

Abstract
Cell morphology dictates response to a wide variety of stimuli, controlling cell metabolism, differentiation, proliferation, and death. Epithelial-mesenchymal transition (EMT) is a developmental process in which epithelial cells acquire migratory characteristics, and in the process convert from a “cuboidal” epithelial structure into an elongated mesenchymal shape. We had shown previously that matrix metalloproteinase-3 (MMP3) can stimulate EMT of cultured mouse mammary epithelial cells through a process that involves increased expression of Rac1b, a protein that stimulates alterations in cytoskeletal structure. We show here that cells treated with MMP-3 or induced to express Rac1b spread to cover a larger surface, and that this induction of cell spreading is a requirement of MMP-3/Rac1b-induced EMT. We find that limiting cell spreading, either by increasing cell density or by culturing cells on precisely defined micropatterned substrata, blocks expression of characteristic markers of EMT in cells treated with MMP-3. These effects are not caused by general disruptions in cell signaling pathways, as TGF-β-induced EMT is not affected by similar limitations on cell spreading. Our data reveal a previously unanticipated cell shape-dependent mechanism that controls this key phenotypic alteration and provide insight into the distinct mechanisms activated by different EMT-inducing agents.
Keywords: Cell Morphology, Cell Spreading, Matrix Metalloproteinase, Epithelial-Mesenchymal Transition, Rac1b, TGFβ, Micropatterned Substrata, Mammary Epithelial Cells, Reactive Oxygen Species
Epithelial-mesenchymal transition (EMT) is a phenotypic alteration in which epithelial cells detach from their neighbors and the underlying basement membrane and become more motile and migratory [Thiery, 2002; Radisky, 2005]. EMT is critical for metazoan embryonic development: during gastrulation, the primitive embryonic epithelium forms the primary mesenchyme, and in vertebrates, multipotent migratory neural crest cells delaminate from the neural ectoderm and form diverse tissue derivatives [Shook and Keller, 2003].
There is an increasing awareness that EMT-related processes are activated under pathological conditions as well, including fibrosis, tumor progression, and metastatic invasion [Kalluri and Neilson, 2003; Petersen et al., 2003; Thiery, 2003; Radisky et al., 2007], and this recognition has triggered intensive investigations into the mechanisms involved in the activation of EMT. In cultured cells, EMT can be induced by cytokines, growth factors, and matrix metalloproteinases (MMPs) [Thiery and Sleeman, 2006; Stallings-Mann and Radisky, 2007]. We have found that MMP3 (previously stromelysin-1) induces EMT in mouse mammary epithelial cells [Lochter et al., 1997a,b; Sternlicht et al., 1999; Radisky et al., 2005], through a signaling mechanism that involves cleavage of E-cadherin and induction of Rac1b, a highly activated splice isoform of the Rac1 GTPase. Rac1b increases the levels of cellular reactive oxygen species (ROS), which in turn activate the EMT transcriptional program, including downregulation of epithelial cytokeratins and increased expression of mesenchymal markers including Snail, vimentin, and α-smooth muscle actin [Radisky et al., 2005]. Cells treated with MMP3 also significantly alter their cytoskeletal structure and morphology by losing cortical actin, developing large lamellapodia and increasing their spreading on the underlying substratum [Radisky et al., 2005].
Modulation of cell morphology can dramatically alter cellular behaviors, such as: glucose uptake and metabolism, [Bissell et al., 1977], cell division [Folkman and Moscona, 1978], proliferation and apoptosis [Chen et al., 1997], differentiation [Watt et al., 1988; Roskelley et al., 1994], nuclear organization [Le Beyec et al., 2007], and morphogenesis [Nelson et al., 2005, 2006]. EMT was first defined [Greenburg and Hay, 1982] as a morphological reshaping of the epithelium that precedes mesenchymal invasion [Kalluri and Neilson, 2003]. Culturing epithelial cells at low density can induce some features of EMT, including a more spread morphology, downmodulation of epithelial keratins, and upregulation of mesenchymal vimentin [Ben-Ze'ev, 1984; Maeda et al., 2005; Sarrio et al., 2008]. Increased cell spreading is an early feature of MMP3-treated mammary epithelial cells [Radisky et al., 2005]; we thus hypothesized that this change in morphology might be necessary for MMP-3 to induce EMT. Here, we tested this hypothesis in mouse mammary epithelial cells. We find that MMP-3-induced cell spreading is required for the downstream induction of EMT. Cells treated with TGFβ also increase their spreading against the underlying substratum, but that spreading is not required for TGFβ-induced EMT. These results reveal differences between the EMT pathways downstream of MMP-3 and TGFβ, and provide insight into the causal relationship between morphology and EMT.
