

Abstract
Type I IFN are strongly induced upon engagement of certain pattern recognition receptors (PRR) by microbial products, and play key roles in regulating innate and adaptive immunity. It has become apparent that the endoplasmic reticulum (ER) stress-induced unfolded protein response (UPR), in addition to restoring ER homeostasis, also influences the expression of certain inflammatory cytokines. However, the extent to which UPR signaling regulates type I IFN remains unclear. Here we show that cells undergoing an UPR respond to TLR4 and TLR3 ligands, and intracellular dsRNA, with log-fold greater IFN-β induction. This synergy is not dependent on autocrine type I IFN signaling, but unexpectedly requires the UPR transcription factor XBP-1. Synergistic IFN-β induction also occurs in HLA-B27/human β2m transgenic rat macrophages exhibiting an UPR as a consequence of HLA-B27 upregulation, where it correlates with activation of XBP-1 splicing. Together these findings indicate that the cellular response to endogenous ‘danger’ that disrupts ER homeostasis is coupled to IFN-β induction by XBP-1, which has implications for the immune response and the pathogenesis of diseases involving the UPR.
Keywords: Interferons, protein misfolding, unfolded protein response, HLA-B27
Introduction
Initially identified as antiviral cytokines, type I interferons (IFN-α/β) are now recognized to have diverse immunoregulatory effects activating macrophages and NK cells, promoting T cell survival and dendritic cell maturation, and increasing the production of Th1-polarizing cytokines to mediate innate and adaptive immune responses [1]. Type I IFN also exert biological effects at low and even constitutive levels of expression [2]. For example, weak IFN-β-mediated signaling augments IFN-α/β induction in response to LPS through positive feedback involving autocrine/paracrine upregulation of interferon regulatory factor 7 (IRF7). In addition, low levels of IFN-α/β prime cells to respond to IFN-γ and IL-6 by providing a phosphorylated type I IFN receptor ‘niche’ for commonly used signaling molecules in proximity to other cytokine receptor complexes [3, 4]. Mice deficient in IFN-β are more susceptible to viral infection, have lower numbers of macrophages and mature B cells, and exhibit reduced bone mass [5] as IFN-β is a positive regulator of bone formation through inhibition of osteoclast differentiation [6]. Thus, even constitutive levels of this cytokine are important in osteo-immune homeostasis.
IFN-β is produced by most cell types upon viral infection, and in large amounts by macrophages and dendritic cells, following engagement of pattern recognition receptors (PRR) by conserved molecular structures referred to as pathogen-associated molecular patterns [7, 8]. PRR that mediate IFN-β induction include cell surface TLR4 and endosomal TLR3 for LPS and dsRNA, respectively [9]. In both instances increased IFN-β gene transcription occurs via Toll/IL-1R domain-containing adapter inducing IFN-β (TRIF)-dependent IRF3 and NF-κB activation. PRR in the RIG-like helicase family, retinoic acid-inducible gene-I (RIG-I) and melanoma-associated differentiation antigen-5 (MDA5), recognize cytoplasmic dsRNA, and mediate IFN-β induction through IFN-β promoter stimulator-1 (IPS-1) and downstream IRF3 and NF-κB activation [10]. In addition, in plasmacytoid and myeloid DCs, engagement of endosomal TLR9 family members results in robust induction of type I IFN via MyD88-IRF7 and MyD88-IRF1 (respectively) rather than IRF3 [11, 12].
Even as cells of the innate immune system are poised to respond to microbial ‘danger’ signals via PRR, there are also mechanisms in place to deal with intracellular threats. When ER homeostasis is disrupted by alterations in redox status, depletion of calcium or nutrients, cholesterol loading, increased polypeptide load, viral infection, or accumulation of proteins that misfold, there is a coordinated response known as the ‘unfolded protein response’ (UPR) [13, 14]. The UPR results in a rapid but transient reduction in protein translation, and accelerated decay of mRNAs encoding membrane and secreted proteins [15], thus reducing the load on the ER. This is followed by a second phase of increased transcription of genes that enhance protein folding and secretion, and ER-associated degradation (ERAD), that act to restore homeostasis.
The UPR is activated when ER transmembrane proteins protein kinas receptor (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring-1 (IRE1) are released from immunoglobulin binding protein (BiP or GRP78), which is normally bound to their lumenal domains. ATF6 is proteolytically processed, releasing the active transcription factor. PERK and IRE1 are activated by homodimerization and trans-autophosphorylation. Active PERK phosphorylates eukaryotic initiation factor 2 alpha (eIF2α) resulting in attenuation of protein synthesis, while certain mRNAs are preferentially translated including ATF4. Active IRE1 is an endonuclease that recognizes a hairpin structure in the constitutively expressed X-box binding protein-1 (XBP-1) mRNA and removes 26 nucleotides. This causes a translational frame shift that bypasses a stop codon and results in the synthesis of transcriptionally active XBP-1. Active ATF6, ATF4, and XBP-1 increase the transcription of UPR target genes [14].
XBP-1 is a ubiquitously expressed transcription factor that is required for hepatocyte and plasma cell differentiation, and in the absence of XBP-1 mouse embryos die in utero [16, 17]. As an integral part of the UPR, XBP-1 is responsible for upregulation of a subset of UPR target genes, including several involved in ERAD [18]. In B cells XBP-1 mRNA splicing is triggered by increased immunoglobulin synthesis, which results in differentiation into plasma cells with expansion of the ER compartment, thus enabling production and secretion of large amounts of immunoglobulin [19]. XBP-1 also has a post-translational role in sustaining IgM synthesis in B cells [20].
Links between UPR activation and cytokine production have also been observed. XBP-1 influences the production of IL-6 in plasma cells, a process that can be initiated by LPS or CD40 ligation [19]. Cholesterol loading of macrophages activates the UPR and induces TNF-α and IL-6 via effects on NF-κB, JNK1/2, p38, and Erk1/2 activation, with the last pathway dependent on UPR-induced C/EBP homologous protein 10 (CHOP or GADD153) [21].
