

Abstract
NF-κB signaling is known to induce the expression of antiapoptotic and proinflammatory genes in endothelial cells (ECs). We have shown recently that Ca2+ influx through canonical transient receptor potential (TRPC) channels activates NF-κB in ECs. Here we show that Ca2+ influx signal prevents thrombin-induced apoptosis by inducing NF-κB-dependent A20 expression in ECs. Knockdown of TRPC1 expressed in human umbilical vein ECs with small interfering RNA (siRNA) suppressed thrombin-induced Ca2+ influx and NF-κB activation in ECs. Interestingly, we observed that thrombin induced >25% of cell death (apoptosis) in TRPC1-knockdown ECs whereas thrombin had no effect on control or control siRNA-transfected ECs. To understand the basis of EC survival, we performed gene microarray analysis using ECs. Thrombin stimulation increased only a set of NF-κB-regulated genes 3- to 14-fold over basal levels in ECs. Expression of the antiapoptotic gene A20 was the highest among these upregulated genes. Like TRPC1 knockdown, thrombin induced apoptosis in A20-knockdown ECs. To address the importance of Ca2+ influx signal, we measured thrombin-induced A20 expression in control and TRPC1-knockdown ECs. Thrombin-induced p65/RelA binding to A20 promoter-specific NF-κB sequence and A20 protein expression were suppressed in TRPC1-knockdown ECs compared with control ECs. Furthermore, in TRPC1-knockdown ECs, thrombin induced the expression of proapoptotic proteins caspase-3 and BAX. Importantly, thrombin-induced apoptosis in TRPC1-knockdown ECs was prevented by adenovirus-mediated expression of A20. These results suggest that Ca2+ influx via TRPC channels plays a critical role in the mechanism of cell survival signaling through A20 expression in ECs.
Keywords: transient receptor potential canonical channel-1, nuclear factor-κB
thrombin generated at the site of injury plays an important role in the coagulation cascade. Besides its role as a procoagulant protease, thrombin activates a variety of cell types including platelets, endothelial cells (ECs), fibroblasts, and smooth muscle cells (7, 26). Thrombin elicits multiple phenotypic changes in ECs (26). For example, thrombin induces change in cell shape, permeability increase, leukocyte trafficking, and gene expression (32–35, 39). Thrombin mediates these effects by activating G protein-coupled receptors [i.e., protease-activated receptors (PARs)] expressed on the EC surface (7, 12, 13). ECs express PAR family members PAR-1, PAR-2, PAR-3, and PAR-4 (7, 19, 26); however, studies using a gene knockout mouse model suggest that thrombin-induced endothelial activation responses are primarily attributed to thrombin activation of PAR-1 (19, 26, 40).
Nuclear factor-κB (NF-κB) signaling is known to play a critical role in host defense, immune response, cell survival, and vascular inflammation and injury (17, 18, 24). NF-κB is composed of dimers of five proteins (p50, p52, p65/RelA, RelB, c-Rel) that exist in the cytoplasm in inactive forms bound to the inhibitory protein IκB (17, 18). Activation of NF-κB requires the activity of IκB kinase (IKK) complex, containing the kinases IKKα and IKKβ and the regulatory protein IKKγ/NF-κB essential modifier (NEMO) (17, 18). Interaction of IKKγ/NEMO with IKKα and IKKβ is critical for the formation of IKK complex, which in turn leads to recruitment of IκB proteins and stimulation of IKKβ activity (17, 18). A variety of agonist-induced signals activate IKKα and IKKβ, which phosphorylate serines 32 and 36 of IκBα and serines 19 and 23 of IκBβ, respectively (17, 18). Phosphorylation of IκBα and IκBβ leads to their proteolytic degradation, and thus the NF-κB dimer translocates to the nucleus to induce target gene expression (17, 18). NF-κB activation induces within hours the expression of A20 (5, 20), which is a potent endogenous negative regulator of NF-κB activation (6). A20 (also known as TNFAIP3) was first identified as an antiapoptotic protein in human umbilical vein ECs (HUVECs) (28). Studies have shown that A20 overexpression in different cell types prevents TNF-α-induced apoptosis (8, 15, 16, 23). Lee et al. (21) first generated A20-deficient mice and showed that they were born at normal Mendelian ratios but died shortly after birth because of massive multiorgan inflammation. Furthermore, they showed that loss of A20 expression increased the lethality of TNF-α and lipopolysaccharide because of excessive activation of NF-κB (2, 21). Further studies have shown that targeted cardiac overexpression of A20 improved the clinical outcome of experimental myocardial infarction in mice by suppressing inflammation (22). These findings collectively suggest that A20 signaling plays a critical role in the signaling mechanism of cell survival.
Thrombin concentration is increased to abnormal levels at sites of vascular injury, in the vascular sites of thrombus, in patients with coronary syndromes, and in sepsis (14, 26). Thrombin-induced signaling prevents cell death in myoblasts, neurons, and astrocytes (26). Chalmers et al. (3) have shown that thrombin activation of PAR-1 in CC139 fibroblasts prevents serum withdrawal-induced apoptosis by inducing the expression of Bcl-2-interacting mediator of cell death (Bim). Another study recently demonstrated that the PAR-1-selective antagonist SCH79797 inhibited cell proliferation and induced apoptosis in NIH 3T3, A375, and HEK-293 cells (11). However, the signaling mechanism by which thrombin prevents EC death is poorly understood.
