

Abstract
Amyloid is associated with debilitating human ailments including Alzheimer’s and prion diseases. Biochemical, biophysical, and imaging analyses revealed that fibers produced by Escherichia coli called curli were amyloid. The CsgA curlin subunit, purified in the absence of the CsgB nucleator, adopted a soluble, unstructured form that upon prolonged incubation assembled into fibers that were indistinguishable from curli. In vivo, curli biogenesis was dependent on the nucleation-precipitation machinery requiring the CsgE and CsgF chaperone-like and nucleator proteins, respectively. Unlike eukaryotic amyloid formation, curli biogenesis is a productive pathway requiring a specific assembly machinery.
Bacteria express a variety of cell-surface proteinacious filaments that can promote colonization of an epithelial surface, entry into host cells, exchange of DNA between bacteria, and development of bacterial communities organized as biofilms, colonies, or multicellular fruiting bodies. Curli are a class of highly aggregated, extracellular fibers expressed by Escherichia and Salmonella spp. that are involved in the colonization of inert surfaces and biofilm formation (1, 2) and mediate binding to a variety of host proteins (3-5).
Polymerized curli appear as 4- to 7-nm-wide fibers of varying lengths by negative-stain electron microscopy (EM) (6). Under high-resolution EM, curli appeared as a tangled and amorphous matrix surrounding the bacteria (Fig. 1A) (7). At higher magnifications, curli fibers appeared as ~6- to 12-nm-wide fibers of varying lengths (Fig. 1, B and C).
Fig. 1.
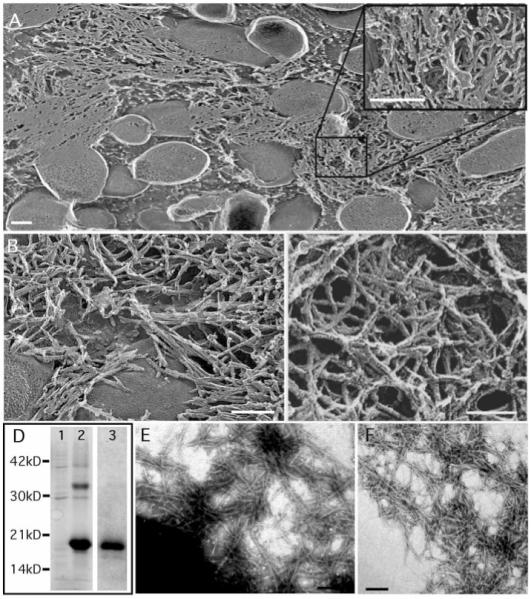
High-resolution deep-etch EM micrographs of curliated E. coli and purification of curli fibers. (A and B) Representative freeze-fractured micrographs that have been rotary shadowed with platinum. The inset in (A) shows curli fibers. (C) MC4110 was absorbed onto glass and deep-etched without being fractured before rotary shadowing with platinum. (D) Coomassie stain SDS-PAGE of curli samples isolated from E. coli strain MC4100. Lanes 1 and 2 contain 40 μg of S6 wild-type curli without and with FA treatment, respectively. Lane 3 contains 20 μg of FA-treated GP curli. Molecular size markers (in kilodaltons) are indicated on the left. (E) Negative-stain EM micrographs of MC4100 grown on YESCA plates at 26°C for 48 hours. (F) Negative-stain EM micrograph of purified wild-type S6 curli. Bars: (A), 400 nm; (B) and (C), 60 nm; (E) and (F), 200 nm.
Curli were purified from MC4100 by sequential differential centrifugation (S6) and analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) (8). Resolution of CsgA, the major structural component of curli, required brief treatment with 90% formic acid (FA) to depolymerize the CsgA polymers into a ~17.5-kD protein and two minor proteins that migrated at ~30 and 32 kD (Fig. 1D). Only the 17.5- and 32-kD bands were unique to FA-treated samples, and these bands were recognized by antibodies to CsgA (anti-CsgA) (9). The migration of these proteins is consistent with monomer and dimer sizes of CsgA (10, 11). By EM, non-FA–treated S6 curli were indistinguishable from those presented naturally on the bacterial surface, appearing as aggregated fibers of varying lengths and widths (compare Fig. 1, E and F). Circular dichroism (CD) analysis indicated that these fibers were rich in β-sheet secondary structure with a minimum peak at ~218 nm (Fig. 2A).
Fig. 2.