ER stress and the UPR have been implicated in the pathogenesis of several diseases including those that involve protein misfolding [14]. HLA-B27 is an MHC-encoded class I protein that is linked to the development of spondyloarthritis (SpA) including ankylosing spondylitis [22, 23], and when expressed in rats along with human β2m (B27-Tg) results in the development of an inflammatory disease that recapitulates many features of human SpA [24]. A proportion of HLA-B27 misfolds in the ER, resulting in increased degradation by ERAD [25], and formation of disulfide-linked and BiP-bound complexes [26–28]. Upregulation of HLA-B27 exacerbates misfolding and activates the UPR in macrophages from transgenic rats [29, 30]. HLA-B27 misfolding has been correlated with disease in these animals [28], but links between protein misfolding and the development of the immune dysregulation have not been established.
Although much is known about innate immune responses to external threats, and in parallel, ER signaling pathways that orchestrate the response to internal stress, little is known about how these pathways intersect. Here, we show that pharmacologic agents and proteins that misfold and activate the UPR synergize strongly with certain PRR agonists in the induction IFN-β, and that XBP-1 is required for this effect. These findings link ER stress with activation of the innate immune response, and have implications for diverse diseases where inflammation and the UPR have been separately implicated.
Results
Induction of type I IFN in macrophages experiencing ER stress
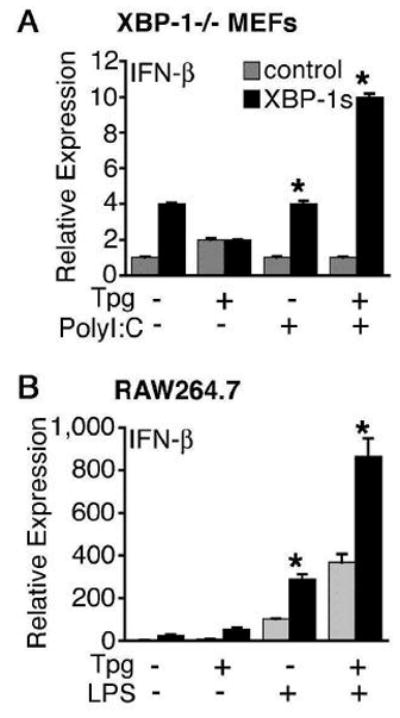
We previously observed that BM macrophages derived from B27-Tg rats exhibited increased UPR target gene expression and a low-level IFN-response signature in the absence of exogenous IFN [30], which was accompanied by a 2–3-fold increase in IFN-β transcripts in these cells (unpublished data). A similar increase in IFN-β mRNA was reported in MEFs treated with tunicamycin (Tm), which activates the UPR by inhibiting N-linked glycosylation of nascent ER-synthesized proteins [18]. To further investigate the relationship between UPR activation and IFN-β induction, we treated mouse macrophages (RAW264.7 cells) with Tm or thapsigargin (Tpg) (which activates the UPR by inhibiting ER Ca2+-ATPase). This resulted in the expected low-level induction of IFN-β, but when cells were also stimulated with LPS, IFN-β was synergistically induced (Fig. 1A,B). (Note that low-level IFN-β induction with ER stress is not apparent in this and other figures due to the scale used to display synergistic effects.) This effect occurs over a broad range of LPS (Fig. 1D) and Tpg concentrations (data not shown), and results in the accumulation of substantially more immunoreactive IFN-β protein in culture supernatants, despite potential impairment of ER function by Tpg (Fig. 1C). Enhanced induction of IFN-β mRNA is apparent as early as 1 h after the addition of LPS (Fig. 1E), while increases in IFN-α lag behind (Fig. 1F). The synergy is most striking 2–4 h after the addition of LPS, where type I IFN mRNA is 100-fold greater in cells primed with Tpg for 1 h prior to the addition of LPS. Qualitatively similar results have been obtained in RAW264.7 macrophages (Fig. 1A–D), primary BM-derived macrophages (Fig. 1E,F), human PBL-derived macrophages (unpublished data), and MEFs (see below), although type I IFN induction in response to TLR stimulation is strongest in macrophages. These data demonstrate a robust, synergistic induction of type I IFN by TLR4 stimulation in cells exposed to chemical agents that cause ER stress.
Figure 1.
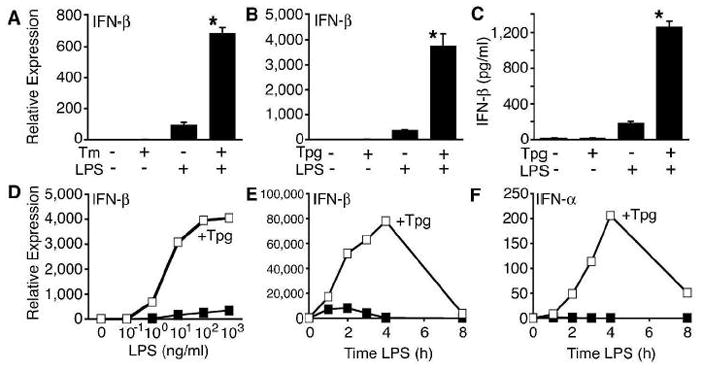
Synergistic induction of type I IFN in macrophages undergoing an unfolded protein response. RAW267.4 macrophages were pretreated with Tm for 8 h (A) or Tpg for 1 h (B) then stimulated with LPS (10 ng/ml) for 2 h prior to RNA isolation and quantitative PCR (qPCR) analysis for IFN-β transcripts. (C) RAW264.7 macrophages were treated with Tpg for 1 h, then LPS was added for an additional 24 h prior to collection of supernatants for IFN-β ELISA. (D) RAW264.7 cells were pretreated with Tpg for 1 h then dilutions of LPS for 3 h. (E, F) Rat BM derived macrophages were pre-treated with Tpg for 1 h then LPS for the times indicated. In (C, E, F), ν represents LPS only, and  indicates Tpg plus LPS. IFN transcript levels were measured with qPCR and are shown normalized to GAPDH. Results shown are means ±SD (A–C) and are representative of 4 (Tm) or 5 (Tpg) experiments. Asterisks (*) indicate p<0.05 for average fold induction for LPS plus Tpg (or Tm) vs. LPS alone, from combined experiments. Concentration curve and time course (D–F) are representative of two experiments.
indicates Tpg plus LPS. IFN transcript levels were measured with qPCR and are shown normalized to GAPDH. Results shown are means ±SD (A–C) and are representative of 4 (Tm) or 5 (Tpg) experiments. Asterisks (*) indicate p<0.05 for average fold induction for LPS plus Tpg (or Tm) vs. LPS alone, from combined experiments. Concentration curve and time course (D–F) are representative of two experiments.
