

Abstract
Oxidative stress is a primary trigger of cachectic muscle wasting, but the signaling pathway(s) that links it to the muscle wasting processes remains to be defined. Here, we report that activation of p38 mitogen-activated protein kinase (MAPK) (phosphorylation) and increased oxidative stress (trans-4-hydroxy-2-nonenal protein modification) in skeletal muscle occur as early as 8 h after lipopolysaccharide (1 mg/kg) and 24 h after dexamethasone (25 mg/kg) injection (intraperitoneal) in mice, concurrent with upregulation of autophagy-related genes, Atg6, Atg7, and Atg12. Treating cultured C2C12 myotubes with oxidant hydrogen peroxide (4 h) resulted in increased p38 phosphorylation and reduced FoxO3 phosphorylation along with induced Atg7 mRNA expression without activation of NF-κB or FoxO3a transcriptional activities. Furthermore, inhibition of p38α/β by SB202190 blocked hydrogen peroxide-induced atrophy with diminished upregulation of Atg7 and atrogenes [muscle atrophy F-box protein (MAFbx/Atrogin-1), muscle ring finger protein 1 (MuRF-1), and Nedd4]. These findings provide direct evidence for p38α/β MAPK in mediating oxidative stress-induced autophagy-related genes, suggesting that p38α/β MAPK regulates both the ubiquitin-proteasome and the autophagy-lysosome systems in muscle wasting.
Keywords: skeletal muscle, atrophy, cachexia
skeletal muscle atrophy occurs as a consequence of numerous pathological conditions, including cachexia and disuse (reviewed in Refs. 35 and 41). Cachectic muscle wasting is a complex process that proceeds via a rapid decline in protein synthesis and a large, sustained increase in protein degradation. In concert with other pathological and functional changes, such as decreases in mitochondrial function, capillary supply, and contractile force production, cachectic muscle wasting poses a profound negative impact on physical performance and metabolism resulting in diminished quality of life and increased mortality. Understanding the molecular and signaling mechanisms underlying cachectic muscle wasting is of critical value to improving survival and quality of life in these patient populations.
Many cellular signaling events contribute to skeletal muscle atrophy (20, 27, 37, 39, 43, 45, 50), and research in these areas has begun to explore the molecular signaling responsible for tipping the homeostatic balance of protein metabolism toward proteolysis and suggested the importance of two major proteolysis systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALS) (37, 43, 50). In particular, activation of the forkhead box O (FoxO) class of transcription factors, which includes forkhead homolog 1 in rhabdomyosarcoma/forkhead box O1 (FKHR/FoxO1), forkhead homolog in rhabdomyosarcoma like 1/forkhead box O3 (FKHR-L1/FoxO3), and AFX/FoxO4 (43), leads to enhanced expression of the muscle-specific ubiquitin ligases in the ubiquitin-proteasome-dependent protein degradation process (37, 43) as well as the autophagy-related genes in the autophagy-lysosomal proteolytic process (27, 50). These findings present FoxO-mediated gene regulation as a nodal point of the two major proteolytic systems in atrophying skeletal muscle. Of course, many other signaling pathways, such as the NF-κB and the p38 MAPK pathways, have also been shown to be functionally involved in cachetic muscle wasting (6, 9, 14, 25, 46, 47). However, the precise mechanisms of each of the signaling pathways and the interrelationship among them in the process of cachectic muscle wasting remain to be fully defined.
Oxidants, free radicals and their derivatives, including hydrogen peroxide (H2O2), superoxide (O2−), hydroxyl radical (·OH), and peroxynitrite (ONOO−), play important roles in the initiation and progression of skeletal muscle atrophy in various diseases or inactivity states, and administration of antioxidants attenuate myofiber atrophy in both disease and disuse models (29, 33, 40). A few studies have suggested that the p38 MAPK pathway mediates oxidative stress-induced muscle-specific E3 ligase muscle atrophy F-box protein (MAFbx) gene expression and ubiquitin-conjugating activity in skeletal muscle (18, 25), but its role in the autophagy-lysosome system has not been investigated. It is particularly intriguing that oxidative stress-induced activation of p38 MAPK signaling and upregulation of the downstream genes in ubiquitin-proteasome system-mediated protein degradation appears to be independent of the NF-κB and the FoxO pathways (9, 18). These findings raised the possibility that there is also a link between oxidative stress and the autophagy-lysosome system-mediated protein degradation through the p38 MAPK pathway.
We designed this study to examine in detail whether cachectic factor-induced activation of the p38 MAPK signaling-transcription cascade contributes functionally to the oxidative stress stimulated atrophic process. We specifically tested the hypothesis that cachectic factor mediated-activation of the p38 MAPK pathway induces skeletal muscle wasting through transcriptional upregulation of autophagy-related genes.
METHODS
Experimental animals.