Materials and Methods
Cell Culture and Reagents
Use of SCp2 mouse mammary epithelial cells and treatment with MMP3 were as previously described [Radisky et al., 2005], with a 3-day time point for all experiments. The following reagents were used at the given concentrations: H2O2 (25 μM, Sigma); NAC (10 mM, Sigma); GM6001 (40 μM, Calbiochem). For measurements of projected cell area, phase contrast images of individual cells were outlined and processed with Image J software. Transfections were performed with lipofectamine-2000 (Invtrogen) as previously published [Radisky et al., 2005]. For luciferase assays, cells were transfected with plasmids expressing 3TP-luciferase and CMV-renilla in a 10:1 ratio, and relative luciferase was assessed using the Dual-Glo Luciferase Assay System (Promega) and a Veritas Microplate Luminometer (Turner Biosystems). Plasmids for expression of YFP-Rac1b, YFP-Rac1N17, and CMV-Rac1b and methods for selective siRNA knockdown of Rac1b and for assessing ROS using DCFDA (Invitrogen) were described previously [Radisky et al., 2005]; pEYFP-C1 (Clontech) was used as a control for transfection experiments. Recombinant human TGF-β1 (240-B, R&D Systems) was used at 25 ng/ml final concentration.
Quantitative Real-Time PCR Analysis
Transcript levels were measured using RT/PCR by isolating RNA (Tri-pure; Roche Diagnostics), synthesizing cDNA and performing quantitative real-time PCR using ABI protocols and software (Lightcycler, Roche). Expression levels were assessed using TaqMan assays (Roche): sm-actin, Mm01546133_m1; GAPDH, Mm99999915_g1. We assessed Rac1b using a custom assay (forward primer, 5′-TGGACAAGAAGATTATGACAGATTGC-3′; reverse primer, 5′-CCCTGGAGGGTCTATCTTTACCA-3′; probe, 5′-CCGCAGACAGTTGGAGA-3′).
Micropatterning
Micropatterned substrata containing fibronectin-coated islands surrounded by non-adhesive regions were created as described [Tan et al., 2004]. Briefly, poly(dimethylsiloxane) (PDMS; Sylgard 184, Ellsworth Adhesives, Germantown, WI) elastomeric stamps containing a relief of 20 or 40 μm squares were coated with fibronectin (25 μg/ml in PBS; BD Biosciences) for 2 h, washed with water, and dried under a stream of nitrogen. Flat PDMS-coated substrata were UV-oxidized for 7 min (UVO cleaner, Jelight Co., Irvine, CA), stamped with fibronectin, blocked with 1% pluronic F108 (BASF Corp., Florham Park, NJ) in PBS for 1 h, and rinsed in PBS before seeding SCp2 cells. Cells were allowed to attach to the patterned islands for approximately 30 min before rinsing off remaining non-adherent cells. Antibodies for immunofluorescence were anti cytokeratin wide screening (Z0622, Dako) and anti vimentin (V5255, Sigma).
Results
The tumors that arise as the result of inappropriate MMP3 expression in mammary glands of transgenic mice exhibit EMT [Sternlicht et al., 1999]. We showed that treatment with MMP3 is directly responsible for induction of EMT in mouse cells in culture [Lochter et al., 1997a,b; Radisky et al., 2005]. The MMP3-induced EMT is characterized by cell scattering, down-regulation of epithelial markers such as cytokeratins and E-cadherin, and upregulation of mesenchymal markers including vimentin, α-smooth muscle actin, and the transcription factor Snail [Radisky et al., 2005]. Additionally, MMP3-induced EMT is accompanied by altered cellular morphology, with loss of colonial morphology, increased lamellipodia (Fig. 1a), and substantial increase in cell spreading (as measured by projected cell area) against the underlying substratum (Fig. 1b). The catalytic activity of MMP3 is required for increased cell spreading and EMT since both are blocked by treatment with the broad spectrum MMP inhibitor, GM6001.
Fig. 1.
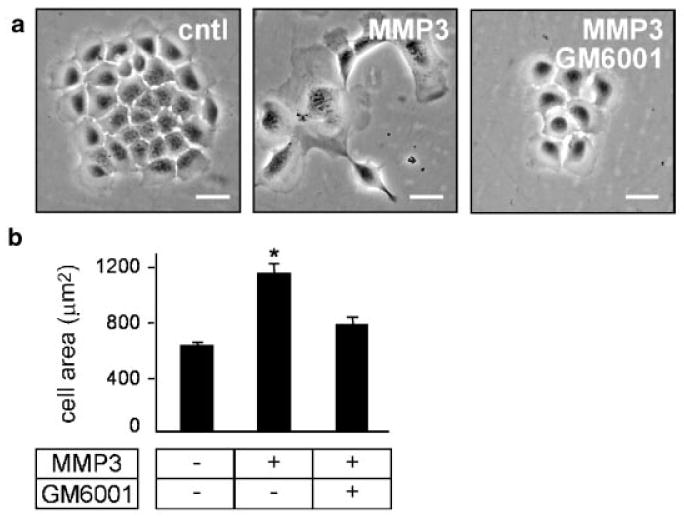
MMP3-mediated EMT correlates with increased cell spreading. a: Phase contrast images of SCp2 cells with and without MMP3 and GM6001. b: Quantification of projected area of SCp2 cells with and without MMP3 and GM6001. *P < 0.01. Scale bar, 25 μm.