ER stress triggered by free cholesterol loading of macrophages has been shown to induce the cytokines TNF-α and IL-6, with ~5-fold increases in their mRNAs after 8 h [21]. Therefore, we asked whether ER stress augmented induction of these cytokines by LPS. Tpg alone (0 ng/ml LPS) had a minimal effect after 4 h compared to LPS or both agents combined. This is not readily apparent in Fig. 2, because of the magnitude of the LPS or LPS/Tpg effect. The combination of Tpg and LPS resulted in similar induction of RANTES and IL-6, whereas TNF-α is about 4–5-fold greater than LPS alone (Fig. 2), in contrast to the ~30-fold synergy seen with IFN-β. Thus, at least at early times, the synergy is most striking for IFN-β.
Figure 2.
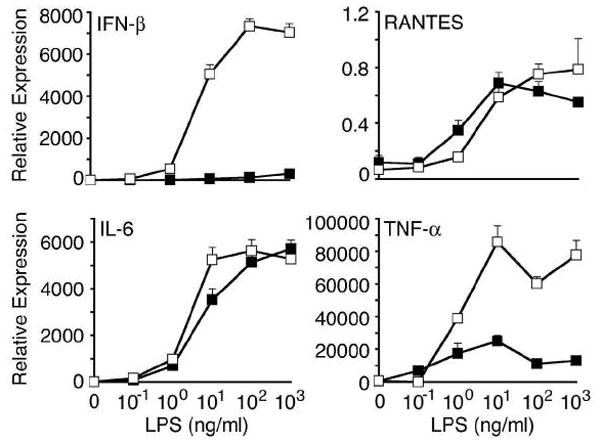
Specificity of ER stress effects on cytokine induction. BM-derived mouse macrophages were pre-treated with Tpg for 1 h then LPS for an additional 3 h. Tpg alone was present for 4 h, and LPS 3 h. Relative expression of cytokine mRNAs was determined by qPCR and is shown normalized to GAPDH. Results shown are mean ±SD and are representative of 3 independent experiments. Asterisks (*) indicate p<0.05 for fold induction with LPS plus Tpg vs. LPS alone from combined experiments.
ER stress enhances IFN-β production in response to multiple PRR
Engagement of TLR4 or TLR3 results in TRIF-dependent IFN-β production, whereas TLR2 and TLR9 use MyD88 as an adaptor. TLR2 does not significantly induce IFN-β in macrophages [31]. We therefore asked whether ER stress influences IFN-β induction in response to other TLR agonists. Tpg pre-treatment of RAW264.7 macrophages had no effect on the response to TLR9 (CpG DNA) or TLR2 (Pam3CysSK4) agonists (Fig. 3A). In contrast, TLR3 engagement with poly(I:C) resulted in robust synergy (Fig. 3A). To determine whether ER stress also augments cytoplasmic dsRNA-mediated induction of IFN-β, we took advantage of HEK293 cells that are unresponsive to exogenous poly(I:C) [32, 33], but produce type I IFN following transfection [34]. Similar to the response to TLR4/3 agonists, there is strong synergistic IFN-β induction by transfected dsRNA in cells experiencing ER stress (Fig. 3B). These data indicate that ER stress can augment IFN-β induction through TRIF-dependent TLR4/TLR3 signaling, as well as intracellular dsRNA recognition, which is dependent on MDA5 [10] and IPS-1 [34].
Figure 3.

Specificity of synergistic IFN-β induction. (A) RAW267.4 macrophages were pretreated with Tpg for 1 h, then LPS (TLR4), CpG (TLR9), exogenous poly(I:C) (TLR3), or Pam3CysSK4 (TLR2), and RNA analyzed by qPCR for IFN-β transcripts. (B) HEK293 cells were pre-treated with Tpg for 1 h, then transfected with dsRNA (poly(I:C)) as indicated. Cells were harvested at 7 h. Relative expression of IFN-β mRNA is shown normalized to GAPDH. Results shown are mean ±SD and are representative of 2 independent experiments. Asterisks (*) indicate p<0.05 for fold induction with TLR agonist plus Tpg vs. TLR agonist alone from combined experiments.
Synergistic IFN-β induction in macrophages does not require autocrine type I IFN
Secreted IFN-β can stimulate the IFN-α/β receptor (IFNAR) resulting in increased transcription of IFN-responsive genes, including IRF7. Increased IRF7 can augment transcription of the IFN-β gene, and is required for the transcription of several subtypes of IFN-α. To investigate whether the synergistic induction of IFN-β in cells experiencing ER stress was dependent on autocrine priming, BM-macrophages from IFNAR1−/− mice were examined. Induction of ER stress with Tpg pre-treatment still had a strong synergistic effect on IFN-β induction by LPS in IFNAR−/− cells, although the maximum induction was reduced approximately 50% (Fig. 4). This suggests that, as expected, there is some positive feedback effect of type I IFN, but the majority of synergy is independent of this mechanism. Consistent with this, IRF7 mRNA was induced in WT cells treated with Tpg and LPS (Fig. 4), while no such response is seen in IFNAR−/− cells. In fact, IRF7 expression is virtually extinguished in IFNAR−/− cells, consistent with known effects of constitutive type I IFN expression [35]. These data indicate that the primary mechanism of synergy does not require autocrine type I IFN signaling and upregulation of IRF7.