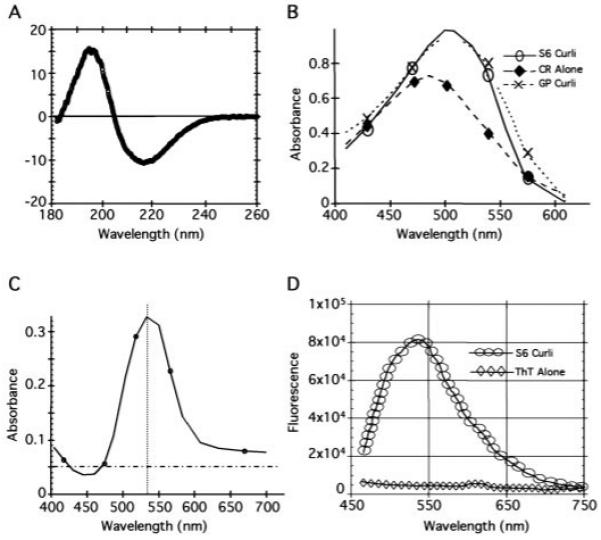
Amyloid-like properties of curli. (A) The CD spectrum of wild-type S6 curli was measured with 300 μg of protein in 10 mM tris (pH 7.4) with a 0.02-cm cell in a JASCO J715 spectropolarimeter at 25°C. S6 and GP curli gave similar CD spectra. (B) A 10-μM solution of CR prepared in 10 mM tris (pH 7.4) and 100 mM NaCl was filtered through a 0.2-μm filter and mixed with 50 μl of buffer [10 mM tris (pH 7.4)] (◆), S6 curli (4 mg/ml stock) (○), or GP curli (4 mg/ml stock) (×) in a final volume of 1 ml. All spectra were normalized against the relevant non-CR–containing solutions. (C) Spectra representing the difference of CR with S6 curli and CR alone. (D) Fluorescence of 5 μM ThT alone (◇) or mixed with 40 μg of S6 curli (○) after excitation at 450 nm on an AlphaScan PTI fluorometer with a slit width of 4 nm. GP and S6 curli gave indistinguishable fluorescence results.
Like other amyloid fibers, S6 curli induced a spectral change of a 10 μM Congo red (CR) solution with a maximum difference in absorbance between CR alone and CR bound to curli fibers at ~541 nm (Fig. 2, B and C) (12). The curli used in these assays contained a small amount of contaminating proteins (Fig. 1D). Pure, intact curli (called GP curli for “gelpurified”) were isolated as described (10) (Fig. 1D). GP curli retained the ability to bind CR and cause the red shift, demonstrating that curli were sufficient to augment the absorbance of CR (Fig. 2B). Addition of purified S6 curli to a 5 μM solution of thioflavin T (ThT) resulted in a fluorescence emission maximum at 482 nm (Fig. 2D), which is identical to the fluorescence induced by other amyloid proteins (13, 14).
Amyloid formation in eukaryotic cells is thought to be the result of a misguided protein-folding pathway. In contrast, E. coli possesses a specific nucleation-precipitation machinery encoded by the csgAB and csgDEFG operons to assemble curli. CsgB is thought to nucleate CsgA fibers (15). The csgDEFG genes encode CsgD, a FixJ-like transcriptional regulator, and three putative curli assembly factors, CsgE, CsgF, and CsgG. The lipoprotein CsgG localizes to the inner leaflet of the outer membrane and may serve as a curli assembly platform (16).
A nonpolar csgF− deletion mutant (MHR592) (17) resulted in aberrant CR binding properties. Wild-type bacteria stained CR-positive after 30 hours of growth on YESCA plates (9). Strain MHR592 (csgF−) was CR-negative after 30 hours of growth and only slightly CR-positive after 48 hours of growth (9). EM analysis of csgF− bacteria showed that fibers were less abundant but were otherwise indistinguishable from those produced by wild-type bacteria (Fig. 3A).
Fig. 3.