We previously showed that MMP3 induces EMT through a cascade involving the generation of Rac1b, a highly active splice variant of Rac1 [Radisky et al., 2005]. We now find that MMP3-induced cell spreading is a consequence of the induction of Rac1b. Exogenous expression of Rac1b induces cell scattering (Fig. 2a), increases expression of EMT markers such as α-smooth muscle actin (Fig. 2b) and increases cell spreading (Fig. 2c); knockdown of Rac1b by siRNA blocks both MMP-3-induced expression of α-smooth muscle actin (Fig. 2d) and cell spreading (Fig. 2e). Our previous studies showed that Rac1b led to the production of cellular reactive oxygen species (ROS), which led to upregulation of the EMT-inducing transcription factor, Snail [Radisky et al., 2005]. Now here we show that cell spreading is upstream of ROS. We found that cells treated simultaneously with MMP-3 and the ROS quenching agent N-acetyl cysteine (NAC) failed to scatter and did not express EMT markers [Radisky et al., 2005], but still showed increased cell spreading (Fig. 2f). We also found that ROS-induced EMT can occur in the absence of cell spreading, as treatment with H2O2 alone induces cell scattering and EMT without increasing cell spreading (Fig. 2f). These data show that MMP3-induced cell spreading is downstream of Rac1b but upstream of, or parallel to, ROS induction of EMT. To evaluate whether MMP3/Rac1b-induced cell spreading was specifically necessary for the induction of EMT, we assessed the effect of MMP3 treatment and Rac1b expression in cells cultured at different densities. We selected cell density values so that there was substantial cell–cell contact even at the low cell density (50,000 cells/cm2) and maintenance of healthy overall culture appearance and cell viability (by trypan blue exclusion, data not shown) at high cell density (150,000 cells/cm2) (Fig. 3a). We found that the limitation of cell spreading at high density blocked the induction of EMT markers including α-smooth muscle actin (Fig. 3b) and that cells plated at low density were able to spread, whereas those at high density were not (Fig. 3c). This effect was not due to an inability of the high cell density cultures to respond to MMP3, as cells in both densities increased expression of Rac1b in response to MMP3 (Fig. 3d). We also found that cells cultured at high density were unable to activate EMT in response to exogenous expression of Rac1b (Fig. 3e; exogenous Rac1b expression levels were similar in the low and high cell density cultures, data not shown). These data show that blocking cell spreading by plating cells at high density effectively inhibits MMP3/Rac1b induction of EMT-related gene expression. To determine whether MMP3/Rac1b-induced cell spreading is necessary for induction of ROS, we assessed induction of DCFDA fluorescence in cells treated with MMP3 or transfected with Rac1b and cultured at high and low densities. We found that while MMP3 treatment or Rac1b expression caused significantly increased levels of ROS in cells cultured at low density, plating at high density to block cell spreading also inhibited induction of ROS by MMP3 (Fig. 3f) or Rac1b expression (Fig. 3g). These results demonstrate that blocking cell spreading by plating cells at high density effectively inhibits MMP3/Rac1b induction of cellular ROS.
Fig. 2.