Figure 4.
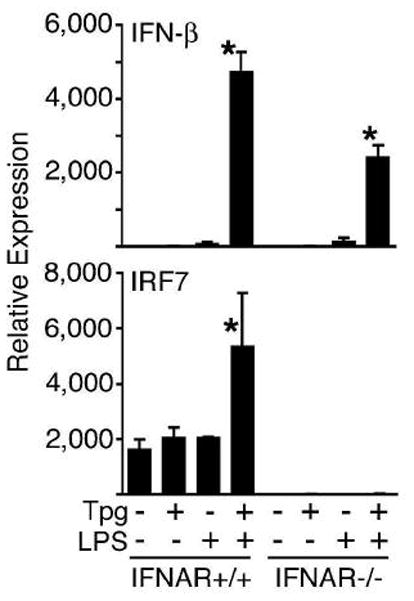
Synergy does not require type I IFN receptor or IRF7 induction. BM-derived macrophages from WT control mice (IFNAR+/+) or ifnar-deficient mice (IFNAR−/−) were treated with Tpg for 1 h then LPS for 3 h. IFN-β (top) and IRF7 (bottom) transcript levels were assessed by qPCR. Relative mRNA expression normalized to GAPDH is shown. Results shown are mean ±SD and are representative of 3 independent experiments. Asterisks (*) indicate p<0.05 for fold induction with LPS plus Tpg vs. LPS alone from combined experiments.
Contribution of UPR signaling pathways to IFN-β synergy
There are three known pathways mediated by PERK, IRE1, and ATF6 by which cells sense ER stress and initiate the UPR [14]. We investigated the role of PERK and XBP-1 in synergistic IFN-β induction using MEFs homozygous for disrupted PERK or XBP-1 genes. In PERK-deficient MEFs, generation of ER stress with Tpg pre-treatment causes synergistic induction of IFN-β that was at least as large as in WT cells (Fig. 5A). In contrast, the absence of XBP-1 completely prevented synergistic induction of IFN-β (Fig. 5B). We confirmed that the XBP-1-deficient cells responded to Tpg and poly(I:C) by examining induction of IL-6, which was equivalent to WT MEFs (unpublished data). Since the ATF6 gene has not been targeted, to investigate its potential role in synergistic IFN-β induction, we took advantage of a serine protease inhibitor (AEBSF) reported to block ATF6 cleavage and activation in cells treated with Tpg [36]. AEBSF pre-treatment of RAW264.7 macrophages caused no inhibition of synergistic IFN-β induction (unpublished observations). Together these data indicate that while PERK and ATF6 activation are not required, synergistic induction of IFN-β in cells experiencing ER stress is completely dependent on XBP-1.
Figure 5.

XBP-1 is required for synergistic IFN-β induction. (A) WT (PERK+/+) and PERK-deficient (PERK−/−) MEFs were stimulated with Tpg for 1 h then LPS for 2 h. IFN-β mRNA expression was assessed by qPCR. (B) WT (XBP-1+/+) or XBP-1-deficient (XBP-1−/−) MEFs were incubated with Tpg for 1 h, then poly(I:C) for 6h. IFN-β mRNA expression is shown normalized to GAPDH. Results shown are mean ±SD and are representative of 2 independent experiments. Asterisks (*) indicate p<0.05 for combined experiments comparing fold induction with LPS or PolyI:C plus Tpg vs. TLR agonist alone, and in B PolyI:C plus Tpg in XBP-1+/+ vs. XBP-1−/−.
To independently confirm the requirement for XBP-1 in mediating synergistic IFN-β induction, small interfering RNA (RNAi) was used to knock down XBP-1 mRNA (4–7-fold) in HEK293 cells stably expressing CD14 and TLR4 (Fig. 6). These cells exhibit synergistic IFN-β induction to the combination of Tpg and LPS, but this response is eliminated when XBP-1 mRNA is reduced. In contrast, XBP-1 RNAi had no effect on IL-8 induction, either in the presence or absence of Tpg, indicating that LPS-responsiveness was intact (data not shown).
Figure 6.
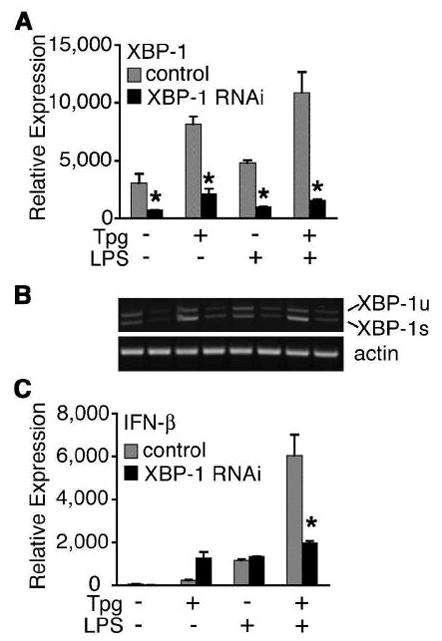
Knockdown of XBP-1 expression prevents synergistic IFN-β induction. (A) HEK293 cells expressing TLR4 and CD14 were transiently transfected with MD2 and control (■) or XBP-1 (ν) RNAi. At 24 h post transfection, cells were pretreated with Tpg for 1 h then stimulated with LPS for 3 h. Relative expression of XBP-1 mRNA by qPCR is shown normalized to GAPDH. (B) Semiquantitative PCR for XBP-1 and actin were performed on samples from (A), showing knockdown of both XBP-1u and XBP-1s. A representative gel used for quantitation is displayed with samples in the same order as in (A). Relative expression of IFN-β is shown normalized to GAPDH. Results shown are mean ±SD and are representative of 2 independent experiments. Asterisk (*) indicates p<0.05 for combined experiments comparing control vs. XBP-1 RNAi for XBP-1 expression (A) and IFN-β expression (B).