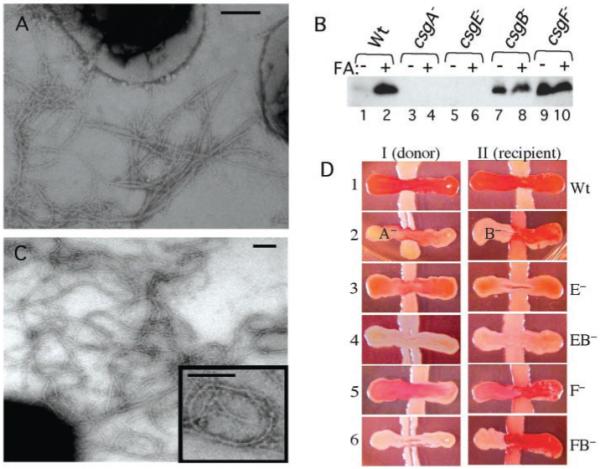
Curli biogenesis in the absence of CsgE and CsgF. (A) Negative-stain EM micrographs of MHR592 (csgF−) bacteria grown on YESCA plates at 26°C for 48 hours. (B) CsgA visualized by Western analysis with anti-CsgA (16) and bacteria grown at 26°C on YESCA plates for 48 hours. Circular plugs of 8 mm, including cells and underlying agar (to collect soluble, unpolymerized and secreted CsgA), were collected and resuspended in 200 μl of 1.5× SDS loading buffer either with or without prior FA treatment. The etracts loaded in each lane are as follows: lanes 1 and 2, MC4100 (wild type); lanes 3 and 4, LSR10 (csgA−); lanes 5 and 6, MHR480 (csgE−); lanes 7 and 8, MHR261 (csgB−); and lanes 9 and 10 MHR592 (csgF−). (C) Negative-stain EM micrographs of MHR480 (csgE−) bacteria grown on YESCA plates at 26°C for 48 hours. These fibers often looped into imperfect circles (see inset). (D) Interbacterial complementation and CR binding in csgE− and csgF− mutants. The CsgA+ donor strain MHR261 (I) and the CsgB+ recipient strain LSR10 (II) were streaked from the top of the plate to the bottom. The horizontal cross-streaks were made from left to right with the following strains: MC4100 (wild type) (I-1 and II-1), csgA− (I-2), csgB− (II-2), csgE− (I-3 and II-3), csgEB− (I-4 and II-4), csgF− (I-5 and II-5), and csgFB− (I-6 and II-6). Bars in (A) and (C), 200 nm.
The monomeric and polymeric state of CsgA in the absence of CsgF was assessed. Very little SDS-soluble CsgA was present in extracts made from wild-type bacteria (Fig. 3B) because most of the CsgA subunits were assembled into curli as determined by the presence of a 17.5-kD band after pretreatment with FA (Fig. 3B). Similar to a csgB− mutant (Fig. 3B), most CsgA produced by a csgF− mutant remained in an SDS-soluble form after 48 hours of growth (Fig. 3B).
CsgA is secreted in a soluble, assembly-competent form by a csgB− mutant (CsgA+ donor) and can be assembled on the surface of csgA− mutant (CsgB+ recipient) bacteria in a process called interbacterial complementation (Fig. 3D) (18). CsgB+ recipient cells lacking the CsgA protein stained CR-positive whencross-streaked with either csgF− or csgF−B− double-mutant bacteria (Fig. 3D), indicating that CsgA was secreted from csgF− cells and assembled on the CsgB+ recipient cells. In contrast, csgF− and csgF−B− mutants were unable to accept CsgA from a CsgA+-donating strain (Fig. 3D). Thus, the curli assembly defect in csgF− mutants was a nucleation defect because CsgA produced by these cells was assembly competent. CsgF may work independently or in concert with CsgB to guide in vivo extracellular nucleation of CsgA.
A nonpolar csgE− deletion mutant (MHR480) (17) produced pale, non CR–binding colonies similar to those produced by a csgA mutant (9). Despite the pale-colony phenotype, MHR480 (csgE−), but not a csgE−A− double mutant, produced fibers that reacted with anti-CsgA. However, these structures were less abundant than wild-type curli and were architecturally distinct in that they tended to arrange into rings (Fig. 3C). In the csgE− cells, no SDS-soluble CsgA could be detected, and the total amount of SDS-insoluble CsgA was markedly reduced (CsgA was detected in the FA-treated sample only after extended exposure) (Fig. 3B). A csgE− mutant was unable to donate CsgA subunits when cross-streaked against the CsgB+ recipient (Fig. 3D). However, a csgE−mutant retained the ability to act as a recipient and guide CsgA polymerization, because it stained CR-positive when cross-streaked against a CsgA+ donor (Fig. 3D). This staining was weaker than that observed on a CsgB+ recipient cross-streaked against a CsgA+ donor (Fig. 3D), suggesting that in addition to the CsgA stability defect, csgE− bacteria are also partially defective in their ability to nucleate exogenous CsgA. A csgB−E− double mutant was unable to accept or donate CsgA (Fig. 3D).