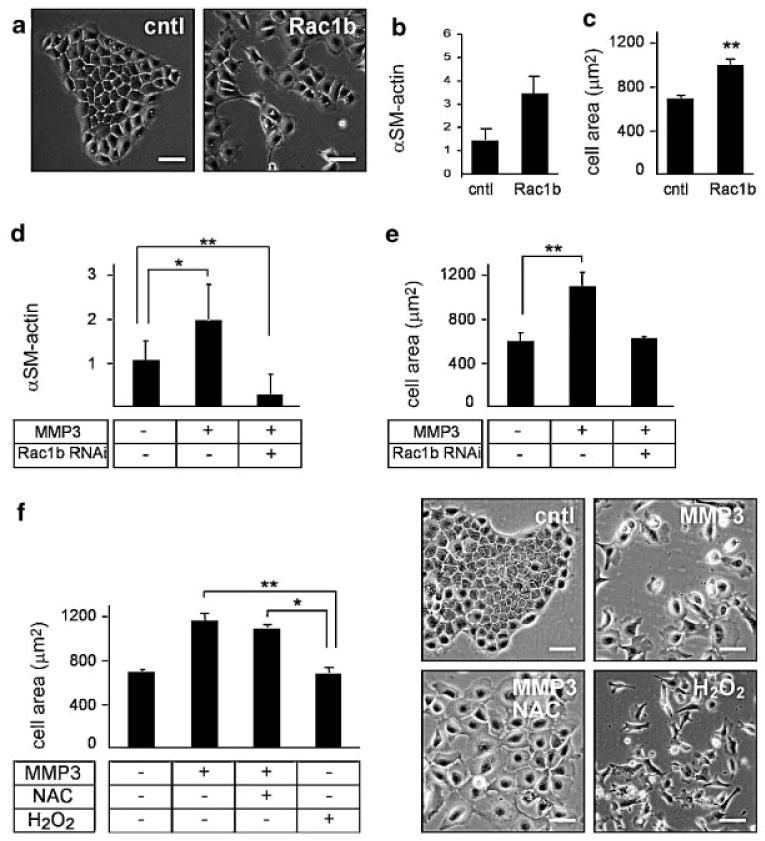
MMP3-induced cell spreading is upstream of EMT-associated induction of ROS. a: Phase contrast images of SCp2 cells transfected with Rac1b or control vector YFP. b: Graph showing RT/PCR analysis for α-smooth muscle actin (αSM-actin) in SCp2 cells transfected with YFP or YFP-Rac1b, expressed relative to GAPDH expression. c: Quantification of projected cell area in SCp2 cells transfected with YFP (cntl) or YFP-Rac1b (Rac1b). d: Expression of α-smooth muscle actin in untreated cells, cells treated with MMP3, or cells transfected with siRNA reagents selectively targeting Rac1b and treated with MMP3. e: Quantification of projected area of cells treated as control or treated with MMP3, or transfected with Rac1b siRNA and treated with MMP3. f: Quantification of area (left) and representative images (right) of MMP3-treated SCp2 cells with and without 10 mM NAC, or in SCp2 cells treated with 25 μM H2O2. *P < 0.01; **P < 0.001. Scale bar, 50 μm.
Fig. 3.
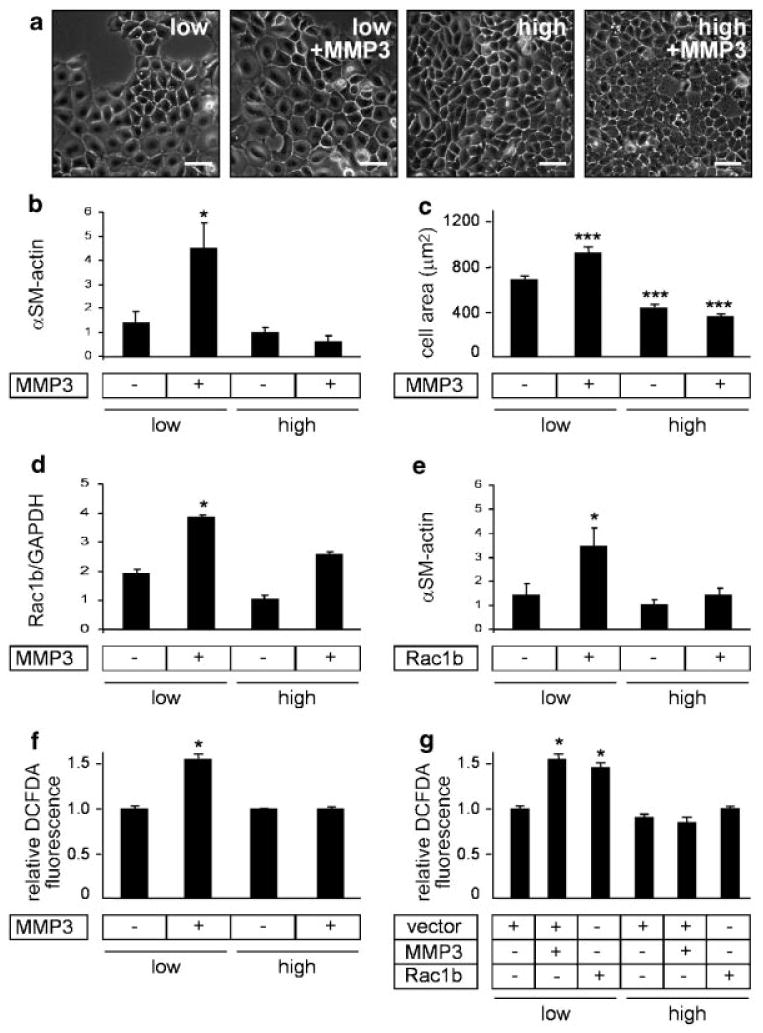
MMP3-induced EMT and cell spreading, but not Rac1b expression, depend on cell density. a: Phase contrast images of MMP3-treated SCp2 cells at different initial seeding densities. b: Graph quantifying projected cell area of MMP3-treated SCp2 cells as a function of initial seeding density. c: Expression of α-smooth muscle actin, normalized to GAPDH, in MMP3-treated SCp2 cells at different initial seeding densities. d: Rac1b expression (relative to GAPDH) in MMP3-treated SCp2 cells as a function of initial seeding density. e: Expression of α-smooth muscle actin, relative to GAPDH, in Rac1b-expressing cells at different initial seeding densities. f: Induction of ROS by MMP3 treatment is blocked by growth of cells at high density (assessed by increased DCFDA fluorescence). g: Induction of ROS by Rac1b transfection is blocked by growth of cells at high density. *P < 0.01; ***P < 0.0001. Scale bar, 50 μm.