Spliced XBP-1 and IFN-β induction
The unspliced XBP-1 mRNA (XBP-1u) is expressed constitutively, but has an in-frame stop codon resulting in the production of a truncated protein that is rapidly degraded [37]. When IRE1 is activated it splices the constitutive XBP-1 mRNA (XBP-1s), causing a frame shift that enables full-length XBP-1 protein to be made. To determine whether XBP-1s was sufficient to restore synergistic induction of IFN-β, we expressed an XBP-1s cDNA in XBP-1-deficient MEFs using transient transfection. Although IFN-β induction in cells subjected to nucleofection was lower overall (vs. Fig. 5), there was 10-fold greater IFN-β expression in cells expressing XBP-1s when treated with poly(I:C) and Tpg compared to cells transfected with empty vector (Fig. 7A). Expression of XBP-1s mRNA in transfected cells was confirmed by RT-PCR (unpublished data). A similar experiment was performed using RAW264.7 macrophages, confirming that overexpression of XBP-1s enhances LPS-induced IFN-β, and that the maximal effect still requires activation of ER stress (Fig. 7B). These data confirm a role for XBP-1s in coupling ER stress to the synergistic induction of IFN-β, and also indicate that while XBP-1 is necessary, it is not sufficient for the maximal response indicating that an additional effect of ER stress is likely to play a role.
Figure 7.
Spliced XBP-1 enhances IFN-β induction. XBP-1−/− MEFs (A) and RAW264.7 macrophages (B) were transfected with XBP-1s (ν) or vector control (■). At 24 h post transfection, MEFs were treated as in Figure 6. RAW264.7 cells were pretreated with 1 μM Tpg for 1 h then LPS for 3 h. IFN-β mRNA expression is shown normalized to GAPDH. Results shown are mean ±SD and are representative of 3 independent experiments. Asterisks (*) indicate p<0.05 for combined experiments comparing control vs. XBP-1s for the conditions indicated.
HLA-B27 misfolding-induced ER stress and IFN-β production
Since Tm and Tpg can dramatically disrupt ER function, these pharmacologic agents may not faithfully reproduce the effects of limited protein misfolding. HLA-B27 misfolding results in UPR activation in macrophages from HLA-B27 Tg rats, but with a lesser magnitude than pharmacologic agents [29, 30]. To determine whether HLA-B27-induced UPR activation is sufficient to impact IFN-β induction, we compared B27-Tg and WT BM macrophages derived from 4-week old rats (prior to the development of inflammatory disease). Cells were treated with IFN-γ for 20 h to upregulate HLA-B27 and activate the UPR, which resulted in ~5-fold induction of BiP and a small increase in XBP-1 splicing (Fig. 8A, ~7% at 0 h, and unpublished observations). IFN-γ (20–24 h) does not cause UPR activation in WT macrophages or cells expressing HLA-B7 [30]. IFN-γ-primed macrophages were then treated with LPS, resulting in a much greater increase in IFN-β mRNA (5-fold) in B27-Tg macrophages experiencing ER stress, compared to WT cells (Fig. 8B). Interestingly, LPS treatment was accompanied by a striking increase in XBP-1 splicing in HLA-B27-expressing but not WT cells (Fig. 8A).
Figure 8.
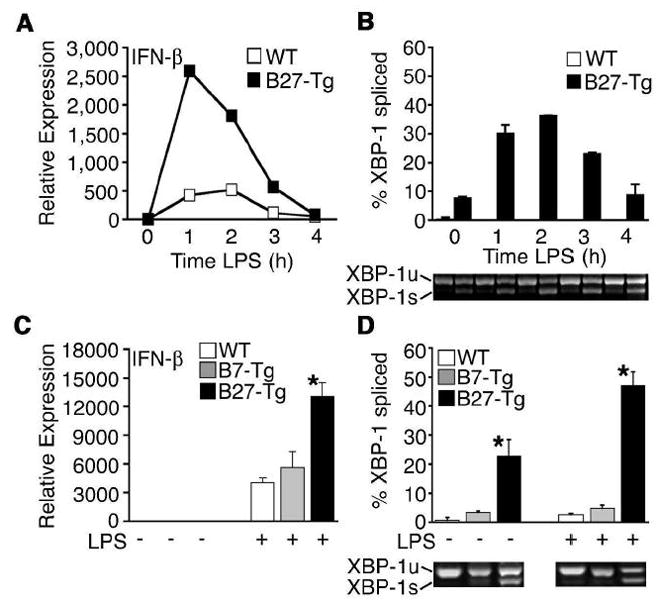
Synergistic IFN-β induction occurs in B27-Tg macrophages and correlates with increased XBP-1 splicing. BM-derived macrophages from F344 B27-Tg (ν) or WT control rats () were activated with IFN-γ for 20 h, then stimulated with 10 ng/ml LPS for 1–4 h. (A) Relative IFN-β transcripts were measured by qPCR. In (B), XBP-1 splicing in WT () and B27-Tg (ν) macrophages was determined as described in the legend to Figure 6. IFN-γ activated macrophages from Lewis WT (), B7-Tg(■), or B27-Tg (ν) rats, were stimulated with LPS for 2 h. IFN-β message levels were determined by qPCR (C) and XBP-1 splicing by PCR (D) as in (B). Results in A are the average of duplicates and are representative of 3 experiments. Results shown in B-D are mean ±SD and are representative of at least 2 experiments. In (B) p<0.05 for B27-Tg vs. WT, where it is barely detected. In (C and D) asterisks (*) indicate p<0.05 for B27-Tg vs. B7-Tg and WT. Representative XBP-1 splicing gel lanes in B and D are in the same sequence as the quantitative data.
We also examined rat macrophages overexpressing HLA-B7, an allele that does not misfold or activate the UPR [26, 28], even with upregulation [30]. XBP-1 splicing and IFN-β induction in response to LPS were no different in B7-Tg cells than WT controls (Fig. 8C,D). These data demonstrate that UPR activation as a consequence of protein misfolding causes synergistic upregulation of IFN-β, and that the concomitant increase in XBP-1 splicing is consistent with previous data (Figs. 5–7) implicating a role for XBP-1s in coupling ER stress to IFN-β production.