To understand the requirements for subunit polymerization, we purified CsgA in a soluble form and analyzed its polymerization in vitro. A six-histidine–tagged version of csgA (CsgA-his) was cloned behind the IPTG (isopropyl-β-D-thiogalactopyranoside)-inducible trc promoter in pHL3 (17), to create pMC3 (19). Plasmid-derived CsgA-his complemented fiber formation and CR binding in a csgA− mutant (9). To produce soluble, nonpolymerized CsgA, we transformed pMC3 (csgA-his) into LSR6 (C600:ΔcsgDEFG;ΔcsgBA) (19). However, attempts to detect CsgA-his protein expressed in LSR6 were unsuccessful (Fig. 4A). When LSR6/pMC3 (csgA-his) was transformed with pMC5 (csgEFG) or pMC1 (csgG), CsgA-his could be detected in the culture supernatants (Fig. 4A). Under these growth conditions (logarithmic growth in LB broth), only CsgG, and not CsgE, was required for efficient CsgA stabilization and secretion.
Fig. 4.
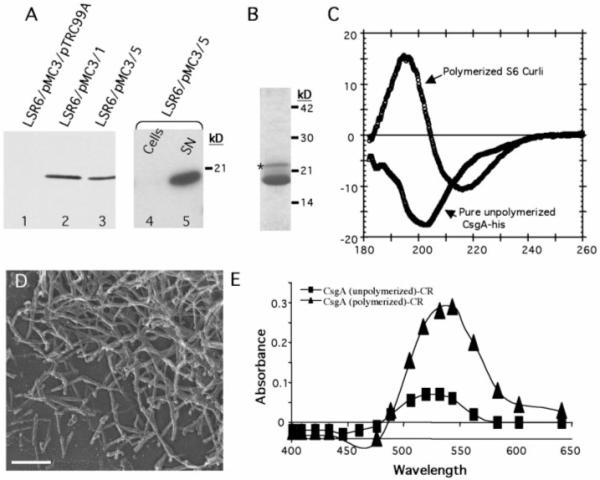
Purification and in vitro assembly of CsgA-his. (A) Western blot with anti-his (Covance, Richmond, California) to determine expression and cellular location of overexpressed CsgA-his. Log-phase cultures containing (lane 1) pMC3 (csgA-his) and pTrc99A (empty vector); (lane 2) pMC3 and pMC1 (csgG); or (lane 3) pMC3 and pMC5 (csgEFG) were induced with 0.5 mM IPTG for 1 hour; samples were removed and mixed with an equal amount of 2× SDS-PAGE dye and heated to 95°C before gel electrophoresis. CsgA-his expression with pTrc99A was detected only after overexposure of the blot. The cellular and supernatant fractions from cultures containing pMC3 and pMC5 were separated by centrifugation and loaded in lanes 4 and 5, respectively. (B) CsgA-his was purified from cleared LSR6/pMC3/pMC5 supernatants filtered through a 0.2-μm filter before loading on a disposable column packed with nickel NTA-agarose beads (Qiagen, Chatsworth, California). The column was washed with 10 column volumes of 10 mM tris (pH 7.4), 100 mM NaCl, and CsgA-his was eluted with 5 ml of wash buffer plus 100 mM imidazole and analyzed by Coomassie stain SDS-PAGE. Both the major band migrating at ~17.5 kD and the higher molecular weight, minor band (indicated by an asterisk) interacted with anti-CsgA (9). (C) The CD spectrum of wild-type S6 curli compared with that of 300 μg of soluble unpolymerized CsgA-his assayed immediately after purification. (D) High-resolution EM of pure CsgA-his preparations after a 1-week incubation at 4°C. Bar, 140 nm. (E) Absorbance of a 10 μM solution of CR with 100 μg of pure, unpolymerized CsgA-his (■) or pure polymerized CsgA-his (▲) after subtracting the absorbance of CR alone.