To assess whether high cell density blocks all inducers of EMT, we tested the response of high and low density cultures to treatment with TGFβ (Fig. 4a). We evaluated the response of cells to 25 ng/ml TGFβ, a concentration which produces amounts of cell scattering comparable to that of cells treated with MMP3 (not shown). We found that TGFβ increased cell spreading in the low density but not the high-density cultures (Fig. 4b). However, TGFβ induced comparable activation of the SMAD-responsive reporter vector 3TP-luc (assessed as relative to expression of co-transfected Renilla luciferase reporter vector; Fig. 4c) and comparable induction of EMT markers such as α-smooth muscle actin in both densities, when assessed as fold increase over untreated cells (Fig. 4d). These results demonstrate that TGFβ-induced EMT is not blocked by limiting cell spreading.
Fig. 4.
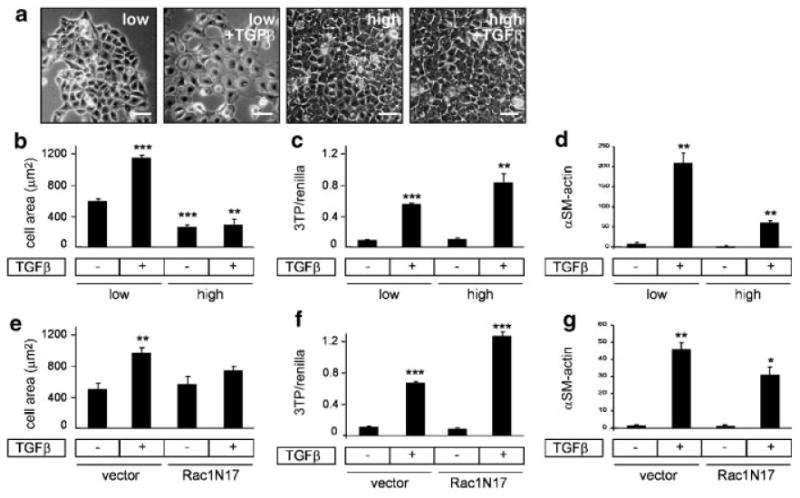
TGFβ-induced EMT can occur at high density. a: Phase contrast images of TGFβ-treated SCp2 cells at different initial seeding densities. b: Quantification of projected cell area of TGFβ-treated SCp2 cells as a function of initial seeding density. c: Activation of 3TP-luc expression construct (relative to co-transfected Renilla luciferase expression) in TGFβ-treated SCp2 cells grown at low and high density. d: Expression of a smooth muscle actin in TGFβ-treated SCp2 cells at different initial seeding densities. e: Quantification of projected cell area of TGFβ-treated SCp2 cells transfected with YFP (vector) or YFP-Rac1N17. f: Activation of 3TP-luc expression construct (relative to co-transfected Renilla luciferase expression) in TGFβ-treated SCp2 cells transfected with YFP or YFP-Rac1N17. g: Expression of α-smooth muscle actin in TGFβ-treated SCp2 cells transfected with YFP or YFP-Rac1N17. *P < 0.01; **P < 0.001; ***P < 0.0001. Scale bar, 50 μm.
MMP3-induced EMT was inhibited by siRNA knockdown of Rac1b (Fig. 2c). Whereas TGFβ does not induce Rac1b in SCp2 cells (data not shown), previous studies have shown that it can activate Rac1 signaling in some cell types [Chiu et al., 2001; Groth et al., 2005]. This prompted us to investigate whether TGFβ might be inducing EMT in mammary cells through an action of Rac1 that does not depend upon altered cell morphology. We assessed the response of cells transfected with dominant negative Rac1 (Rac1N17), plated at low initial seeding density and treated with TGFβ. We found that unlike Rac1b, Rac1N17 did not significantly affect cell spreading (Fig. 4e), did not block activation of the 3TP-luc reporter vector (Fig. 4f) and did not significantly affect activation of EMT markers such as α-smooth muscle actin (Fig. 4g).