A rise in intracellular Ca2+ concentration ([Ca2+]i) was found to signal the activation of NF-κB (9, 10). PAR-1 activation in ECs mediates increase in [Ca2+]i by activating the Gq/11-phospholipase C pathway that results in depletion of endoplasmic reticulum (ER) Ca2+ stores and the subsequent store depletion-activated Ca2+ entry across the plasma membrane (37). This accounts for the sustained rise in [Ca2+]i required for the increase in endothelial permeability (37). We have shown (30, 38) that the members of the transient receptor potential canonical (TRPC) family of channels are essential for Ca2+ entry induced by PAR-1 agonist in ECs. The members of the TRPC family include seven isoforms (TRPC1 to 7). Recent studies have shown that TRPC1 forms a complex with STIM1 and Orai1 and is involved in store-operated Ca2+ entry (4, 27). Our studies have shown (30, 31) that human ECs predominantly express TRPC1 and contribute to thrombin-induced Ca2+ entry. Mouse lung ECs predominantly express TRPC4, which contributes to thrombin-induced Ca2+ entry (1, 38). Furthermore, we have demonstrated (1) that Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-κB activation in ECs through AMP-activated protein kinase and protein kinase Cδ. In this study we investigated whether NF-κB activation induced by Ca2+ entry via TRPC channels is also responsible for A20 expression and thus prevents thrombin-induced EC death. Our results suggest that Ca2+ influx-dependent A20 expression is critical for EC survival.
MATERIALS AND METHODS
Materials.
Human α-thrombin was obtained from Enzyme Research Laboratories (South Bend, IN). Endothelial growth medium-2 (EGM-2) was purchased from Cambrex BioScience (Walkersville, MD). Fetal bovine serum was from Hyclone (Logan, UT). Hanks’ balanced salt solution (HBSS), l-glutamine, trypsin, Vybrant apoptosis staining kit, TRIzol reagent, and Taq DNA polymerase were from Invitrogen. NF-κB oligonucleotides and polyclonal anti-phospho-ERK1/2 and anti-ERK1/2 antibodies were from Promega (Madison, WI). PCR primers and A20 promoter-specific NF-κB oligonucleotides were custom synthesized by IDT (Coralville, IA). [γ-32P]ATP (specific activity 6,000 Ci/mmol) was obtained from ICN Biomedical (Irvine, CA). 2-Aminoethoxydiphenyl borate (2-APB) and A20 monoclonal antibody (MAb) were purchased from Calbiochem-Novabiochem (San Diego, CA). TRPC1 small interfering RNA (siRNA), scrambled siRNA (Sc-siRNA), A20 siRNA, and anti-p65, anti-p50, and anti-TRPC1 polyclonal antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti-Bax and anti-caspase-3 antibodies were from Millipore (Billerica, MA). p65-siRNA was from Dharmacon (Denver, CO). PAR-1 agonist (TFLLRNPNDK) peptide was synthesized in-house as the COOH-terminal amide (Biotechnology Center, University of Illinois at Urbana-Champaign).
Cell culture.
Primary HUVECs obtained from Cambrex BioScience were grown in EGM-2 supplemented with 10% FBS as described previously (30, 31). ECs between passages 4 and 7 were used for all described experiments.
Cytosolic Ca2+ measurement.
Thrombin-induced increase in [Ca2+]i was measured with the Ca2+-sensitive fluorescent dye fura-2 AM (30, 31).
siRNA transfection.
ECs grown to ∼70% confluence were transfected with or without indicated concentrations of TRPC1-siRNA, A20-siRNA, or Sc-siRNA with Santa Cruz Biotechnology transfection reagents. Forty-eight hours after transfection, cells were used for the experiments.
Immunoblotting.
ECs grown to confluence were serum starved for 2 h in 1% FBS. After inhibitor or agonist treatment, cells were lysed in cell lysis buffer. Equal amounts of protein were resolved by SDS-PAGE and subsequently transferred to polyvinylidene difluoride (PVDF) membrane. Membranes were incubated in blocking buffer (5% nonfat milk in 10 mM Tris·HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween 20) for 60 min at room temperature. Membranes were then incubated with the indicated antibody overnight at 4°C. After two washes the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 60 min at room temperature. Protein bands were detected by enhanced chemiluminescence.
Microarray analysis.
ECs were incubated in medium containing 1% FBS for 2 h and then treated with or without thrombin (50 nM) for 4 h at 37°C. Total RNA was isolated with TRIzol (Invitrogen). Total RNA isolated was used for preparation of cDNA. cDNA hybridization with human NF-κB signaling Pathway Microarray EHS-025 was carried out by SuperArray Bioscience (Frederick, MD). Each array experiment was performed in duplicate. Microarray results were quantified densitometrically by SuperArray Bioscience. The gene induction fold increase was calculated over basal with housekeeping gene GAPDH expression levels.
Nuclear protein extraction.
ECs grown to confluence were incubated with 1% FBS-containing medium for 2 h before exposure to different agonists for various time intervals. Nuclear extracts were prepared from ECs by the method described previously (1).
Electrophoretic mobility shift.
Oligonucleotide containing the NF-κB consensus sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and A20 promoter-specific NF-κB oligonucleotide (−41 to −57; 5′-CCGTGGAAAT CCCCGGG-3′) were labeled with [γ-32P]ATP with the use of T4 polynucleotide kinase for 20 min at 37°C in the presence of 50 μg of poly(dI-dC) and 10 mM Tris·HCl buffer, pH 7.5, containing 50 mM NaCl, 0.5 mM EDTA, 0.5 mM dithiothreitol, 4% (wt/vol) glycerol, and 1 mM MgCl2. The nuclear extracts (15 μg of protein) were incubated with the radiolabeled NF-κB oligonucleotide (80,000 cpm/reaction) and subjected to electrophoresis on a 4% native gel, dried on Whatman paper, and then exposed to autoradiography. For supershift assays, nuclear extracts were incubated with indicated antibodies for 30 min subsequent to the addition of radiolabeled oligonucleotide probe.