CsgA-his was purified from the supernatant of LSR6/pMC3/pMC5 and LSR6/pMC3/pMC1 by standard nickel affinity chromatography (Fig. 4B). Immediately after elution from the nitrilotriacetic acid (NTA) agarose column, solutions containing purified CsgA-his were clear with no evidence of aggregation, and EM of this material revealed no fibers (9). CD analysis of this material indicated that soluble CsgA-his, unlike curli, was not rich in β-sheet secondary structure (Fig. 4C). However, after prolonged incubation (4°C for 4 to 12 hours), the CsgA-his solutions became opaque and noticeably viscous. EM analysis revealed that fibers had formed that were similar to those produced by wild-type bacteria (Fig. 4D). The in vitro–assembled CsgA-his fibers, but not the CsgA-his soluble precursors, were able to bind CR and cause a red shift (Fig. 4E), signifying that they had adopted the cross β structure conserved in all amyloid fibers. CsgA purified from cells expressing csgEFG or only csgG formed CR-binding fibers with indistinguishable kinetics (9). Thus, although CsgB and CsgEFG are required to facilitate efficient polymerization in vivo, they are not required for polymerization to proceed under these in vitro conditions. A critical function of the nucleation-precipitation assembly machinery may be to prevent CsgA polymerization within the cell and accelerate it at the cell surface.
Our demonstration that E. coli can produce extracellular amyloid-like fibers increases the recognized functional repertoire of amyloid fibers and provides a useful model system to study their formation. Purified amyloid fiber subunits associated with human diseases, such as the Aβ protein associated with Alzheimer’s disease, readily polymerize when incubated at high concentrations in vitro (20). However, the accessory proteins and conditions that facilitate in vivo polymerization of Aβ are incompletely understood. Understanding the regulation of curli subunit polymerization in E. coli will offer insight into the formation of eukaryotic amyloids. This work also raises the intriguing possibility that bacterial amyloids could play a role in certain human neurodegenerative and amyloid-related diseases. Future experiments will further examine the role of CsgB, CsgE, and CsgF during the in vivo polymerization of curli, and their function will be used as a model to understand the formation of other amyloids.
Acknowledgments
We thank members of the Hultgren lab and especially K. Dodson for helpful comments during the preparation of this manuscript. This work was supported by the Alzheimer’s Disease Research Center grant NIA P50 AG05681-17 and NIH grants AI29549, DK51406, and AI48689 (S.J.H.). S.N. acknowledges grants from the Swedish Medical Research Council (16x-10843) and Swedish Natural Science Research Council (3373-309). M.R.C. was supported by a Keck fellowship and by NIH fellowship 1 F32 AI10502-01A1.
References and Notes
- 1.Vidal O, et al. J. Bacteriol. 1998;180:2442. doi: 10.1128/jb.180.9.2442-2449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin JW, Sanders G, Kay WW, Collinson SK. FEMS Microbiol. Lett. 1998;162:295. doi: 10.1111/j.1574-6968.1998.tb13012.x. [DOI] [PubMed] [Google Scholar]
- 3.Sjobring U, Pohl G, Olsen A. Mol. Microbiol. 1994;14:443. doi: 10.1111/j.1365-2958.1994.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 4.Olsen A, Arnqvist A, Hammar M, Normark S. Infect. Agents Dis. 1993;2:272. [PubMed] [Google Scholar]
- 5.Ben Nasr A, Olsen A, Sjobring U, Muller-Esterl W, Bjorck L. Mol. Microbiol. 1996;20:927. doi: 10.1111/j.1365-2958.1996.tb02534.x. [DOI] [PubMed] [Google Scholar]
- 6.Olsen A, Jonsson A, Normark S. Nature. 1989;338:652. doi: 10.1038/338652a0. [DOI] [PubMed] [Google Scholar]
- 7.MC4100 was grown on YESCA plates at 26°C for 50 hours, washed with 1× phosphate-buffered saline, placed on aldehyde-fixed slices of rabbit lung, and then quickly frozen by abruptly pressing the samples against a copper block cooled to 4 K with liquid helium. Frozen samples were fractured (where indicated), then deep-etched by exposure to a vacuum for 2.5 min, and replicas were made by rotary shadowing with a mixture of platinum and carbon (21).
