

Abstract
Nitroalkene derivatives of nitro-oleic acid (OA-NO2 ) are endogenous lipid products with potent anti-inflammatory properties in vitro. The present study was undertaken to evaluate the in vivo anti-inflammatory effect of OA-NO2 in mice given LPS. Two days before LPS administration, C57BL/6J mice were chronically infused with vehicle (LPS vehicle) or OA-NO2 (LPS OA-NO2) at 200 μg·kg−1·day−1 via osmotic minipumps; LPS was administered via a single intraperitoneal (ip) injection (10 mg/kg in saline). A third group received an ip injection of saline without LPS or OA-NO2 and served as controls. At 18 h of LPS administration, LPS vehicle mice displayed multiorgan dysfunction as evidenced by elevated plasma urea and creatinine (kidney), aspartate aminotransferase (AST) and alanine aminotransferase (ALT; liver), and lactate dehydrogenase (LDH) and reduced ejection fraction (heart). In contrast, the severity of multiorgan dysfunction was less in LPS OA-NO2 animals. The levels of circulating TNF-α and renal TNF-α mRNA expression, together with renal mRNA expression of monocyte chemoattractant protein-1, ICAM-1, and VCAM-1, and with renal mRNA and protein expression of inducible nitric oxide synthase and cyclooxygenase 2, and renal cGMP and PGE2 contents, were greater in LPS vehicle vs. control mice, but were attenuated in LPS OA-NO2 animals. Similar patterns of changes in the expression of inflammatory mediators were observed in the liver. Together, pretreatment with OA-NO2 ameliorated the inflammatory response and multiorgan injury in endotoxin-induced endotoxemia in mice.
Keywords: lipopolysaccharide, TNF-α, sepsis, acute kidney injury, COX-2
sepsis is a systemic inflammatory response to a blood-borne infection that is associated with an extremely high rate of morbidity and mortality. The incidence of septic shock is increasing, affecting 750,000 patients each year, with an overall mortality rate of 20–50% (28). It is the second leading cause of death among patients in noncoronary intensive care units (ICUs) (32) and the tenth leading cause of death overall in the United States (19). The high mortality often results from the failure of multiple organs, including the kidney and liver. In particular, acute kidney injury (AKI) secondary to sepsis is a highly prevalent diagnosis in the ICU setting in which the mortality rate can reach as high as 70% (24, 40); the mortality rate for septic patients with acute renal failure (ARF) is approximately double compared with patients with sepsis alone (29). ARF is considered a critical prognostic factor in sepsis (37), while the management of sepsis and sepsis-induced ARF is largely supportive. Therefore, novel therapies to prevent or treat this devastating disease are urgently required.
Recently, nitrated free fatty acids (NO2-FA), notably nitroalkene derivatives of linoleic acid (nitrolinoleic acid; LNO2) and nitro-oleic acid (OA-NO2), are identified as endogenous molecules with several attractive signaling properties. These derivatives are formed via nitric oxide (NO)-dependent oxidative reactions (30, 36). Plasma concentrations are ∼0.5 μM for LNO2 (3) and ∼0.6 μM for OA-NO2 (3) in healthy human blood, and combined blood levels of these two fatty acid derivatives exceed 1 μM, indicating their capability to act in physiological concentration ranges. To date, there are three major mechanisms by which nitroalkenes appear to mediate cell signaling. First, LNO2 and OA-NO2, at physiological concentrations, serve as potent ligands for proxisome proliferator-activated receptor subtype-γ (PPARγ) (39). The second is as a NO-storage form; thus nitroalkenes can mediate vessel relaxation through a cGMP-dependent pathway (38). Aqueous decay and release of NO by nitroalkenes occur via a modified Nef reaction and leads to the regulation of NO-sensitive cell-signaling pathways (38). Last, nitroalkenes are electrophiles and thus can nitroalkylate proteins and small peptides such as glutathione through reaction with cysteine thiols and histidine (4).