Our results had shown that inhibiting cell spreading by plating cells at high density blocked MMP3/Rac1b-induced expression of EMT markers, but did not affect TGFβ-induced expression of EMT markers. However, it is an important consideration that altering cell density changes parameters in addition to cell spreading. In particular, cells cultured at high density form more stable adherens junctions; E-cadherin was found to be a substrate for MMP3 and loss of E-cadherin is a key step in the induction of EMT in a number of cell and tissue types [Thiery, 2002]. To isolate the effects of cell spreading from other parameters, we used a micropatterning approach to restrict the ability of single cells to spread further upon treatment with MMP3 or TGFβ. We cultured single SCp2 cells on micropatterned substrata that contained either 20-μm square or 40-μm square islands of fibronectin surrounded by non-adhesive regions. When cultured on 40-μm islands, treatment with MMP3 or TGFβ induced both additional cell spreading and EMT, assessed as reduced expression of epithelial keratins and increased expression of mesenchymal vimentin (Fig. 5a). However, cells cultured on 20-μm islands were prevented from spreading in response to MMP3 or TGFβ; these cells were refractory to EMT after MMP3 treatment, but were responsive to TGFβ for induction of EMT (Fig. 5a). Quantification of keratin positive epithelial cells showed that the inhibition of MMP3-induced EMT by blocking cell spreading was significant (Fig. 5b). These data show that inhibition of MMP3-induced EMT by limitation of cell spreading on micropatterned substrata is not due to a general inhibition of cellular processes, and demonstrate that changes in cell shape are required for MMP3-induced EMT, but not TGFβ-induced EMT, and additionally demonstrate that MMP3 and TGFβ induce EMT through distinct and separable pathways (Fig. 5c).
Fig. 5.
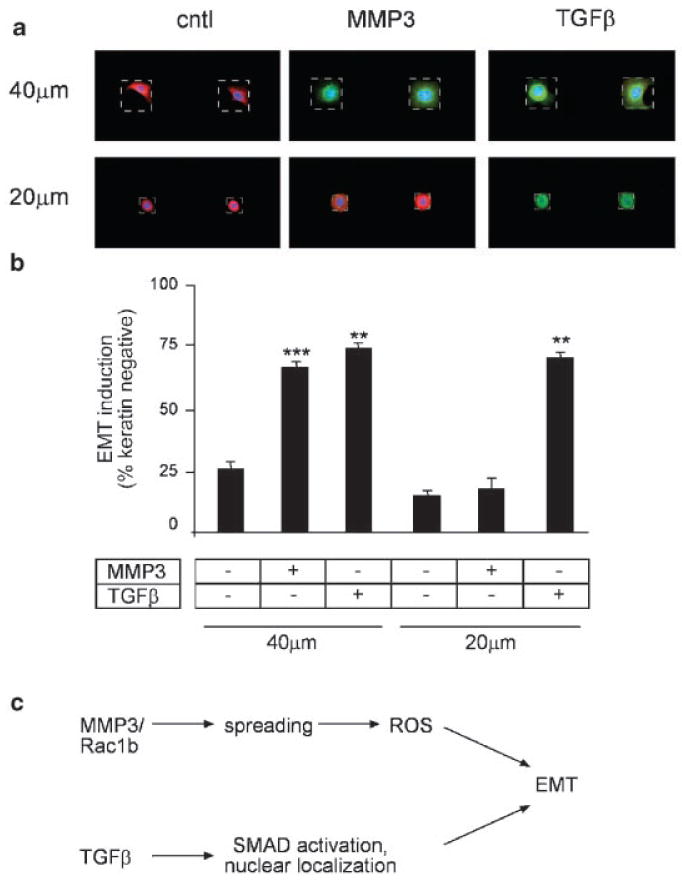
Restricting cell spreading prevents MMP3-induced EMT but not TGFβ-induced EMT. a: Immunofluorescence images of cytokeratins (red) and vimentin (green) in SCp2 cells cultured on small (20 μm) and large (40 μm) squares, and untreated (cntl) or exposed to MMP3 or TGFβ. Stamped islands outlined by white dotted line. b: Graph quantifying loss of cytokeratin expression in MMP3 and TGFβ-treated SCp2 cells on 20- and 40-μm squares. **P < 0.005; ***P < 0.0005. c: Scheme outlining distinct EMT induction pathways for MMP3 and TGFβ: MMP3 stimulates increased expression of Rac1b, which causes cell spreading, which leads to ROS-induced EMT, while TGFβ stimulates activation and nuclear localization of SMADs in a cell spreading-independent EMT induction pathway.