- 8.MC4100 was spread as a lawn on 10 150-mm YESCA plates and grown at 26°C for 60 hours before being scraped from the plates and suspended in 300 ml of 10 mM tris (pH 7.4). Bacteria were blended five times on ice with an Omni-Mixer homogenizer for 1 min at 3-min intervals. The bacteria were pelleted by centrifuging two times at low speed (5000g, 10 min). The supernatant was made 150 mM NaCl and the curli pelleted by centrifuging at 13,000g. The pellet was resuspended in 300 ml of 10 mM tris (pH 7.4), 150 mM NaCl, and incubated on ice for 30 min before being centrifuged at 13,000g. This procedure was repeated three times. The pellet was then resuspended in 30 ml of 10 mM tris (pH 7.4) and pelleted as described above. The pellet was again suspended in 30 ml of 10 mM tris (pH 7.4) and centrifuged at 35,000g. For the gel-purified curli, 2.5 ml of the S6 fraction was mixed with an equal amount of 2× SDS loading buffer and subjected to electrophoresis for 5 hours on a 12% SDS-PAGE gel. The curli remaining in the slot after electrophoresis were recovered as described (10).
- 9.Chapman MR, Robinson LS, Hultgren SJ. unpublished observations. [Google Scholar]
- 10.Collinson SK, Emody L, Muller KH, Trust TJ, Kay WW. J. Bacteriol. 1991;173:4773. doi: 10.1128/jb.173.15.4773-4781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsen A, Arnqvist A, Hammar M, Sukupolvi S, Normark S. Mol. Microbiol. 1993;7:523. doi: 10.1111/j.1365-2958.1993.tb01143.x. [DOI] [PubMed] [Google Scholar]
- 12.Klunk WE, Jacob RF, Mason RP. Methods Enzymol. 1999;309:285. doi: 10.1016/s0076-6879(99)09021-7. [DOI] [PubMed] [Google Scholar]
- 13.LeVine H., III Methods Enzymol. 1999;309:274. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 14.LeVine H. Protein Sci. 1993;2:404. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bian Z, Normark S. EMBO J. 1997;16:5827. doi: 10.1093/emboj/16.19.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loferer H, Hammar M, Normark S. Mol. Microbiol. 1997;26:11. doi: 10.1046/j.1365-2958.1997.5231883.x. [DOI] [PubMed] [Google Scholar]
- 17.Hammer M. Karolinska Institute; 1997. [Google Scholar]
- 18.Hammer M, Bian Z, Normark S. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6562. doi: 10.1073/pnas.93.13.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MC4100 chromosomal DNA was amplified with the primers 5′-CATATGAAACT T T TAAAAGTAGCAGCAATTG and 5′-GAATTCTAATGGTGATGGTGATGGTGGTACTGATGAGCGATCG, and the amplicon was cloned into the Nde I and Eco RI sites of pHL3 to create pMC3 by standard techniques. The csgEFG genes were similarly amplified and cloned into the Nco I and Bam HI sites of pTrc99A to create pMC5 (Pharmacia, Pisca-taway, NJ) with the primers 5′-CCATGGCGAAACGTTATTTACGCTG and 5′-GGATCCTCAGGATTCCGGTGGAACCGAC. Knockouts of csgA or of the entire curli locus were made by homologous recombination as described (22). The kanr cassette from pKD13 was am-plified by polymerase chain reaction (PCR) with primers containing 5′-homology arms corresponding to sequences flanking the regions to be deleted. Primers used to create LSR10 (ΔcsgA) were 5′-gttaatttccattcgacttttaaatcaatccgatgggggttttacGTGTAGGCTGGAGCTGCTTC and 5′-agggcttgcgccctgtttctgtaatacaaatgatgtATTCCGGGGATCCGTCGACC (lower-case letters correspond to csg sequences). The primers used to generate LSR5 (ΔcsgDEFG;ΔcsgBA), were 5′-agggcttgcgccctgtttctgtaatacaaatgatgtATTCCGGGGATCCGTCGACC and 5′-gccgacatcaggcacagcataacaggttcgttcgagGTGTAGGCTGGAGCTGCT TC. PCR products were electroporated into MC4100-expressing Red recombinase proteins from pKD46 (22). The resulting Kanr strains were confirmed by PCR and failed to bind CR when grown on YESCA plates. The mutation from LSR5 was transferred into C600 by standard P1 transduction, creating LSR6.
- 20.Barrow CJ, Yasuda A, Kenny PT, Zagorski MG. J. Mol. Biol. 1992;225:1075. doi: 10.1016/0022-2836(92)90106-t. [DOI] [PubMed] [Google Scholar]
- 21.Hueser JE, Muscle J. Res. Cell Motil. 1987;8:303. doi: 10.1007/BF01568887. [DOI] [PubMed] [Google Scholar]
- 22.Datsenko KA, Wanner BL. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]