Discussion
In this study we demonstrate a novel link between ER stress and the innate immune response to microbial products that induce type I IFN. Specifically, UPR activation synergistically augments IFN-β induction by TLR3 and TLR4 agonists as well as cytoplasmic dsRNA. This effect is robust in that it occurs in multiple cell types, including primary macrophages, macrophage-like cell lines (RAW264.7), fibroblasts, and HEK293 cells. Synergy does not require autocrine IFN-α/β, but is dependent on XBP-1, implying a previously unrecognized role for this transcription factor. Moreover, we show that synergistic IFN-β induction occurs in rat macrophages expressing HLA-B27, an MHC class I allele that has been shown to misfold, and is strongly linked to spondyloarthritis. These results have implications for the pathogenesis of protein misfolding diseases.
Several lines of evidence support a critical role for XBP-1 in synergistic IFN-β induction. Genetic ablation or RNAi-mediated knockdown of XBP-1 mRNA prevents synergy, and XBP-1 splicing is dramatically increased during LPS stimulation of stressed B27-Tg macrophages. Furthermore, restoring expression of XBP-1s in XBP-1−/− cells permits synergy, and overexpression in RAW264.7 macrophages enhances IFN-β induction in response to LPS. While these data clearly implicate XBP-1s in the UPR component of synergistic IFN-β induction, it should be noted that overexpression of XBP-1s does not substitute entirely for ER stress, indicating that it is necessary but not sufficient.
XBP-1u encodes a truncated protein lacking the transactivation domain found in XBP-1s, and turns over rapidly during non-stress situations [20, 37]. However, there is evidence that XBP-1u protein can act as a dominant negative when forced to accumulate by inhibiting proteasomal degradation [37], and as a negative feedback regulator of XBP-1s protein during the recovery phase of ER stress when it accumulates naturally [38]. While our data imply that XBP-1s protein mediates synergistic upregulation of IFN-β, whether or not XBP-1u plays any role is not clear. In preliminary experiments we do not observe any inhibition of IFN-β expression or upregulation when XBP-1u is transiently overexpressed (unpublished observations). However, it is not possible to draw conclusions about effects of XBP-1u overexpression on synergistic upregulation since these experiments require UPR activation, and IRE1-mediated XBP-1u mRNA splicing is strongly activated under these conditions.
In macrophages undergoing a UPR, synergistic IFN-β induction was observed with activators of TLR3, TLR4, and MDA5, but not TLR2 or TLR9. TLR3/4 use the TRIF-dependent pathway to activate IRF3 via TBK-1 (Tank binding kinase 1) and induce IFN-β, whereas TLR2 signals solely through MyD88, and does not cause significant upregulation of type I IFN [39, 40]. MDA5 recognizes cytoplasmic dsRNA (poly(I:C)) and activates IRF3 via IPS-1 and TBK-1, suggesting that ER stress exerts its effect downstream of TRIF [10, 34]. TLR9 agonists (e.g. Type B CpG DNA) stimulate robust IFN-β induction in myeloid DCs and macrophages through MyD88 and IRF-1 independently of IRF3 or IRF7 [12, 41]. Lack of synergy with the TLR9 agonist is consistent with specificity of IFN-β synergy for the IRF3 activating pathways.
IFN-β expression is regulated at transcriptional and post-transcriptional levels. Transcriptional regulation is complex, requiring promoter access via deacetylation of histones and coordinated assembly of several transcription factors including IRF3, IRF7, CEBP/p300, AP-1 (ATF-2/c-Jun), and NF-κB [42–45]. We have not found any XBP-1 binding sites in the IFN-β gene promoter region using TFSEARCH and the TRANSFAC database, suggesting that XBP-1 is unlikely to directly activate IFN-β gene transcription. Nevertheless, in preliminary experiments using HEK293 cells expressing CD14 and TLR4, we find some increased activation of an IFN-β promoter reporter construct with the combination of ER stress and LPS stimulation (unpublished observations). Another possibility is that UPR activation prolongs IFN-β mRNA stability, which is regulated by an AU-rich element in the 3′ untranslated region and sequences in the coding region [46, 47]. Further studies will be necessary to define the molecular mechanism of synergistic induction of IFN-β.
The sensitization of cells by ER stress to PRR agonists has implications for diseases where the UPR plays a role in pathogenesis. For example, ischemia-reperfusion, which results in inflammatory injury, is associated with induction of UPR target genes and XBP-1 splicing [48, 49]. In the liver, damage has been shown to occur through a TLR4-mediated IRF3-dependent mechanism implicating endogenous TLR4 ligands, and raising the possibility that synergistic IFN-β induction might contribute to ischemia reperfusion injury [50]. The UPR is also activated in inflammatory myopathies, where there is evidence for an IFN-induced gene expression signature that may contribute to disease chronicity [51, 52]. In addition, infection with many viruses activates the UPR, which could enhance cytoplasmic dsRNA-induced type I IFN production through MDA5/RIG-I. It is noteworthy that Hepatitis C and human CMV have evolved mechanisms to inhibit XBP-1 activity [53, 54]. This has been hypothesized to inhibit viral protein degradation by ERAD, but it might also limit type I IFN production and thus be an important viral strategy for subverting the innate immune response.
Our demonstration that HLA-B27-induced UPR activation can enhance TLR-induced IFN-β has implications for the pathogenesis of SpA. In the rat model, inflammation begins in the gastrointestinal tract shortly after weaning and requires normal flora [55], which may provide a rich source of TLR ligands. IFN-β has recently been shown to play a crucial role in promoting macrophage survival after TLR stimulation [56]. Thus, we suggest that enhanced type I IFN production by HLA-B27-expressing macrophages could promote survival of activated macrophages via autocrine and paracrine effects, thus sustaining production of cytokines that promote T cell activation and IFN-γ synthesis [57]. This in turn could contribute to a positive feedback loop where IFN-mediated upregulation of HLA-B27 further enhances misfolding, UPR activation, and type I IFN production.