Discussion
In this study, we found that treatment of mouse mammary epithelial cells with MMP3 leads to increased cell spreading in a Rac1b-dependent fashion, and that this cell spreading is required for the induction of EMT by MMP3 or Rac1b. While we observed that TGFβ also causes increased cell spreading against the substratum at lower cell densities, we found that this cell spreading is not necessary for TGFβ-induced EMT: culturing cells at high density or on micropatterned substrata to limit cell spreading did not block TGFβ-induced EMT. These results reveal an important difference between the mechanisms by which these extracellular mediators exert their effects (Fig. 5c). The growing evidence that EMT-related processes are involved in fibrosis and cancer [Kalluri and Neilson, 2003] has prompted a large number of studies designed to elucidate the cytosolic signaling pathways that control EMT. Whereas these studies have used a variety of different models and a number of different inducing agents, the fact that relatively few have examined interactions between the different signaling pathways has complicated the generation of global models of EMT-associated signaling networks. Our results demonstrate that alterations in cell morphology, an often underappreciated aspect of experimental procedures, are critical for the induction of EMT. We found that Rac1b-induced cell spreading was specifically required for MMP3-mediated EMT in SCp2 cells, as knockdown of Rac1b using siRNA oligos that do not target Rac1 blocked cell spreading and induction of EMT. Some studies have shown that morphological alterations are sufficient to induce characteristics of EMT in MCF10A human breast cancer cells and in bovine kidney cells [Ben-Ze'ev, 1984; Maeda et al., 2005; Sarrio et al., 2008]. We suggest that Rac1b may be expressed at higher levels in these cell types, or alternatively that other intrinsic factors that induce cell spreading can substitute for Rac1b to induce EMT. Identifying these factors would provide additional information about the relationship between cell morphology and important events in development and progression of cancer.
Our results also give insight into specific differences between TGFβ signaling pathways and those initiated by MMP3. In the canonical signaling pathway, binding of TGFβ to cell surface receptors stimulates phosphorylation of cytosolic SMAD proteins; phosphorylated SMADs translocate to the nucleus where they cause alterations in gene expression [Shi and Massague, 2003]. This direct connection from the cell surface to the nucleus may circumvent the cell spreading requirement of MMP3/Rac1b-induced EMT. While we have elucidated many intracellular signals required for MMP-3 to induce EMT, the exact cell-surface target(s) of MMP3 that are instrumental in Rac1b production have not been defined conclusively. We showed previously that MMP3 cleaves E-cadherin [Lochter et al., 1997a,b], a critical target of EMT and tumor progression [Thiery and Sleeman, 2006]. Our identification that MMP3 can induce EMT in isolated cells on micropatterned substrata suggests that E-cadherin cleavage is not the cause of EMT, but that it may be necessary to allow cell spreading after breakdown of cell–cell junctions. Since we show also that in high density, MMP-3 induces Rac1b but is not sufficient to induce EMT, additional/alternative targets of MMP3 could play a role.
Previous studies have demonstrated that active Rac1 can stimulate the production and release of mitochondrial superoxide [Kheradmand et al., 1998; Werner and Werb, 2002], and that mitochondrial depolarization correlates with cell spreading [Bereiter-Hahn et al., 1990]. Our results demonstrate that changes in cell shape are required for the activation of mitochondrial ROS-dependent EMT. Since Rac1 is normally localized to the plasma membrane [Michaely et al., 1999], it is possible that alterations in cell spreading facilitate translocation of Rac1b to the mitochondria leading to release of superoxide. Alternatively, Rac1b could be acting indirectly, as the increased cytoskeletal tension associated with cell spreading is known to modulate mesenchymal differentiation [McBeath et al., 2004].
MMP3 also induces EMT, tissue fibrosis, and development of tumors when over-expressed in the mammary glands of transgenic mice [Sternlicht et al., 1999]. Our study could explain in part the relatively late appearance of tumors in these animals–the generation of ROS downstream of MMP3 and Rac1b requires cellular distention. A prerequisite for cellular distention is stiffening of the surrounding tissue in vivo. Increased tissue stiffness is a hallmark of fibrosis [Desmouliere et al., 2005], and has recently been noted to facilitate tumor progression and metastatic invasion [Paszek et al., 2005; Provenzano et al., 2006]. Given that MMP3 was shown to increase collagen deposition in vivo [Thomasset et al., 1998], and induces genomic instability in culture also through induction of ROS, our data would suggest that normal tissue architecture protects against both pathological EMT and genomic instability. Identification of the specific processes by which cell spreading facilitates MMP3-mediated effects may be a useful avenue for identifying therapeutic targets for blocking MMP-mediated fibrosis and malignancy; the results presented here provide an experimental framework to begin these investigations.