Increasing in vitro evidence demonstrates that nitroalkenes exert potent anti-inflammatory actions. For example, nitroalkenes reduce human neutrophil superoxide generation, degranulation, and integrin expression and thrombin-induced Ca2+ elevations and platelet aggregation (9, 10, 26). Moreover, in cultured RAW264.7 cells, a murine macrophage cell line, nitroalkenes attenuate the LPS-elicited inflammatory response via diverse mechanisms involving activation of mitogen-activated protein kinase phosphatase 1 (21) and nitroalkylation of NF-κB p65 (11). Compared with the detailed in vitro analysis of signaling properties of nitroalkenes, there are few studies that investigate their biological function in vivo. We previously demonstrated that OA-NO2 had protective effects on renal injury in the ischemia-reperfusion mouse model (27). The present study seeks to examine the potential therapeutic effects of OA-NO2 in LPS-induced inflammation and renal injury.
MATERIALS AND METHODS
Materials.
9-Nitro-oleic acid and 10-nitro-oleic acid are two regioisomers of OA-NO2, which are formed by nitration of oleic acid (OA) in approximately equal proportions in vivo (3). The two compounds were purchased from Cayman Chemical (Ann Arbor, MI) (9-nitro-oleic acid: catalog no. 10008042; 10-nitro-oleic acid: catalog no. 10008043), dissolved in DMSO, and used as a 1:1 mixture of the isomers. OA was purchased from the same company (catalog no. 90260) and dissolved in the same solvent. All other reagents were purchased from Sigma-Aldrich unless otherwise specified. All protocols employing mice were conducted in accordance the principles and guidance of the University of Utah Institutional Animal Care and Committee.
Animal protocol.
Male C57BL/6 mice (8–10 wk old) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were maintained on a standard rodent chow and had free access to water. OA-NO2 was dissolved in 100% DMSO at 1 μg/ml. Mice were pretreated for 48 h with DMSO (LPS vehicle) or OA-NO2 (LPS OA-NO2) at 0.2 mg·kg−1·day−1 via a micro-osmotic pump (DURECT, Cupertino, CA), and then both groups were treated with a single intraperitoneal (ip) injection of LPS at 10 mg/kg (Escherichia coli 0127:B8, Sigma, St. Louis, MO; dissolved in saline). The infusion rate of the pump was 6 μl/day. A third group received an ip injection of saline only and served as controls. Functional studies were done at 18 h after LPS injection. A separate experiment was performed to compare the anti-inflammatory effects of OA-NO2 vs. OA; OA was delivered at the same dose (0.2 mg·kg−1·day−1) via the same route as OA-NO2.
Measurement of body temperature and hematocrit.
Rectal temperature was measured before and 18 h after LPS injection using a digital thermometer. At 18 h after LPS injection, hematocrit was determined as previously described (49). Briefly, 5–10 μl of blood was collected from a tail cut using a 10-μl capillary glass (Idaho Technology). One side of the tube was sealed with Hemato-Seal and then centrifuged for 4 min in a Thermo IEC microcentrifuge. The total height of the sample and height of the red blood cell column were measured. The hematocrit reflects the ratio between the red blood cell column and total height.
Echocardiography.
In vivo cardiac function was assessed using echocardiography. The animals were lightly anesthetized with isofluorane and were imaged in the left lateral decubitus position with a linear 13-MHz probe (Vivid V echocardiograph, General Electric). Digital images were obtained at a frame rate of 180 images/s. Two-dimensional images were recorded in parasternal long- and short-axis projections with guided M-mode recordings at the midventricular level in both views.
Blood pressure measurements.
Mean arterial pressure (MAP) was determined by telemetry. The radiotelemetric device was implanted in mice through catheterization of the carotid artery (model TA11PA-C20, DSI, St. Paul, MN). Animals were allowed to recover from surgery for 1 wk. MAP was recorded 2 days before and 18 h after LPS injection.
Measurement of circulating TNF-α.
Circulating TNF-α was measured by using a commercially available enzyme immunoassay kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Measurement of biochemical parameters.
Blood samples from anesthetized mice were collected by puncturing the vena cava using a 1-ml insulin syringe containing 50 μl of 1 mM EDTA in the absence of protease inhibitors. Plasma levels of both urea and creatinine were measured by spectrophotometry using Fusion 5.1 (Johnson & Johnson) at the University Hospital.