Reverse transcription-PCR.
ECs grown to confluence were incubated in 1% FBS-containing medium for 2 h. After this period, the cells were treated with or without thrombin (50 nM) in 1% FBS-containing medium for various time intervals. After treatment, total RNA was isolated with TRIzol reagent. Reverse transcription (RT) was performed with oligo(dT) primers and SuperScript reverse transcriptase (Invitrogen) according to the manufacturer's recommendation. A20 transcript expression was amplified with the following primer sets: human A20 forward 5′-GAGGCTGCTGAGACTGAACAT-3′ and reverse 5′-TACTCCCTGTGGTCACGAACT-3′; the PCR product was 459 bp for A20. The reaction conditions for A20 included an initial 95°C for 5 min and then 30 cycles of 95°C for 30 s, 54°C for 30 s, and 68°C for 45 s. For GAPDH the primers used were forward 5′-TATCGTGGAAGGACTCATGACCC0-3′ and reverse 5′-TACATGGCAACTGTGAGGGG-3′. Reaction conditions were 30 cycles of 94°C for 45 s, 60°C for 45 s, and 72°C for 2 min. PCR product were resolved by 1.5% agarose gel and identified by ethidium bromide staining. Normalization of A20 expression was achieved by comparing the expression of GAPDH for the matching sample.
Apoptotic assay.
Chromatin condensation was detected by nucleus staining of ECs with the Vybrant apoptosis assay kit from Invitrogen. After exposure to thrombin for different time intervals, culture medium was collected and centrifuged at 700 g to pellet the cells. The cell pellet was suspended and mixed with the cells harvested by trypsinization. The pooled ECs were washed with ice-cold phosphate-buffered saline (PBS) and then stained with Hoechst 33342 (5 μg) for 5 min at 4°C. Apoptosis was quantified by flow cytometric analysis (36).
Preparation of adenoviral expression constructs.
A 2,998-bp human A20 coding sequence (GenBank accession no. NM_006290.2) in pBluescriptR was released and subcloned into adenoviral expression vector, and amplification of recombinant A20 adenovirus was carried out by Vector Biolabs (Philadelphia, PA).
Infection of ECs with adenovirus.
ECs grown to ∼80 confluence were infected with adeno-null virus (Ad-null) or adeno-A20 virus [100 plaque-forming units (pfu) per cell]. At 12 h after infection cells were washed with serum-free medium, followed by incubation of cells with 1% FBS-containing medium for 2 h before treatment with or without thrombin (50 nM) at different time intervals.
Statistical analysis.
Statistical comparisons were made with two-tailed Student's t-test. Experimental values are reported as means ± SE. Differences in mean values were considered significant at P < 0.05.
RESULTS
TRPC1 knockdown prevents thrombin-induced Ca2+ influx and NF-κB activation and induces apoptosis in ECs.
To study the importance of TRPC1 expression in signaling EC survival, we silenced TRPC1 expression by transfecting ECs with siRNA specific to TRPC1 and measured thrombin-induced Ca2+ influx and NF-κB activation. We observed that in TRPC1 siRNA-transfected ECs TRPC1 protein expression was suppressed (Fig. 1A), whereas in control or Sc-siRNA transfected cells TRPC1 expression was not altered. Thrombin-induced Ca2+ influx was markedly reduced in TRPC1-knockdown ECs compared with control ECs (Fig. 1B). These results are in agreement with our previous reports (1, 30) indicating that TRPC1 expression is required for Ca2+ influx in response to thrombin in ECs. To address the effect of TRPC1 knockdown in ECs, we measured thrombin-induced NF-κB activation by electrophoretic mobility shift assay (EMSA). We found that TRPC1 knockdown prevented thrombin-induced NF-κB-DNA complex formation in ECs (Fig. 1C) whereas TRPC1 knockdown had no significant effect on TNF-α-induced NF-κB activation in ECs (Fig. 1C), suggesting that Ca2+ influx signal is required for thrombin-induced, but not for TNF-α-induced, NF-κB activation in ECs. These results further suggest that stimulus-specific TRPC1 activation signals NF-κB activation in ECs.
Fig. 1.