Acknowledgments
We thank Connie Chen and Dinah Levy for technical assistance. This work was supported by grants from the Office of Biological and Environmental Research of the Department of Energy (DE-AC03-76SF00098 and a Distinguished Fellow Award to MJB), the National Cancer Institute (CA64786 to MJB and CA57621 to MJB and Zena Werb; CA122086 to DCR, CA128660 to CMN and DCR), and the Breast Cancer Research Program of the Department of Defense (an Innovator Award to MJB; W81XWH-04-1-0582 to CMN). CMN holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.
Grant sponsor: Office of Biological and Environmental Research of the Department of Energy; Grant number: DE-AC03-76SF00098; Grant sponsor: National Cancer Institute; Grant numbers: CA64786, CA57621, CA122086, CA128660.
Abbreviations used
- EMT
epithelial-mesenchymal transition
- MMP
matrix metalloproteinase
- TGF-β
transforming growth factor-β
- NAC
N-acetyl cysteine
- ROS
reactive oxygen species
References
- Ben-Ze'ev A. Differential control of cytokeratins and vimentin synthesis by cell–cell contact and cell spreading in cultured epithelial cells. J Cell Biol. 1984;99:1424–1433. doi: 10.1083/jcb.99.4.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Luck M, Miebach T, Stelzer HK, Voth M. Spreading of trypsinized cells: Cytoskeletal dynamics and energy requirements. J Cell Sci. 1990;96(Pt 1):171–188. doi: 10.1242/jcs.96.1.171. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Farson D, Tung AS. Cell shape and hexose transport in normal and virus-transformed cells in culture. J Supramol Struct. 1977;6:1–12. doi: 10.1002/jss.400060102. [DOI] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Chiu C, Maddock DA, Zhang Q, Souza KP, Townsend AR, Wan Y. TGF-beta-induced p38 activation is mediated by Rac1-regulated generation of reactive oxygen species in cultured human keratinocytes. Int J Mol Med. 2001;8:251–255. [PubMed] [Google Scholar]
- Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–339. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth S, Schulze M, Kalthoff H, Fandrich F, Ungefroren H. Adhesion and Rac1-dependent regulation of biglycan gene expression by transforming growth factor-beta. Evidence for oxidative signaling through NADPH oxidase. J Biol Chem. 2005;280:33190–33199. doi: 10.1074/jbc.M504249200. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- Le Beyec J, Xu R, Lee SY, Nelson CM, Rizki A, Alcaraz J, Bissell MJ. Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp Cell Res. 2007;313:3066–3075. doi: 10.1016/j.yexcr.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997a;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Srebrow A, Sympson CJ, Terracio N, Werb Z, Bissell MJ. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J Biol Chem. 1997b;272:5007–5015. doi: 10.1074/jbc.272.8.5007. [DOI] [PubMed] [Google Scholar]
- Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118:873–887. doi: 10.1242/jcs.01634. [DOI] [PubMed] [Google Scholar]
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- Michaely PA, Mineo C, Ying YS, Anderson RG. Polarized distribution of endogenous Rac1 and RhoA at the cell surface. J Biol Chem. 1999;274:21430–21436. doi: 10.1074/jbc.274.30.21430. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Jean RP, Tan JL, Liu WF, Sniadecki NJ, Spector AA, Chen CS. Emergent patterns of growth controlled by multicellular form and mechanics. Proc Natl Acad Sci USA. 2005;102:11594–11599. doi: 10.1073/pnas.0502575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, Bissell MJ, Ronnov-Jessen L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC. Epithelial-mesenchymal transition. J Cell Sci. 2005;118:4325–4326. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–839. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci USA. 1994;91:12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120:1351–1383. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Stallings-Mann M, Radisky D. Matrix metalloproteinase-induced malignancy in mammary epithelial cells. Cells Tissues Organs. 2007;185:104–110. doi: 10.1159/000101310. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JL, Liu WF, Nelson CM, Raghavan S, Chen CS. Simple approach to micropattern cells on common culture substrates by tuning substrate wettability. Tissue Eng. 2004;10:865–872. doi: 10.1089/1076327041348365. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Thomasset N, Lochter A, Sympson CJ, Lund LR, Williams DR, Behrendtsen O, Werb Z, Bissell MJ. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am J Pathol. 1998;153:457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM, Jordan PW, O'Neill CH. Cell shape controls terminal differentiation of human epidermal keratinocytes. Proc Natl Acad Sci USA. 1988;85:5576–5580. doi: 10.1073/pnas.85.15.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]