Male C57BL/6 mice (8–9 wk old) from the Jackson Laboratory (Bar Harbor, ME) were maintained in light- (12:12-h light-dark cycle) and temperature-controlled quarters (21°C) provided with water and chow (Purina, Richmond, IN) ad libitum. Animals were injected intraperitoneally (ip) with Escherichia coli LPS (1 mg/kg; Sigma, St. Louis, MO) or normal saline 8 h, or dexamethasone (Dex; 25 mg/kg; Sigma) or normal saline 24 h before being euthanized humanely by isoflurane-induced anesthesia and cervical dislocation. LPS and Dex injection did not result in morbidity or mortality for the time point utilized. At the end of the experiment, oxidative soleus and glycolytic white vastus lateralis muscles were harvested for further analyses. All animal protocols were approved by the Duke University Institutional Animal Care and Use Committee.
Assessment of oxidative stress.
4-HNE (C9H16O2) is an α,β-unsaturated hydroxyalkenal which is produced by lipid peroxidation in cells (42). 4-HNE modification of proteins was analyzed by Western blotting as described (5) as an indicator of oxidative damage in skeletal muscles.
Myogenic cell line.
Myoblasts derived from mouse skeletal muscle (C2C12 cells; American Type Culture Collection, Rockville, MD) were cultured on six-well dishes in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum, 1% penicillin-streptomycin, and 0.1% fungizone at 37°C in 5% CO2 until 80% confluence was reached as visualized by light microscopy. Myotube differentiation was then initiated by replacing the growth medium with differentiation medium: DMEM supplemented with 2% heat-inactivated horse serum, 1% penicillin-streptomycin, and 0.1% fungizone at 37°C in 5% CO2 for 7 days. The muscle cells were examined for evidence of myotube formation by light microscopy. Hydrogen peroxide treatment of C2C12 cells involved dilution in differentiation media to final desired concentrations and standard incubation (37°C in 5% CO2). H2O2-treated media were replaced at 12-h intervals for incubation times >12 h. Dex treatment of C2C12 cells involved dilution in differentiation media to final desired concentration (10 μM) and standard incubation (37°C in 5% CO2) for 24 h as previously described (43).
Myotube atrophy.
Morphological analysis for myotube diameter was performed as previously described (30) with the following modifications. Briefly, images of myotubes after the appropriate treatment were obtained via phase contrast at ×100 magnification on an inverted microscope (Carl Zeiss Axiovert 200) camera system. The diameters were measured in ∼100 myotubes from at least 10 random fields, a number chosen by determination of no additional change in standard deviation, using computerized image analysis (Scion Image, Frederick, MD). Each myotube analyzed was measured at three points along the length of the myotube in a blinded fashion, and results are expressed as percentage of the control treatment diameter.
Transient transfection and luciferase reporter gene analysis.
The NF-κB-GL3 reporter plasmid was obtained from Dr. Steffan Ho and the Foxo3a-GL3 reporter was obtained from Dr. Alex Toker (Beth Israel Deaconess Medical Center, Boston, MA). Both have been previously used and described (19, 39). Plasmid DNA was prepared and isolated using an Endotoxin-Free Maxi or Mega Prep Kit (Qiagen, Valencia, CA). Differentiating myotubes (Day 3) were transiently transfected in six-well plates using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions as previously described (28). Briefly, firefly luciferase and Renilla luciferase control plasmid DNAs were diluted in Opti-MEM and transfected using Lipofectamine 2000 reagent. The diluted Lipofectamine 2000 reagent was incubated for 5 min and then added to the DNA mixture at a final volume of 500 μl per well. The lipid/plasmid mixtures were allowed to complex for 20 min during which the cells were rinsed twice with Opti-MEM and bathed in a final volume of 2 ml Opti-MEM per well. The lipid:plasmid complexes were then applied to the cells (final transfection volume of 2.5 ml). Separate experiments were performed using equivalent amounts of lipid reagent and empty vector DNA. Cultures were rocked every 2 h for the initial 6 h of incubation and incubated with the transfection mixture for a total of 24 h. After 24 h, transfection medium was removed and replaced with fresh differentiation medium. Cultures were maintained for an additional 72 h before H2O2 treatment (total of 7 days of differentiation). Following cell lysis in a passive lysis buffer (Promega, Madison, WI) and centrifugation for 20 min at 5,000 g, 20 μl of the supernatant was added to 100 μl of luciferase reagent (Promega) for determination of total cell luciferase activity as corrected for Renilla luciferase activity, using an LMax II microplate luminometer (Molecular Devices, Sunnyvale, CA). Reporter gene activities are presented as relative light units, corrected for Renilla luciferase activity.
Western blot analysis.
Upon completion of the appropriate incubation time, cells were rinsed twice in ice-cold 1× PBS and scraped for protein isolation in 130 μl nondenaturing lysis buffer (1% Triton X-100, 300 mM NaCl, 50 mM Tris-base, 5 mM EDTA, 3.1 mM sodium azide, 95 mM NaF, and 22 μM Na3VO4), vortexed, incubated at 4°C for 25 min, and centrifuged at 1,000 g for 5 min. The supernatants were subsequently assayed for protein using the Bradford method (Sigma). Whole muscle homogenates or cellular lysates were separated by polyacrylamide gel electrophoresis and transferred (100 V for 3 h at 4°C) to nitrocellulose membranes for Western blotting. As verification of equal loading and transfer, the resulting membrane was stained with Ponceau S as well as probing for β-actin as loading references. Membranes were then probed for 4-HNE (ab48506, Abcam, Cambridge, MA), phosphorylated FoxO3 (9465, 9466), total FoxO3 (9467), phosphorylated p38 MAPK (9216), total p38 MAPK (9212), Atg7 (2631), Beclin-1 (3738), Bnip3 (3769), LC3b (2775), and Atg12–5 (2011): (all purchased from Cell Signaling Technology, Carlsbad, CA), and polyubiquitin (clone FK1, Biomol International, Plymouth Meeting, PA).