It should be emphasized that the expression of HLA-B27 in transgenic rats surpasses normal levels found in humans carrying this allele. However, rat SpA is not a consequence of class I overexpression per se, since HLA-B7 transgenic rats expressing comparable amounts of heavy chain remain healthy [24]. There is evidence that the UPR is activated in cells from the synovial fluid of patients with spondyloarthritis [58]. However, more studies will need to be done to determine the conditions leading to this response in cells from humans, and the role of HLA-B27 misfolding in disease pathogenesis.
In summary, we demonstrate that intracellular stress can magnify IFN-β induction in response to exogenous and endogenous danger signals recognized by Toll-like and viral dsRNA receptors. These results suggest an additional novel function for XBP-1 coupling UPR signaling to IFN-β induction, and support the idea that ER homeostasis may play an important role in influencing the production of inflammatory cytokines. A better understanding of how the various processes encompassed by the integrated stress response impact the immune system may aid in elucidating the pathogenesis of a variety of disease states.
Materials and methods
Animals
Inbred B27-Tg (33.3 line) and WT F344 rats were purchased from Taconic and housed in the conventional animal facility at Cincinnati Children’s Research Foundation [30]. HLA-B7 transgenic rats (120–4 line) were kindly provided by J. Taurog. Type I IFN receptor deficient (IFNAR−/−) and 129S7/SvEvBrd control mice were purchased from B&K Universal. All animal experiments were approved by the Cincinnati Children’s Research Foundation Institutional Animal Care and Use Committee.
Bone marrow derived macrophages
Rat BM macrophages were derived as described [30]. For comparisons of WT and B27-Tg cells macrophages were washed with PBS, cultured for 20 h with rat rIFN-γ (R&D Systems) and then stimulated with Salmonella enteridis LPS (Sigma) for 1–4 h. For mouse macrophages, low-density BM cells were isolated by Ficoll histopaque 1083 (Sigma) and plated with CMG 12–14 conditioned media (kindly provided by D. Williams) containing M-CSF.
Cell lines and expression vectors
RAW267.4 macrophages were maintained in RPMI (Mediatech) with 10% FBS (Gibco) and standard supplements. PERK-deficient MEFs were kindly provided by D. Ron [13], and XBP-1-deficient MEFs by L. Glimcher [18], and maintained in DMEM with supplements described above. The MD2 expression vector and HEK293 cells expressing with TLR4 and CD14, were kindly provided by C. Karp [59]. Human XBP-1 stealth-select RNAi (HS111392) and low GC RNAi control were purchased from Invitrogen. The pcDNA3.1 vector containing XBP-1s was provided by L. Glimcher [19]. Plasmids were prepared endotoxin free (Qiagen) and determined to have < 0.1 endotoxin U/ml by Limulus assay (Cambrex).
Transfections
RAW264.7 cells and XBP-1−/− MEFs were transfected with 4–5 μg XBP-1s plasmid by AMAXA nucleofection using programs U24 and A23, solutions V and MEF2, and 2 and 1 million cells per condition, respectively. HEK293 cells expressing TLR4 and CD14 were transfected using Lipofectamine 2000 (Invitrogen) with RNAi (100 pmol per well) and MD2 (0.5 μg per well) in 6-well plates. Transiently transfected cells were stimulated 24 h post-transfection.
In vitro stimulation assays
To induce a UPR, cells were pre-treated with Tm (10 μg/ml) (Sigma) for 5–8 h or Tpg (Sigma) (1 μM) for 1 h, except the XBP-1−/− MEFs and XBP-1s transfected RAW264.7 cells, which were pre-treated for 1 h with 0.25 μM Tpg. The longer pre-treatment with Tm is to accommodate the difference in how UPR is induced (see results). LPS (Sigma) was used at 10 ng/ml except where noted; poly(I:C) (Amersham) at 100 μg/ml, ODN 1826 CpG (InvivoGen) at 1 μg/ml for 6 h, Pam3CysSK4 (EMC Microcollections) at 1 μg/ml for 3 h. HEK293 cells were transfected with 1 μg poly(I:C) using Lipofectamine 2000 (Invitrogen). Following stimulation, cells were lysed in Trizol (Invitrogen) and RNA isolated. RNA was pre-treated with DNase (Invitrogen) prior to RT reactions. Oligo-dT primers and reverse transcriptase kits (Invitrogen) were used to generate cDNA for quantitative PCR. Culture supernatants were collected at 24 h and analyzed by ELISA (PBL Biomedical Laboratories) for mouse IFN-β.
Quantitative PCR and XBP-1 splicing assay
Quantitative PCR utilized Syber green (Bio-Rad) and was run on an I-cycler (Bio-Rad). Individual primer sequences are available upon request. Experimental results were normalized to actin and/or GAPDH, as indicated in the figure legends. Data are shown as averages with error bars representing standard deviation. For XBP-1 splicing, RT-PCR reactions were run on a 4% agarose gel to visualize unspliced and spliced XBP-1 products. Ethidium bromide signal was measured on a Typhoon 9400 scanner (Amersham) and quantitated using Imagequant software. Percent splicing refers to optical density of spliced band divided by total optical density (spliced plus unspliced) × 100.
Statistical analysis
Statistical analysis was performed using Student’s t-test. Differences were considered statistically significant when p values were less than 0.05.
Acknowledgments
We thank Dihua Hong for excellent technical assistance, David Ron, Laurie Glimcher, David Williams, and Chris Karp for providing cell lines or reagents, and Joel Taurog for HLA-B7 transgenic rats. Supported by NIH grants AR46177, AR048372, and AR48929 to RAC, and the Arthritis Foundation to JAS.
