubMed] [Google Scholar]
- 78.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 79.Zhang L, Insel PA. J Biol Chem. 2004;279:20858–20865. doi: 10.1074/jbc.M310643200. [DOI] [PubMed] [Google Scholar]
- 80.Labied S, Kajihara T, Madureira PA, Fusi L, Jones MC, Higham JM, Varshochi R, Francis JM, Zoumpoulidou G, Essafi A, Fernandez dM, Lam EW, Brosens JJ. Mol Endocrinol. 2006;20:35–44. doi: 10.1210/me.2005-0275. [DOI] [PubMed] [Google Scholar]
- 81.Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, Iwashita S, Kako K, Kishi T, Kasuya Y, Fukamizu A. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 82.Cai B, Xia Z. Apoptosis. 2008;13:803–810. doi: 10.1007/s10495-008-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang XZ, Ron D. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 84.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 85.Zhang CC, Shapiro DJ. J Biol Chem. 2000;275:479–486. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- 86.Korus M, Mahon GM, Cheng L, Whitehead IP. Oncogene. 2002;21:4601–4612. doi: 10.1038/sj.onc.1205678. [DOI] [PubMed] [Google Scholar]
- 87.Sarnico I, Lanzillotta A, Boroni F, Benarese M, Alghisi M, Schwaninger M, Inta I, Battistin L, Spano P, Pizzi M. J Neurochem. 2009;108:475–485. doi: 10.1111/j.1471-4159.2008.05783.x. [DOI] [PubMed] [Google Scholar]
- 88.Gilley J, Coffer PJ, Ham J. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 90.Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. J Biol Chem. 2004;279:50375–50381. doi: 10.1074/jbc.M408493200. [DOI] [PubMed] [Google Scholar]
- 91.Houseknecht KL, Vanden Heuvel JP, Moya-Camarena SY, Portocarrero CP, Peck LW, Nickel KP, Belury MA. Biochem Biophys Res Commun. 1998;244:678–682. doi: 10.1006/bbrc.1998.8303. [DOI] [PubMed] [Google Scholar]
- 92.Yu Y, Correll PH, Vanden Heuvel JP. Biochim Biophys Acta Mol Cell Biol Lipids. 2002;1581:89–99. doi: 10.1016/s1388-1981(02)00126-9. [DOI] [PubMed] [Google Scholar]
- 93.Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui YZ, Hennighausen L, Gonzalez F, Rohrer J, Benninghoff AU, Hontecillas R. Gastroenterology. 2004;127:777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 94.Han S, Roman J. Mol Cancer Ther. 2006;5:430–437. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]
- 95.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nieto-Miguel T, Fonteriz RI, Vay L, Gajate C, Lopez-Hernandez S, Mollinedo F. Cancer Res. 2007;67:10368–10378. doi: 10.1158/0008-5472.CAN-07-0278. [DOI] [PubMed] [Google Scholar]
- 97.Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]