RNA isolation and real-time PCR analysis.
Cells harvested for RNA isolation were rinsed twice in ice-cold 1× PBS and scraped in TRIzol Reagent (Life Technologies, Carlsbad, CA), according to the manufacturer's instructions. Total RNA (5 μg) was then reverse transcribed using the Superscript III First-Strand Synthesis System for RT-PCR (Life Technologies) using oligo(dT)20 primers and the protocol outlined by the manufacturer. cDNA (1 μg) was added to a 25-μl PCR reaction for real-time PCR using Taqman chemistry and the ABI Prism 7000 Sequence Detection System (ABI, Foster City, CA). Quantitative analysis of gene expression in C2C12 cells was performed using the comparative computed tomography method (ABI, User Bulletin no. 2). This method uses a single sample, the calibrator sample (hprt; GenBank NM_013556.2), for comparison of every unknown sample. ΔΔCT [ΔCT(calibrator) − ΔCT(sample)] was then calculated for each sample, and relative quantity was calculated as 2ΔΔCT. Hypoxanthine-guanine phosphoribosyltransferase (Hprt), an enzyme involved in purine catabolism, was chosen as the reference gene based on initial experiments showing unchanged expression with our experimental manipulations. Fivefold dilution curves were assayed on selected samples to confirm the validity of this quantitation method for each gene. MAFbx (NM_026346.2), muscle ring finger protein 1 (MuRF-1; NM_001039048.2), Nedd4 (NM_010890.3), Atg7 (NM_028835.3), Bnip3 (NM_009760.4), Bnip3L (NM_009761.3), and Maplc3b (NM_026160.4) mRNA transcripts were assayed by using predesigned mouse primer and probe sequences commercially available from Applied Biosystems (Assays-on-Demand).
Statistical analysis.
A priori analysis was utilized to determine differences between in vivo fiber types at baseline. For comparisons between two groups within the fiber type, Student's t-test was performed. Comparisons between the in vitro treatments were made by a one-way analysis of variance, and, when appropriate, a Tukey honestly significant difference test was performed post hoc. Significance was established at P < 0.05 for all the experiments in this study. Values are reported as means ± SE.
RESULTS
Endotoxin induces oxidative stress and p38 MAPK activation.
Our previous findings suggest that endotoxin challenge leads to increased oxidative stress in skeletal muscle with enhanced protein carbonylation in glycolytic muscles (48). Here, we assayed 4-HNE protein modification by Western blot as a measure of skeletal muscle oxidative stress and found that less 4-HNE modification of muscle protein in glycolytic muscle versus oxidative muscle at baseline, but LPS injection increased 4-HNE protein modification in both soleus (+129%) and white vastus lateralis (+309%) skeletal muscles (Fig. 1A).
Fig. 1.

Endotoxin induces oxidative stress and muscle p38 MAPK activation. Whole muscle homogenates were obtained from oxidative soleus muscles (soleus, black bars) and glycolytic white vastus lateralis muscles (WV, gray bars) 8 h after intraperitoneal (ip) injection of LPS (1 mg/kg body wt) and were assayed by Western blot for α,β-unsaturated hydroxyalkenal [4-hydroxynonenal (trans-4-hydroxy-2-nonenal); 4-HNE, C9H16O2; 4-HNE, lipid peroxidation by-product] modification of proteins, phosphorylated p38 MAPK (Thr180/Tyr182), and total p38 MAPK. A: 4-HNE modification of proteins. Inset: representative immunoblot of 4-HNE. B: relative phosphorylation of p38 (p-p38 MAPK). Inset: representative immunoblot of p-p38 MAPK, total p38 MAPK, and β-actin as loading reference. C: whole muscle homogenates were obtained from oxidative soleus muscles (black bars) and glycolytic white vastus lateralis muscles (gray bars) 24 h after ip injection of dexamethasone (Dex; 25 mg/kg body wt) and were assayed for relative phosphorylation of p38 (p-p38 MAPK). Inset: representative immunoblot of p-p38 MAPK, total p38 MAPK, and β-actin as loading reference. Values are n = 6 and presented as means ± SE. *Significantly different (P < 0.05) from muscle-matched control. †Significantly different (P < 0.05) from soleus control.