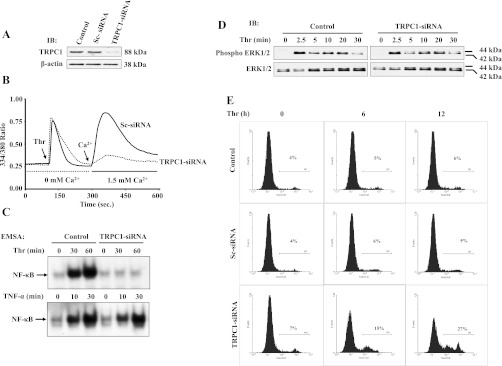
Canonical transient receptor potential channel 1 (TRPC1) knockdown prevents thrombin-induced Ca2+ influx and nuclear factor-κB (NF-κB) activation and induces apoptosis in endothelial cells (ECs). A: ECs were transfected with either 100 nM scrambled small interfering RNA (Sc-siRNA) or TRPC1 siRNA. At 48 h after transfection, cells were lysed and immunoblotted (IB) with anti-TRPC1 polyclonal Ab (top). The membrane was stripped and blotted with anti-β-actin (bottom). B: control ECs and ECs transfected with 100 nM Sc-siRNA or TRPC1-siRNA were used to measure thrombin (Thr)-induced store Ca2+ release and Ca2+ release-activated Ca2+ influx. The results were compared with control cells (not transfected). The experiment was repeated 3 times with similar results. C: control ECs and ECs transfected with TRPC1-siRNA (100 nM) were exposed to thrombin (50 nM) or TNF-α (100 U) for the indicated time periods, and then electrophoretic mobility shift assay (EMSA) was performed. The experiment was repeated 3 times with similar results. D: ECs transfected with either 100 nM Sc-siRNA or TRPC1-siRNA were treated with or without thrombin (50 nM) for various time intervals. The whole cell lysate was immunoblotted with anti-phospho-ERK1/2 and anti-ERK1/2 antibody. E: ECs were transfected with either 100 nM Sc-siRNA or TRPC1-siRNA. At 48 h after transfection, cells were treated with thrombin for 0, 6, and 12 h. After thrombin treatment, cells were collected and stained with 5 μg of Hoechst 33342 and subjected to fluorescence-activated cell sorting (FACS) analysis to determine apoptosis in ECs. The experiment was repeated 3 times. Results from representative experiments are shown.
PAR-1 is functionally coupled to multiple heterotrimeric G proteins (6). PAR-1 activation in ECs induces increase in cytosolic Ca2+ ([Ca2+]i) by activating the Gq/11-phospholipase C pathway that results in depletion of ER Ca2+ stores and the subsequent store depletion-dependent Ca2+ entry in ECs (37, 38). To study whether TRPC1 knockdown had any influence on PAR-1 signaling, we measured thrombin-induced ERK1/2 phosphorylation, which is dependent on PAR-1 functional coupling to Gi/o (6, 12). We observed that TRPC1 knockdown did not alter the thrombin-induced ERK1/2 phosphorylation in ECs (Fig. 1D), suggesting that suppression of TRPC1 expression does not alter PAR-1 activation in ECs.
To address whether Ca2+ influx-dependent NF-κB activation is involved in the mechanism of cell survival, we determined apoptosis in ECs. In this experiment, 48 h after transfection with Sc-siRNA or TRPC1-siRNA ECs were incubated for 2 h with 1% FBS-containing medium and then exposed to thrombin for up to 12 h. After this treatment, ECs were used for fluorescence-activated cell sorting (FACS) analysis to measure apoptosis (see details in materials and methods). We observed no significant apoptosis in control ECs and ECs transfected with Sc-siRNA (Fig. 1E); however, we observed apoptosis in ∼30% of ECs transfected with TRPC1-siRNA, indicating that TRPC1 expression prevents thrombin-induced apoptosis in ECs.
Thrombin induces the expression of only a set of NF-κB-regulated genes in ECs.
To identify NF-κB signaling molecule expression modulated by thrombin stimulation in ECs, we performed NF-κB signaling pathway microarray analysis using ECs stimulated with and without thrombin (see details in materials and methods). The cDNA library used to make the microarray consists of 120 different NF-κB signaling pathway molecules. Of the total 120 NF-κB signaling pathway genes analyzed, thrombin stimulation increased the expression levels of at least 12 genes more than threefold over basal (Fig. 2A). Among these genes, the induction of the antiapoptotic gene A20 was 14-fold over basal (Fig. 2A). To validate the microarray results, we performed semiquantitative RT-PCR and immunoblot analysis to determine A20 expression in response to thrombin in ECs. In this experiment, we exposed ECs to thrombin or the PAR-1 agonist peptide (TFLLRNPNDK) for different time intervals. After this treatment, total RNA isolated was used for RT-PCR to determine A20 transcript expression. We observed that activation of PAR-1 with either thrombin or PAR-1 agonist peptide markedly increased A20 transcript expression in ECs (Fig. 2B). We also observed the induction of A20 protein expression in ECs after PAR-1 activation (Fig. 2C). These results suggest that PAR-1 signaling is essential for the expression of A20 in ECs. Next we addressed whether thrombin-induced A20 expression is necessary for EC survival; we knocked down A20 expression by transfecting ECs with A20-specific siRNA and measured thrombin-induced apoptosis in ECs. In A20-siRNA-transfected ECs, A20 protein expression was prevented; however, A20 protein expression in control or Sc-siRNA-transfected ECs was not altered (Fig. 2D). Interestingly, we observed apoptosis in A20-siRNA-transfected ECs (>20% cell death), and thrombin treatment further enhanced apoptosis (>30% cell death) in A20-siRNA-transfected ECs compared with control or Sc-siRNA-transfected ECs (Fig. 2E). These results suggest that thrombin-mediated cytoprotective effect in ECs may be regulated by TRPC1 signaling in ECs.
Fig. 2.