Abbreviations
- ATF
activating transcription factor
- BiP
immunoglobulin binding protein
- eIF2α
eukaryotic initiation factor 2 alpha
- ERAD
ER-associated degradation
- IRE1
inositol requiring 1
- IRF
interferon regulatory factor
- MDA5
melanoma differentiation-associated gene 5
- MEF
mouse embryo fibroblast
- PRR
pattern recognition receptor
- PERK
PKR-like ER kinase
- TRIF
Toll-like receptor/IL-1 receptor related adaptor protein inducing IFN-β
- Tm
tunicamycin
- Tpg
thapsigargan
- UPR
Unfolded Protein Response
- XBP-1
X-box binding protein 1
- XBP-1s
spliced XBP-1
- XBP-1u
unspliced XBP-1
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
References
- 1.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 3.Mitani Y, Takaoka A, Kim SH, Kato Y, Yokochi T, Tanaka N, Taniguchi T. Cross talk of the interferon-alpha/beta signalling complex with gp130 for effective interleukin-6 signalling. Genes Cells. 2001;6:631–640. doi: 10.1046/j.1365-2443.2001.00448.x. [DOI] [PubMed] [Google Scholar]
- 4.Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 5.Deonarain R, Verma A, Porter AC, Gewert DR, Platanias LC, Fish EN. Critical roles for IFN-beta in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 2003;100:13453–13458. doi: 10.1073/pnas.2230460100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, Wagner EF, Taniguchi T. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- 7.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 8.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 9.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 10.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 11.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M, Adler H, Drexler I, Haas T, Lang R, Wagner H. Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur J Immunol. 2007;37:315–327. doi: 10.1002/eji.200636767. [DOI] [PubMed] [Google Scholar]
- 13.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 14.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 15.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 16.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 17.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 18.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 20.Tirosh B, Iwakoshi NN, Glimcher LH, Ploegh HL. XBP-1 specifically promotes IgM synthesis and secretion, but is dispensable for degradation of glycoproteins in primary B cells. J Exp Med. 2005;202:505–516. doi: 10.1084/jem.20050575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 22.Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DCO, Sturrock RD. Ankylosing spondylitis and HL-A 27. Lancet. 1973;1:904–907. doi: 10.1016/s0140-6736(73)91360-3. [DOI] [PubMed] [Google Scholar]
- 23.Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288:704–706. doi: 10.1056/NEJM197304052881403. [DOI] [PubMed] [Google Scholar]
- 24.Taurog JD, Maika SD, Satumtira N, Dorris ML, McLean IL, Yanagisawa H, Sayad A, Stagg AJ, Fox GM, O’Brien AL, Rehman M, Zhou M, Weiner AL, Splawski JB, Richardson JA, Hammer RE. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–223. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 25.Mear JP, Schreiber KL, Munz C, Zhu X, Stevanovic S, Rammensee H-G, Rowland-Jones SL, Colbert RA. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163:6665–6670. [PubMed] [Google Scholar]
- 26.Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, Ziegler A, Colbert RA. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem. 2002;277:23459–23468. doi: 10.1074/jbc.M110336200. [DOI] [PubMed] [Google Scholar]
- 27.Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem. 2004;279:8895–8902. doi: 10.1074/jbc.M311757200. [DOI] [PubMed] [Google Scholar]
- 28.Tran TM, Satumtira N, Dorris ML, May E, Wang A, Furuta E, Taurog JD. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172:5110–5119. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 29.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56:215–223. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 30.Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, Brandewie JR, Taurog JD, Colbert RA. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 31.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 32.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 33.Rudd BD, Burstein E, Duckett CS, Li X, Lukacs NW. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J Virol. 2005;79:3350–3357. doi: 10.1128/JVI.79.6.3350-3357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 35.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 36.Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- 37.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006;172:565–575. doi: 10.1083/jcb.200508145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 40.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 41.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 42.Shestakova E, Bandu MT, Doly J, Bonnefoy E. Inhibition of histone deacetylation induces constitutive derepression of the beta interferon promoter and confers antiviral activity. J Virol. 2001;75:3444–3452. doi: 10.1128/JVI.75.7.3444-3452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 44.Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J Biol Chem. 2003;278:15495–15504. doi: 10.1074/jbc.M212940200. [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur J Biochem. 2004;271:3693–3703. doi: 10.1111/j.1432-1033.2004.04310.x. [DOI] [PubMed] [Google Scholar]
- 46.Paste M, Huez G, Kruys V. Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3′-untranslated region and an instability sequence in the coding region. Eur J Biochem. 2003;270:1590–1597. doi: 10.1046/j.1432-1033.2003.03530.x. [DOI] [PubMed] [Google Scholar]
- 47.Whittemore LA, Maniatis T. Postinduction repression of the beta-interferon gene is mediated through two positive regulatory domains. Proc Natl Acad Sci U S A. 1990;87:7799–7803. doi: 10.1073/pnas.87.20.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- 49.Emadali A, Nguyen DT, Rochon C, Tzimas GN, Metrakos PP, Chevet E. Distinct endoplasmic reticulum stress responses are triggered during human liver transplantation. J Pathol. 2005;207:111–118. doi: 10.1002/path.1798. [DOI] [PubMed] [Google Scholar]
- 50.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 51.Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, Craft J, Hooft Van Huijsduijnen R, Plotz P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, Chang J, Dwivedi S, Mitsak M, Chen YW, Plotz P, Rosen A, Hoffman E, Raben N. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 53.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem. 2004;279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 54.Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol. 2005;79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE, Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB. Normal luminal bacteria, especially bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human β2 microglobulin transgenic rats. J Clin Invest. 1996;98:945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci U S A. 2006;103:19794–19799. doi: 10.1073/pnas.0609671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sartor RB. Colitis in HLA-B27/beta 2 microglobulin transgenic rats. Int Rev Immunol. 2000;19:39–50. doi: 10.3109/08830180009048388. [DOI] [PubMed] [Google Scholar]
- 58.Gu J, Rihl M, Marker-Hermann E, Baeten D, Kuipers JG, Song YW, Maksymowych WP, Burgos-Vargas R, Veys EM, De Keyser F, Deister H, Xiong M, Huang F, Tsai WC, Yu DT. Clues to the pathogenesis of spondyloarthropathy derived from synovial fluid mononuclear cell gene expression profiles. J Rheumatol. 2002;29:2159–2164. [PubMed] [Google Scholar]
- 59.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]