Since the p38 MAPK pathway has been implicated in mediating oxidative stress-induced muscle-specific E3 ligase MAFbx gene expression and ubiquitin-conjugating activity in skeletal muscle (18, 25), which could occur in a fiber type-specific manner (48), we investigated p38 MAPK signaling in glycolytic and oxidative skeletal muscles from endotoxin (LPS, 1 mg/kg ip)-treated mice utilizing an abbreviated time frame (8 h). We measured the relative phosphorylation of p38 in skeletal muscles (presumably α/β- and/or γ-isoform) and found that phosphorylation of p38 increased in both glycolytic white vastus lateralis muscle (+82%) and oxidative soleus muscle (+41%) compared with control mice injected with saline (Fig. 1B). Body weight and muscle weight (soleus, plantaris, and gastrocnemius) did not show any significant differences from the values in the control mice injected with saline, demonstrating that the time frame chosen is early in the atrophic process before the actual development of muscle atrophy (not shown). To verify whether the p38 MAPK pathway is activated in response to other cachectic stimuli, mice were also injected with Dex (25 mg/kg ip) or saline, and the relative phosphorylation of p38 was analyzed 24 h after the injections. Dex induced p38 phosphorylation in the white vastus muscle (+38%) as compared with no alteration the soleus (Fig. 1C).
Endotoxin induces enhanced expression of autophagy-related genes and protein ubiquitinylation in both glycolytic and oxidative muscles.
The autophagy-lysosome system functions in degradation of membrane and extracellular imported proteins, damaged organelles (macromolecular autophagy), and other cellular cytoplasmic proteins (38) with critical proteolytic role in the progression of muscle atrophy (2, 27, 50). To begin to understand whether oxidative stress enhances autophagy-related gene expression in cachectic muscle wasting, we analyzed the abundances of several autophagy-related proteins by Western blot. Basal levels of autophagy related proteins, Atg6 (Beclin-1), Atg7, Atg12, were generally lower in glycolytic skeletal muscle versus oxidative skeletal muscle except for Bnip3. LPS injection resulted in significantly increased expression of Atg7 in both oxidative (+30%) and glycolytic (+50%) muscles and similar degrees of induced expression of Beclin-1 and Atg12 (Fig. 2A).
Fig. 2.
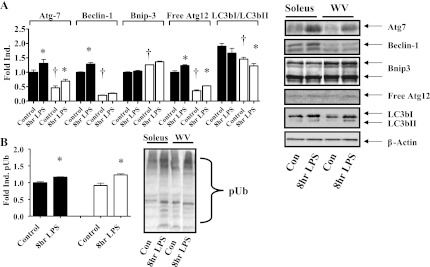
Endotoxin induces autophagy-related protein expression and protein polyubiquitinylation in mouse oxidative and glycolytic muscles. Whole muscle homogenates were obtained from oxidative soleus muscles (black bars) and glycolytic white vastus lateralis muscles (white bars) 8 h after ip injection of LPS (1 mg/kg body wt) and were assayed by Western blot for Atg7, Beclin-1, Bnip3, and Atg12-5 proteins, as well as polyubiquitinylated proteins. A: autophagy-related protein expression. Inset: representative immunoblots with β-actin as loading reference. Bnip3 antibody recognizes endogenous levels of total Bnip3 protein in two distinct band regions: 22–28 kDa and 50–55 kDa. Con, control. B: polyubiquitinylated (pUb) proteins. Inset: representative immunoblot. Values are n = 6 and presented as means ± SE. *Significantly different (P < 0.05) from muscle-matched control. †Significantly different (P < 0.05) from soleus control.
Enhanced expression of E3 ubiquitin ligases and consequently protein ubiquitinylation are key steps in muscle wasting (18). We have previously shown that LPS injection induces greater expression of E3 ubiquitin ligases expression in glycolytic than oxidative muscles (48). To further confirm that endotoxin challenge induces protein ubiquitinylation, we performed Western blot and showed that the relative level of protein polyubiquitinylation increased in both oxidative (+16%) and glycolytic (+35%) skeletal muscles (Fig. 2B).
Hydrogen peroxide induces early activation of the p38 MAPK and FoxO3 pathways in myotubes.
Our collective findings of enhanced activity of stress signaling cascades by endotoxin administration in both oxidative and glycolytic skeletal muscles prompted us to investigate cultured muscle cells to determine the temporal relationship of the signal transduction pathways in atrophying skeletal muscle. We subjected differentiated myotubes to oxidant, hydrogen peroxide, followed by Western blot analysis to determine the relative activation of both p38 and FoxO3a proteins. Phosphorylation (activation) of p38 increased with 4-h H2O2 treatment and decreased with prolonged exposure (Fig. 3A). The relative phosphorylation (activation) of FoxO3a decreased with 4-h H2O2 treatment, but it was not altered with prolonged exposure (Fig. 3B). To ascertain the functional activity of NF-κB and FoxO3-dependent transcriptional activation, we analyzed the transcriptional activity of NF-κB and FoxO3 in cells transfected with NF-κB and FoxO3 reporter genes, respectively. NFκB reporter gene activity was not induced by either 4- or 12-h H2O2 treatment, whereas FoxO3 reporter gene activity was only induced by H2O2 treatment at 12 h (Fig. 3C).
Fig. 3.