Protease-activated receptor (PAR)-1 signaling-induced A20 expression prevents apoptosis in ECs. A: ECs grown to confluence were challenged with thrombin (50 nM) for 4 h. After thrombin treatment, total RNA isolated was used for microarray analysis. The experiment was repeated twice with duplicates. Results from representative experiments are shown (top). The gene induction fold increase was calculated over basal with housekeeping gene GAPDH expression levels (bottom). B: ECs were treated with or without thrombin (50 nM) or PAR-1 agonist peptide (40 μM) for various time intervals. Total RNA was isolated and RT-PCR was performed to determine A20 and GAPDH expressions. The experiment was repeated 4 times, and results from a representative experiment are shown (left). RT-PCR gel images were quantified, and A20 mRNA induction fold increase over basal was calculated by measuring the ratio of A20 to GAPDH (right). *P < 0.05, significantly different from control. C: after thrombin (50 nM) or PAR-1 agonist peptide (40 μM) treatment, cell lysates were immunoblotted with anti-A20 MAb. The membrane was stripped and reprobed with anti-β-actin Ab. The experiment was repeated 4 times, and results from representative experiment are shown (left). Immunoblots were quantified by densitometry, and A20 protein induction fold increase over basal was calculated by measuring the ratio of A20 to β-actin (right). *P < 0.05, significantly different from control. D: ECs were transfected with either 100 nM Sc-siRNA or A20-siRNA. At 48 h after transfection cells were immunoblotted with anti-A20 MAb. E: ECs were transfected with either 100 nM Sc-siRNA or A20-siRNA as described above. At 48 h after transfection, cells were treated with or without thrombin (50 nM) for 0 and 12 h. After thrombin treatment, cells were used for FACS analysis to determine apoptosis. The experiment was repeated 3 times, and results shown are from a representative experiment.
Pharmacological inhibition of Ca2+ influx or p65/RelA knockdown prevents thrombin-induced A20 expression in endothelial cells.
Inositol 1,4,5-trisphosphate (IP3) receptor antagonist 2-APB is known to inhibit stored Ca2+ release as well as Ca2+ entry (29). In earlier studies (1) we showed that thrombin-induced Ca2+ influx was inhibited by 2-APB in ECs. In light of this observation, we examined the effect of 2-APB on thrombin-induced NF-κB-DNA binding activity by EMSA using A20 promoter-specific NF-κB consensus sequence (see details in materials and methods). Thrombin exposure increased NF-κB-DNA binding activity in control ECs; however, the thrombin effect was abolished in 2-APB-treated ECs (Fig. 3, A and B). A supershift assay was carried out utilizing NF-κB-specific antibodies to identify the NF-κB proteins translocated to the nucleus. The data showed a shift in the presence of anti-p65 antibody in thrombin-stimulated ECs (Fig. 3B). We also determined the effect of 2-APB on thrombin-induced A20 mRNA and protein expression. We observed that 2-APB pretreatment markedly reduced thrombin-induced A20 transcript and protein expression in ECs (Fig. 3, C and D). Next, we used siRNA to knockdown the endogenous p65/RelA expression and determined thrombin-induced A20 expression. We observed that in p65-siRNA-transfected ECs p65/RelA protein expression was suppressed (Fig. 3E), whereas in control or Sc-siRNA-transfected cells p65/RelA expression was not altered. Furthermore, we observed that thrombin failed to induce A20 protein expression in p65/RelA-knockdown ECs but not in control or Sc-siRNA-transfected ECs (Fig. 3F). These results indicate that Ca2+ signaling-dependent p65/RelA activation is essential for thrombin-induced A20 expression in ECs.
Fig. 3.
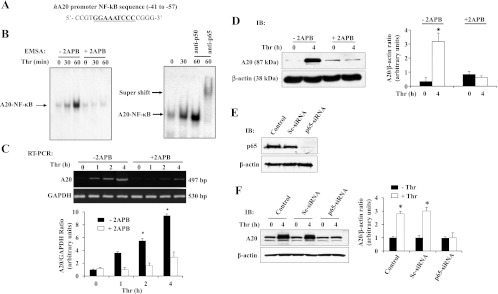
Pharmacological inhibition of Ca2+ influx or knockdown of p65/RelA prevents thrombin-induced A20 expression in ECs. A: human (h)A20 promoter-specific NF-κB oligonucleotide probe used for EMSA. B: ECs grown to confluence were incubated in serum-free medium for 2 h. After this incubation period, cells were exposed to 2-aminoethoxydiphenyl borate (2-APB; 75 μM) for 30 min and then treated with thrombin (50 nM) for up to 60 min. After thrombin treatment, the nuclear extracts prepared were used for EMSA utilizing hA20 promoter-specific NF-κB probe (left). Nuclear extracts prepared from ECs exposed to thrombin for 60 min were incubated with either anti-p65 or anti-p50 antibodies for 30 min before addition of labeled probe (right). The experiment was repeated 3 times, and results shown are from a representative experiment. C: ECs were pretreated with 2-APB (75 μM) for 30 min and then challenged with thrombin (50 nM) up to 4 h. After thrombin treatment, total RNA was isolated and RT-PCR was performed to determine A20 and GAPDH transcript expression levels. The experiment was repeated 4 times, and results from a representative experiment are shown (top). RT-PCR gel images were quantified, and the A20 mRNA induction fold increase over basal was calculated by measuring the ratio of A20 to GAPDH (bottom). *P < 0.05, significantly different from control. D: ECs pretreated with 2-APB as described in C were challenged with thrombin (50 nM) for up to 4 h. After thrombin treatment, cell lysates were immunoblotted with anti-A20 MAb. The membrane was stripped and reprobed with anti-β-actin Ab. A representative blot is shown at left. The blots were quantified as described in Fig. 2C. Results are means ± SE from 4 separate experiments shown (right). *P < 0.05, significantly different from control. E: ECs were transfected with either 50 nM Sc-siRNA or p65-siRNA. At 48 h after transfection, cell lysates were immunoblotted with anti-p65 Ab (top). The membrane was stripped and reprobed with anti-β-actin Ab (bottom). F: ECs transfected with either 50 nM Sc-siRNA or p65-siRNA were challenged with thrombin (50 nM) for up to 4 h, and then cell lysates were immunoblotted with anti-A20 MAb or anti-β-actin Ab. A representative blot is shown at left. The results from 4 separate experiments were quantified by densitometry, and the A20 protein induction fold increase over basal was calculated by measuring the ratio of A20 to β-actin (right). *P < 0.05, significantly different from control.