Hydrogen peroxide challenge induces p38 and forkhead box O (FoxO) signaling in myotubes. Fully differentiated C2C12 myotubes were treated with vehicle (control) or 25 μM H2O2 for 4 h (black bars) or 12 h (gray bars). Cellular lysates were collected and analyzed via Western blot for phosphorylated (Thr180/Tyr182), total p38 MAPK protein, phosphorylated (Ser318/321), and total FoxO3a protein. A: relative phosphorylation of p38 (p-p38 MAPK). Inset: representative immunoblot of p-p38 MAPK, total p38 MAPK, and β-actin as loading reference. B: relative phosphorylation of FoxO3a (p-FoxO3a). Inset: representative immunoblot of p-FoxO3a, total FoxO3a, and β-actin as loading reference. C: C2C12 myotubes were also transiently cotransfected with a NF-κB-luciferase or FoxO3a-luciferase reporter plasmids and Renilla, subsequently allowed to continue differentiating out to day 7, treated with vehicle (control) or 25 μM H2O2 for 4 or 12 h, and analyzed for relative luciferase activity. RLU, relative light units. Values are representative of 5 independent experiments and presented as means ± SE. *Significantly (P < 0.05) different from duration-matched control cells.
Hydrogen peroxide induces early transcriptional activation of genes involved in ubiquitin-proteasome- and autophagy-lysosome-mediated proteolysis in myotubes.
To further investigate oxidative stress-induced expression of genes that may be functionally involved in cachectic muscle wasting, we analyzed the gene expression of key components of atrophic signaling, including autophagy-related genes and atrogenes. The mRNA abundance of Atg7 increased with 4-h H2O2 treatment, whereas mRNA abundances of the other autophagy-related genes Bnip3, Bnip3L and Maplc3b were not increased by 4- or 12-h H2O2 treatment (Fig. 4A). The mRNA abundance of the E3 ubiquitin ligases MAFbx/Atrogin-1 and Nedd4 increased only with 4-h H2O2 treatment, while MuRF-1 mRNA did not increase (Fig. 4B). Consistent with enhanced ubiquitin-proteasome-dependent protein degradation, myotube protein polyubiquitinylation increased 150% with 4-h H2O2 treatment and 190% with 12-h H2O2 treatment (Fig. 4C).
Fig. 4.

Hydrogen peroxide challenge induces transcriptional activation of proteasomal and lysosomal proteolytic genes in myotubes. Fully differentiated C2C12 myotubes were treated with vehicle (control) or 25 μM H2O2 for 4 h (black bars) or 12 h (white bars). mRNA abundance was determined by real-time RT-PCR and is presented as corrected for hprt and normalized to the control cells. A: lysosomal-dependent proteolytic genes (Atg7, Bnip3, Bnip3L, and Maplc3b mRNA abundances). B: proteasomal-dependent proteolytic genes (MAFbx/Atrogin-1, MuRF-1, and Nedd4 mRNA abundances). C: hydrogen peroxide induced polyubiquitinylated myotube proteins. Values are means ± SE and representative of 5 independent experiments. *Significantly different (P < 0.05) from respective duration-matched control cells.
Activity of p38α/β MAPK is required for oxidant-induced expression of autophagy-related genes and myotube atrophy.
The finding that H2O2 treatment induces concurrent activation of autophagy-related genes and the p38 MAPK pathway in the absence of activation of NFκB or FoxO3 reporter genes prompted us to examine whether p38 MAPK mediates the induction of the autophagy-related genes in myotubes. We treated differentiated myotubes for 24 h with 25 μM H2O2 (28) and measured myotube diameter as an indicator of myotube atrophy. Myotube diameter decreased by 28% with H2O2 treatment, which appears to be mainly via autophagy-related proteolysis, because administration of the pharmacological autophagy inhibitor concanamycin A (ConC-A) prevented ∼65% of this atrophy versus the approximate 26% prevented by MG-132 (pharmacological proteasome inhibitor). Inhibition of p38α/β MAPK activity by SB202190 or SB203580 compounds (10 μM) profoundly prevented the myotube atrophy induced by H2O2 treatment (Fig. 5A). Western blot analysis showed attenuated p38 phosphorylation with SB inhibitor administration (data not shown) as previously described (10). However, SB inhibitors (including SB202190) may bind to the ATP pocket of p38 to reversibly block its intrinsic ATPase activity without affecting p38 phosphorylation (8, 23); therefore, this decrease may not be solely attributed to the SB administration. The induction of the ubiquitin ligases MAFbx/Atrogin-1 and Nedd4 as well as the autophagy-related protein Atg7 was also prevented by SB202190 (Fig. 5B).
Fig. 5.

p38 is required for hydrogen peroxide atrophy, ubiquitin ligase, and autophagy-related gene expression in myotubes. A: myotube atrophy. Fully differentiated C2C12 myotubes were treated with vehicle (control), 25 μM H2O2, 25 μM H2O2 and 1 μM MG-132 (protease inhibitor), 25 μM H2O2 and 0.1 μM ConC-A (lysosomal inhibitor), 25 μM H2O2 with 10 μM SB202190 (pharmacological inhibitor of p38), 25 μM H2O2 with 10 μM SB203580, or 10 μM dexamethasone for 24 h. Representative images were acquired (inset) and analyzed for myotube diameters. Values are representative of 5 independent experiments and presented as the percentage of vehicle control values, means ± SE. *Significantly (P < 0.05) different from control. B: p38 regulation of proteasomal and lysosomal proteolytic gene expression. Fully differentiated C2C12 myotubes were treated with vehicle (control), 25 μM H2O2, or 25 μM H2O2 with 10 μM SB202190 (pharmacological inhibitor of p38). mRNA abundance (MAFbx/Atrogin-1, Nedd4, and Atg-7) was determined by real-time RT-PCR and is presented as corrected for hprt and normalized to the control cells. Values are means ± SE and representative of 5 independent experiments. *Significantly different (P < 0.05) from control cells. †Significantly different (P < 0.05) from 25 μM H2O2.
DISCUSSION
A number of new findings emerged from this study. First, we demonstrated that endotoxin challenge in vivo induced p38 activation in both glycolytic and oxidative skeletal muscles along with induced expression of a number of autophagy-related genes during the initiation of cachectic muscle wasting. These findings suggest the possibility that the p38 MAPK pathways play a regulatory role in the autophagy-lysosomal proteolytic function in atrophying skeletal muscle. Second, oxidative stress-induced expression of an autophagy-related gene (Atg7) in the autophagy-lysosomal proteolytic pathway and the E3 ligases (MAFbx/Atrogin-1 and Nedd4) in the ubiquitin-proteasome proteolytic pathway is temporally associated with activation of the p38 MAPK pathway, independent of NF-κB and FoxO-dependent transcriptional activation in culture muscle cells. Finally, we demonstrated that oxidant mediated-myotube muscle wasting involving transcriptional upregulation of atrogenes and autophagy-related genes can be potently blocked by inhibitors specific for the p38α/β MAPK pathway. These findings provide direct evidence for the functional role of the p38α/β MAPK pathway in mediating oxidative stress to autophagy-lysosomal protein degradation in cachectic muscle wasting.
Oxidative stress is an initial signal and a key contributor to the development of skeletal muscle proteolysis and atrophy (35). Several key atrophic signaling cascades have been defined in skeletal muscle, including protease activation (28), repression of the insulin and/or IGF-I activation of the serine/threonine protein kinase B (PKB/Akt) (reviewed in Ref. 20), and activation of FoxO or NF-κB (6, 14, 46). The p38 MAPK pathway has been implicated in mediating oxidative stress-induced atrophy process (18, 25). Our findings in this study that cachectic stimuli causes increased 4-HNE protein modification in both glycolytic and oxidative muscles concurrent with p38 MAPK activation and enhanced autophagy-related gene expression suggest the possible link between oxidative stress and the autophagy-lysosome system-mediated proteolysis through the p38 MAPK pathway.
It is worth noting that the changes in 4-HNE protein modification, p38 phosphorylation, and autophagy-related gene expression all demonstrated similar trends. These parameters were low in the glycolytic muscles to begin with and showed more dramatic increases following endotoxin challenge compared with oxidative muscles (Figs. 1 and 2). These findings are consistent with some of our recent studies where we have shown that oxidative myofibers are more resistant to cachectic stimuli, such as LPS and tumor necrosis factor-α (24), and that the potential mechanisms for the inherent oxidative phenotype-associated resistance to cachectic stimuli include decreased free radical burden due to increased antioxidant expression and attenuated ubiquitin ligase gene expression (48). Altogether, these observations support the notion that the p38 MAPKs serve as an oxidative stress sensor in skeletal muscle.
Few previously published studies have examined the role of p38 MAPK in cachectic skeletal muscle wasting. One study in particular, by Jin and Li (18), expanding on previous in vitro work (25), revealed that p38 is a signaling regulator of MAFbx/Atrogin-1 and MuRF-1 ubiquitin ligase gene expression independent of the Akt and NF-κB pathways. A more recent study has also shown that activation of the p38 MAPK-atrogene transcription is independent of Akt/FoxO signaling axis (47). Our data both confirm and expand these findings and support the concept that p38 MAPK is a key contributor to proteolytic degradation and atrophy in response to cachectic stimuli. In agreement with the in vivo findings, treatment of mature murine C2C12 myotubes with relatively low doses of hydrogen peroxide (25 μM) resulted in a rapid phosphorylation of p38, in agreement with our overall hypothesis, and stimulated the “next-step” analysis of atrogene and autophagy-related gene expression in atrophying myotubes. Activation of the p38 MAPK pathway in vitro occurs at a time (4 h) before FoxO3a and NF-κB are being transcriptionally activated (Fig. 3). More importantly, oxidant treatment induces both autophagy-related gene (Atg7) and atrogene expression (MAFbx/Atrogin-1 and Nedd4) at the same time point. The temporal induction of signaling pathways and gene transcription lend credence to the concept that p38 MAPK may preferentially regulate atrogene and autophagy-related gene expression, at least initially, during cachexia-induced muscle wasting.