TRPC1 expression in ECs is required for thrombin-induced A20 expression.
We showed previously (1) that TRPC1 knockdown prevents thrombin-induced Ca2+ influx and NF-κB activation in ECs (Fig. 1). To address the physiological relevance of TRPC1 expression in ECs, we measured thrombin-induced NF-κB binding to A20 promoter-specific NF-κB consensus sequence in TRPC1-knockdown and control ECs. We observed that thrombin-induced NF-κB binding to A20 promoter-specific NF-κB consensus sequence was abolished in TRPC1-knockdown ECs compared with Sc-siRNA or control ECs (Fig. 4A, top). However, TRPC1 knockdown had no effect on TNF-α-induced NF-κB binding to A20 promoter-specific NF-κB consensus sequence (Fig. 4A, bottom). We also determined A20 transcript and protein expression in TRPC1-knockdown ECs. Thrombin-induced expression of A20 transcript and protein was significantly reduced in TRPC1-knockdown cells compared with control cells (Fig. 4, B and C). To address the specificity of Ca2+ signaling, we determined TNF-α-induced A20 transcript and protein expression in control and TRPC1-knockdown ECs. We observed that TNF-α-induced A20 transcript (Fig. 4D) and protein (not shown) expression were not altered in TRPC-1-knockdown ECs. To further confirm the role of TRPC-1 in regulating the antiapoptotic signaling in ECs, we measured the expression levels of the proapoptotic proteins BAX and caspase-3 in control and TRPC1-knockdown ECs. Surprisingly, in TRPC1-knockdown ECs, thrombin stimulation increased the expression of BAX and caspase-3 compared with control ECs, suggesting that TRPC1 signaling-mediated A20 expression is required for EC survival.
Fig. 4.

TRPC1 expression is required for thrombin-induced A20 expression in ECs. A: control ECs and ECs transfected with 100 nM Sc-siRNA or TRPC1-siRNA were exposed to thrombin (50 nM) or TNF-α (100 U) for the indicated time periods. After this treatment, EMSA was performed with A20 promoter-specific NF-κB probe as shown in Fig. 3B. The experiment was repeated 3 times with similar results, and results from representative experiments are shown. B: ECs were transfected with either 100 nM Sc-siRNA or TRPC1-siRNA. At 48 h after transfection, cells were exposed to thrombin (50 nM) for the indicated time periods. Total RNA was isolated, and RT-PCR was performed to determine A20 and GAPDH transcript expression levels. The experiment was repeated 3 times, and results from representative experiments are shown. C: control ECs and ECs transfected with 100 nM Sc-siRNA or TRPC1 siRNA were exposed to thrombin as described above. After thrombin exposure, cells were lysed and immunoblotted with anti-A20 MAb (top). The membrane was stripped and probed with β-actin Ab (bottom). D: control ECs and ECs transfected with 100 nM Sc-siRNA or TRPC1-siRNA were exposed to TNF-α (100 U) for 0 and 4 h. After TNF-α exposure, cells were used to measure A20 mRNA expression by RT-PCR. The experiment was repeated 3 times, and results from representative experiments are shown. E: control ECs and ECs transfected with TRPC1 siRNA as described above were treated with thrombin (50 nM) for 0, 6, and 12 h. After thrombin treatment, cells were lysed and immunoblotted with anti-BAX and anti-caspase-3 antibodies. The experiment was repeated 3 times, and results from representative experiments are shown.
Adenovirus-mediated A20 overexpression rescues TRPC1-knockdown phenotype in ECs.
Since A20 was identified as an antiapoptotic factor in ECs (28), we determined the effect of adenovirus (Ad)-mediated A20 expression on thrombin-induced apoptosis in TRPC1-knockdown ECs. In this experiment, we first transfected ECs with TRPC1 siRNA for 48 h and then infected ECs with Ad-null virus or Ad-A20 virus (100 pfu/cell) for 12 h. After infection, the ECs were challenged with or without thrombin for 12 h and then used for FACS analysis to measure apoptosis. Figure 5A shows the A20 expression levels in ECs after infection with recombinant Ad-A20 and Ad-null virus. We observed that thrombin induced cell death in TRPC1-knockdown ECs infected with Ad-null virus (Fig. 5B) but failed to induce apoptosis in TRPC1-knockdown ECs infected with Ad-A20 virus (Fig. 5B).
Fig. 5.

Adenovirus (Ad)-mediated A20 expression rescues TRPC1 knockdown phenotype in ECs. A: ECs were transfected with or without 100 nM TRPC1 siRNA as described in Fig. 4. At 48 h after transfection, ECs were infected with either Ad-null or Ad-A20 virus (100 pfu/cell) for 12 h, and then ECs were challenged with thrombin (50 nM) for 0 and 4 h. After thrombin treatment, cell lysates were immunoblotted with anti-A20 MAb. The experiment was repeated 3 times, and results from representative experiments are shown. B: ECs were transfected with or without 100 nM TRPC1-siRNA. At 48 h after transfection, ECs were infected with either Ad-null or Ad-A20 virus (100 pfu/cell) for 24 h, and then ECs were challenged with thrombin (50 nM) for 0 and 12 h. After thrombin treatment, cells were used for FACS analysis. Percentages of apoptotic cells are indicated on the histograms. The experiment was repeated 3 times, and results from representative experiments are shown.