An important question is how much the p38 MAPK contributes to cachetic muscle wasting. In cultured cells, the relative contribution of the ubiquitin-proteasome system to myotube proteolysis in atrophying myotubes may be as low as 20% (50) depending on the type of atrophic stimulus. Specifically, myotube atrophy that relies heavily on the activation of FoxO transcription factors may utilize autophagy preferentially as a means for cellular proteolysis (27, 50). Here, we show that inhibition of the p38 MAPK by SB202190 profoundly blocked oxidative stress-induced muscle atrophy, providing functional evidence of the p38 MAPK in cachectic muscle wasting. In addition, SB202190 treatment attenuated MAFbx/Atrogin-1, Nedd4, and Atg7 mRNA expression, strongly supporting a link between the activity of p38 MAPK and the early transcriptional induction of both atrogene and autophagy-related genes in cachectic muscle wasting.
The p38 MAPK belongs to a complex MAPK family, which includes four subclasses, including ERKs, JNK/SAPK, ERK5/MAPK1 (BMK1), and p38 MAPK (32). During the revision of this article, research was published outlining a specific role for the JNK subclass of MAPK in the regulation of endotoxin-induced diaphragm dysfunction and wasting (44), lending credence to the potential contribution of this convoluted family of signaling proteins in the regulation of catabolism in skeletal muscle. The p38 MAPK subclass has four isoforms, p38α, p38β, p38γ, and p38δ, that share roughly 60% sequence homology (17). While p38α and p38β are expressed ubiquitously (16), p38γ is expressed primarily in skeletal muscle (22, 26) and p38δ is expressed in non-skeletal muscle tissues (21). These proteins have been shown to be involved in inflammation, cell growth and myogenic differentiation, cell cycle regulation, cell death, glucose metabolism, and energy expenditure (16, 31, 36, 49).
Other than the previous studies in which p38 MAPK was implicated in mediating oxidative stress-induced muscle atrophy (25), the precise functional role of p38 MAPK in cachectic muscle wasting remains to be fully defined. Interestingly, contractile activity activates this pathway in skeletal muscle and promotes skeletal muscle mitochondrial biogenesis (1, 3, 4, 15), whereas various other stresses also activate this pathway (11, 25, 32, 34). A recent publication showed that p38 inhibition triggers autophagy-related gene expression via AMPK-FoxO3 axis in colorectal cancer cells (7). All these findings demonstrate the complexity of the p38 MAPK signaling system in mammalian cells. Our data concerning the effect of SB202190, a pyridinyl imidazole derivative that has been reported to have primary inhibitory effects against the ubiquitously expressed p38α and p38β isoforms (12, 13), support the notion that p38α/β MAPK is functionally involved in skeletal muscle atrophic process. Together with previous findings that exercise-induced activation of p38 MAPK promotes mitochondrial biogenesis, it is entirely possible that different p38 isoforms play distinct functions in skeletal muscle in response to physiological and pathological stress signals.
In summary, cachectic skeletal muscle wasting is a complex process that occurs as a consequence of numerous pathological conditions and diseases. Despite skeletal muscle's widespread susceptibility to cachectic muscle wasting and its importance in predicting the morbidity and mortality in patients, the underlying mechanisms remain unclear. These studies were undertaken to provide evidence for a critical role of p38 signaling in the progression of cachectic muscle wasting. Our findings demonstrate that cachectic stimuli result in increased phosphorylation of p38 in vivo and in vitro. This p38 activation occurs in a manner consistent with an initial regulation of both ubiquitin ligases in the ubiquitin-proteasome system-mediated muscle proteolysis and autophagy-related genes in the autophagy-lysosome system-mediated muscle proteolysis. Inhibition of p38 MAPK activity attenuates myotube atrophy in vitro with attenuated ubiquitin ligase and autophagy-related gene expression. Although the increase in autophagy-related genes may not directly match the absolute level of p38 phosphorylation, we contend that the absolute level of p38 phosphorylation may not be linear with its function among different muscles. On the basis of these findings, we propose a model in which oxidative stress-induced p38 MAPK activation initiates and participates in cachectic muscle wasting through both the ubiquitin-proteasome- and the autophagy-lysosome-dependent proteolytic mechanisms.
GRANTS
This work was supported by the National Institutes of Health Grants RO1-HL-072789 (to S. K. Powers) and RO1-AR-050429 (to Z. Yan).
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
All authors on this manuscript have contributed to the conception, design, analysis, and/or interpretation of the data and to drafting/revising the article and have approved the version for publication. The in vitro and in vivo experiments were performed at University of Florida and Duke University Medical Center, respectively.
REFERENCES
- 1.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 280: 19587–19593, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. Int J Biochem Cell Biol 37: 2098–2114, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Boppart MD, Asp S, Wojtaszewski JF, Fielding RA, Mohr T, Goodyear LJ. Marathon running transiently increases c-Jun NH2-terminal kinase and p38 activities in human skeletal muscle. J Physiol 526: 663–669, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boppart MD, Hirshman MF, Sakamoto K, Fielding RA, Goodyear LJ. Static stretch increases c-Jun NH2-terminal kinase activity and p38 phosphorylation in rat skeletal muscle. Am J Physiol Cell Physiol 280: C352–C358, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Braga M, Sinha Hikim AP, Datta S, Ferrini MG, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis 13: 822–832, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ 16: 1203–1214, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J 16: 295–305, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Giovanni S, Molon A, Broccolini A, Melcon G, Mirabella M, Hoffman EP, Servidei S. Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann Neurol 55: 195–206, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Geiger PC, Wright DC, Han DH, Holloszy JO. Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 288: E782–E788, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37: 1974–1984, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Goedert M, Cuenda A, Craxton M, Jakes R, Cohen P. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J 16: 3563–3571, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol 286: R342–R349, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Hunter RB, Kandarian SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest 114: 1504–1511, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA. PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol 284: C1669–C1677, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J Biol Chem 271: 17920–17926, 1996 [DOI] [PubMed] [Google Scholar]
- 17.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem 272: 30122–30128, 1997 [DOI] [PubMed] [Google Scholar]
- 18.Jin B, Li YP. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J Cell Biochem 100: 960–969, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Judge AR, Koncarevic A, Hunter RB, Liou HC, Jackman RW, Kandarian SC. Role for IκBα, but not c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol 292: C372–C382, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33: 155–165, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun 235: 533–538, 1997 [DOI] [PubMed] [Google Scholar]
- 22.Lechner C, Zahalka MA, Giot JF, Moller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci USA 93: 4355–4359, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746, 1994 [DOI] [PubMed] [Google Scholar]
- 24.Li P, Waters RE, Redfern SI, Zhang M, Mao L, Annex BH, Yan Z. Oxidative phenotype protects myofibers from pathological insults induced by chronic heart failure in mice. Am J Pathol 170: 599–608, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19: 362–370, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun 228: 334–340, 1996 [DOI] [PubMed] [Google Scholar]
- 27.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471, 2007 [DOI] [PubMed] [Google Scholar]
- 28.McClung JM, Judge AR, Talbert EE, Powers SK. Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am J Physiol Cell Physiol 296: C363–C371, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClung JM, Kavazis AN, Whidden MA, DeRuisseau KC, Falk DJ, Criswell DS, Powers SK. Antioxidant administration attenuates mechanical ventilation-induced rat diaphragm muscle atrophy independent of protein kinase B (PKB Akt) signalling. J Physiol 585: 203–215, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren PO. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem 105: 353–364, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niu W, Huang C, Nawaz Z, Levy M, Somwar R, Li D, Bilan PJ, Klip A. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J Biol Chem 278: 17953–17962, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal 12: 1–13, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Otis JS, Brown LA, Guidot DM. Oxidant-induced atrogin-1 and transforming growth factor-beta1 precede alcohol-related myopathy in rats. Muscle Nerve 36: 842–848, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel J, McLeod LE, Vries RG, Flynn A, Wang X, Proud CG. Cellular stresses profoundly inhibit protein synthesis and modulate the states of phosphorylation of multiple translation factors. Eur J Biochem 269: 3076–3085, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol 102: 2389–2397, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell 8: 971–982, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott SV, Klionsky DJ. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr Opin Cell Biol 10: 523–529, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 22: 3836–3845, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Servais S, Letexier D, Favier R, Duchamp C, Desplanches D. Prevention of unloading-induced atrophy by vitamin E supplementation: links between oxidative stress and soleus muscle proteolysis? Free Radic Biol Med 42: 627–635, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith IJ, Lecker SH, Hasselgren PO. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab 295: E762–E771, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sompol P, Ittarat W, Tangpong J, Chen Y, Doubinskaia I, Batinic-Haberle I, Abdul HM, Butterfield DA, St Clair DK. A neuronal model of Alzheimer's disease: an insight into the mechanisms of oxidative stress-mediated mitochondrial injury. Neuroscience 153: 120–130, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Supinski GS, Ji X, Callahan LA. The JNK MAP kinase pathway contributes to the development of endotoxin-induced diaphragm caspase activation. Am J Physiol Regul Integr Comp Physiol 297: R825–R834, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wing SS. Control of ubiquitination in skeletal muscle wasting. Int J Biochem Cell Biol 37: 2075–2087, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Wyke SM, Russell ST, Tisdale MJ. Induction of proteasome expression in skeletal muscle is attenuated by inhibitors of NF-kappaB activation. Br J Cancer 91: 1742–1750, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto Y, Hoshino Y, Ito T, Nariai T, Mohri T, Obana M, Hayata N, Uozumi Y, Maeda M, Fujio Y, Azuma J. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-MAP kinase in cardiac myocytes. Cardiovasc Res 79: 89–96, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Yu Z, Li P, Zhang M, Hannink M, Stamler JS, Yan Z. Fiber type-specific nitric oxide protects oxidative myofibers against cachectic stimuli. PLoS ONE 3: e2086, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem 274: 5193–5200, 1999 [DOI] [PubMed] [Google Scholar]
- 50.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483, 2007. [DOI] [PubMed] [Google Scholar]