DISCUSSION
Thrombin is known to provide cytoprotection in various cell types including ECs; however, the signaling mechanisms by which thrombin elicits this effect are poorly understood. Inducible transcription factor NF-κB activation is known to induce both anti-inflammatory and proinflammatory genes in ECs (25, 26, 32, 33). Previous studies have shown that NF-κB signaling-mediated expression of the ubiquitin-editing enzyme A20 prevents apoptosis in ECs (6, 28). We have shown in recent studies (1) that Ca2+ influx through TRPC channels is essential for thrombin-induced NF-κB activation in ECs. Since A20 is a NF-κB-regulated gene, we addressed whether Ca2+ influx through TRPC channels is essential for A20 expression and thus prevents thrombin-induced apoptosis in ECs. Here we provide evidence for the first time that thrombin-induced Ca2+ influx through TRPC channels is critical for p65/RelA activation-mediated A20 expression to prevent apoptosis in ECs.
We have shown (1, 30) that TRPC isoform TRPC1 is essential for thrombin-induced Ca2+ entry in human vascular ECs. In this study, we knocked down TRPC1 with siRNA and studied thrombin-induced EC survival. We observed that TRPC1 knockdown prevented both the thrombin-induced Ca2+ entry and NF-κB activation in ECs. Interestingly, we observed thrombin-induced apoptosis in TRPC1-knockdown ECs, indicating that TRPC1 expression in ECs contributes to cell survival signaling via NF-κB activation.
A20 has been most intensively studied as the key negative regulator of NF-κB signaling (6) and also an antiapoptotic protein (28). In this study, we investigated whether thrombin-induced A20 expression plays an important role in EC survival. To address this question, we first performed gene microarray analysis using ECs challenged with or without thrombin to determine the expression profile of NF-κB signaling pathway genes. Thrombin challenge increased only a set of genes more than threefold over basal in ECs. Interestingly, we observed that thrombin-induced expression of the antiapoptotic gene A20 was the highest (>13-fold over basal) among the upregulated genes. We also observed that IL-8 expression was increased >10-fold over basal in response to thrombin. To validate the microarray results, we measured PAR-1 signaling-induced A20 expression in ECs. We observed that activation of PAR-1 with either thrombin or PAR-1 agonist peptide (TFLLRNPNDK) markedly increased the expression of A20 mRNA as well as A20 protein. Next, to determine whether A20 expression contributes to the cytoprotective effect in ECs, we downregulated A20 expression by transfecting with A20-specific siRNA and measured thrombin-induced apoptosis in ECs. Results showed that thrombin challenge induced cell death in A20-knockdown ECs but not in control or in Sc-siRNA-transfected cells. These results further support the notion that A20 expression may be the critical factor that contributes to the cytoprotective effect in ECs. Moreover, this observation is in agreement with previous reports indicating that A20 signaling prevents apoptosis in ECs (5, 8, 23).
To address the physiological relevance of Ca2+ influx in the signaling mechanism of A20 expression, we first used a pharmacological approach to inhibit Ca2+ influx in ECs and measured thrombin-induced p65/RelA-binding A20 promoter-specific NF-κB sequence and A20 transcript and protein expression in ECs. We observed that prevention of Ca2+ influx in ECs markedly reduced thrombin-induced p65/RelA binding to A20 promoter-specific NF-κB sequence and A20 mRNA and protein expression in ECs. In addition, we observed that thrombin failed to induce A20 expression in p65/RelA-knockdown ECs, suggesting that Ca2+ influx induces A20 expression signaling via p65/RelA activation in ECs. In this study, we also addressed the specific role of TRPC channels in mediating the Ca2+ influx and thereby inducing A20 expression in ECs. In this experiment, we suppressed TRPC1 expression in ECs by transfecting ECs with TRPC1 siRNA. In TRPC1-knockdown ECs, thrombin-induced NF-κB binding to A20-NF-κB sequence was abolished. Furthermore, thrombin-induced A20 transcript and protein expression were suppressed in TRPC1-knockdown ECs. Interestingly, TRPC1 knockdown had no significant effect on TNF-α-induced NF-κB activation and subsequent A20 expression, which differentiates TNF-α induced signaling of NF-κB activation from that of thrombin in ECs.
Studies have shown that A20 overexpression prevents CD40- and TNF-α-mediated apoptosis in ECs (23, 28). To examine the role of TRPC1-mediated A20 expression in preventing thrombin-induced apoptosis in ECs, we ectopically expressed A20 by infecting TRPC1-knockdown ECs with adenovirus encoding A20. In this experiment, we observed that adenovirus-mediated A20 expression rescued the apoptosis in TRPC1-knockdown ECs. These results collectively support the concept that Ca2+ influx through TRPC channels provides cytoprotective effect signaling through expression of the antiapoptotic protein A20 in ECs.
A20 signaling is also known to prevent the induction of proapoptotic protein in ECs (6, 8). To examine whether TRPC1-mediated A20 expression is required to prevent proapoptotic protein expression, we knocked down TRPC1 expression in ECs and determined the effect of thrombin on the induction of proapoptotic proteins BAX and caspase-3 expression in ECs. We observed that thrombin stimulation markedly increased the expression of BAX and caspase-3 in TRPC1-knockdown ECs compared with control ECs. These results suggest that TRPC1 signaling is required for suppressing proapoptotic protein expression in ECs. In this study, we have shown that thrombin-induced Ca2+ influx through TRPC1 signals p65/ReA activation-mediated A20 expression to prevent apoptosis in ECs. Thus our results suggest that TRPC channel expression in ECs is potentially important for maintaining vascular integrity.
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants GM-058531 and P01-HL-077806
REFERENCES
- 1.Bair AM, Thippegowda PB, Freichel M, Cheng N, Ye RD, Vogel SM, Yu Y, Flockerzi V, Malik AB, Tiruppathi C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J Biol Chem 284: 563–574, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Sui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 10: 1052–1060, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Chalmers CJ, Balmanno K, Hadfield K, Ley R, Cook SJ. Thrombin inhibits Bim (Bcl-2-interacting mediator of cell death) expression and prevents serum-withdrawal-induced apoptosis via protease-activated receptor 1. Biochem J 375: 99–109, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem 283: 12935–129340, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem 271: 18068–18073, 1996 [DOI] [PubMed] [Google Scholar]
- 6.Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem 284: 8217–8221, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coughlin SR. Thrombin signaling and protease activated receptor. Nature 40: 258–264, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, Mahiou J, Sun DW, Mottley C, Grey ST, Ferran C. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood 104: 2376–2384, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386: 855–858, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933–936, 1998 [DOI] [PubMed] [Google Scholar]
- 11.Di Serio C, Pellerito S, Duarte M, Massi D, Naldini A, Cirino G, Prudovsky I, Santucci M, Geppetti P, Marchionni N, Masotti G, Tarantini F. Protease-activated receptor 1-selective antagonist SCH79797 inhibits cell proliferation and induces apoptosis by a protease-activated receptor 1-independent mechanism. Basic Clin Pharmacol Toxicol 101: 63–69, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Ellis CA, Malik AB, Gilchrist A, Hamm H, Sandoval R, Voyno-Yasenetskaya T, Tiruppathi C. Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J Biol Chem 274: 13718–13727, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Ellis CA, Tiruppathi C, Sandoval R, Niles WD, Malik AB. Time course of recovery of endothelial cell surface thrombin receptor (PAR-1) expression. Am J Physiol Cell Physiol 276: C38–C45, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol 131: 417–430, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C. A20 inhibits cytokine-induced apoptosis and nuclear factor kappaB-dependent gene activation in islets. J Exp Med 190: 1135–1146, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grey ST, Lock J, Bach FH, Ferran C. Adenovirus-mediated gene transfer of A20 in murine islets inhibits Fas-induced apoptosis. Transplant Proc 33: 577–588, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev 18: 2195–2224, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Hoffman A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev 210: 171–186, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Kataoka H, Hamilton JR, McKemy DD, Camerer E, Zheng YW, Cheng A, Griffin C, Coughlin SR. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood 102: 3224–3231, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor α-inducible zinc finger protein, A20, is mediated by κB elements. J Biol Chem 267: 17971–17976, 1992 [PubMed] [Google Scholar]
- 21.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289: 2350–2354, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, Yang Q, Huang Y, Wei YS, Liu PP, Liu DP, Liang CC. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation 115: 1885–1894, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Longo CR, Arvelo MB, Patel VI, Daniel S, Mahiou J, Grey ST, Ferran C. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation 108: 1113–1118, 2003 [DOI] [PubMed] [Google Scholar]
- 24.May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 289: 1550–1554, 2000 [DOI] [PubMed] [Google Scholar]
- 25.McLaughlin JN, Mazzoni MR, Cleator JH, Earls L, Perdigoto AL, Brooks JD, Muldowney JAS, III, Vaughan DE, Hamm HE. Thrombin modulates the expression of a set of genes including thrombospondin-1 in human microvascular endothelial cells. J Biol Chem 280: 22172–22180, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, Aird WC. Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol 24: 41–53, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS. Relocalization of STIM1 for activation of store-operated Ca2+ entry is determined by the depletion of subplasma membrane endoplasmic reticulum Ca2+ store. J Biol Chem 282: 9105–9116, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Opipari AW, Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem 267: 12424–12427, 1992 [PubMed] [Google Scholar]
- 29.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev 85: 757–810, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Paria BC, Bair AM, Xue J, Yu Y, Malik AB, Tiruppathi C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J Biol Chem 281: 20715–20727, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, Tiruppathi C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem 278: 37195–37203, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Rahman A, Anwar KN, True AL, Malik AB. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol 162: 5466–5476, 1999 [PubMed] [Google Scholar]
- 33.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galphaq and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res 91: 398–405, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca2+ signalling and PKCα activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol 533: 433–45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandoval R, Malik AB, Naqvi T, Mehta D, Tiruppathi C. Requirement for Ca2+ signaling in the mechanism of thrombin-induced increase in endothelial permeability. Am J Physiol Lung Cell Mol Physiol 280: L239–L247, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Steinberg R, Harari OA, Lidington EA, Boyle JJ, Nohadani M, Samarel AM, Ohba M, Haskard DO, Mason JC. A protein kinase C-epsilon-anti-apoptotic kinase signaling complex protects human vascular endothelial cells against apoptosis through induction of Bcl-2. J Biol Chem 282: 32288–32297, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time. Assessment of endothelial barrier function. Proc Natl Acad Sci USA 89: 7919–7923, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel SM, Gao X, Mehta D, Ye RD, John TA, Andrade-Gordon P, Tiruppathi C, Malik AB. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics 4: 137–145, 2000 [DOI] [PubMed] [Google Scholar]