Real-time RT-PCR.
Under isoflurane anesthesia, kidneys were harvested and preserved in RNAlater solution (Ambion, Austin TX). The tissue samples in the RNAlater solution were kept on ice after sampling, stored overnight at 4°C, and kept frozen at −20°C until RNA extraction. Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA), and cDNA was synthesized using SuperScript (Invitrogen). The sequence of oligonucleotides used for real-time PCR (RT-PCR) is listed as follows: TNF-α sense:5′-TCCCCAAAGGGATGAGAAG-3′ and antisense: 5′-CACTTGGTGGTTTGCTACGA-3′; inducible nitric oxide synthase (iNOS) sense: 5′-ACTGTGTGCCTGGAGGTTCT-3′ and antisense: 5′-TCTCTGCCTATCCGTCTCGT-3′; cyclooxygenase 2 (COX-2) sense: 5′-AGGACTCTGCTCACGAAGGA-3′ and antisense: 5′-TGACATGGATTGGAACAGCA-3′; MCP-1 sense: 5′-GCTCTCTCTTCCTCCACCAC-3′ and antisense: 5′-ACAGCTTCTTTGGGACACCT-3′; ICAM-1 sense: 5′-CGCTTCCGCTACCATCAC-3′ and antisense: GGCGGCTCAGTATCCTC-3′; VCAM-1 sense: 5′-GGAGAGACAAAGCAGAAGTGG-3′ and antisense: 5′-AACACAAGCGTGGATTTGG-3′; and β-actin sense: 5′-GCTCTGGCTCCTAGCACCAT-3′ and antisense: 5′-GCCACCGATCCACACAGAGT-3′. RT-PCR was performed using Sybergreen and the ABI Prism 7900 Sequence Detection system. The amplification was carried out for 40 cycles with conditions of 15-s denaturation at 95°C.
Immunoblotting.
The kidney lysates were stored at −80°C until assayed. Protein concentrations were determined using Coomassie reagent. An equal amount of the whole tissue protein (70 μg) was denatured at 100°C for 10 min, separated by SDS-PAGE, and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline (TBS), followed by incubation for 1 h with rabbit polyclonal antibody against COX-2 (Cayman Chemical), or polyclonal rabbit antibody against iNOS (BD Transduction Laboratories, Mississauga, ON). The blots were washed with TBS followed by incubation with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody. Immune complexes were detected using ECL methods. The immunoreactive bands were quantified using the Gel and Graph Digitizing System (Silk Scientific, Tustin, CA).
Measurement of tissue PGE2 content.
The whole kidney was snap-frozen in liquid nitrogen immediately upon collection and then stored at −80°C. The kidney was lysed and homogenized in a buffer containing 0.1 M phosphate, 1 mM EDTA, and 10 μM indomethacin, pH 7.4. Protein concentrations were determined using Coomassie reagent. The tissue PGE2 content was determined using an EIA kit (Cayman Chemical) and normalized by protein concentrations.
Statistical analysis.
Values shown represent means ± SE. Data were analyzed using unpaired t-test or ANOVA followed by a Bonferroni posttest. A P value <0.05 was considered significant.
RESULTS
Body temperature and hematocrit.
The body temperature decreased significantly at 18 h after LPS injection (29.0 ± 0.55 vs. 36.9 ± 0.6°C, P < 0.01) (Fig. 1A). Pretreatment of OA-NO2 for 2 days significantly improved LPS-induced hypothermia (31.76 ± 1.14 vs. 29.0 ± 0.55°C, P < 0.05) (Fig. 1A). LPS is reported to induce erythrocytopenia and leukocytopenia, resulting in a reduction of hematocrit, particularly at the later phase of septic shock (20, 23). We found that at 18 h post-LPS, hematocrit was significantly decreased that was attenuated by pretreatment with OA-NO2 (control: 53.1 ± 2.6%; LPS vehicle: 43.7 ± 1.4%; LPS OA-NO2: 49.5 ± 0.5%).
Fig. 1.
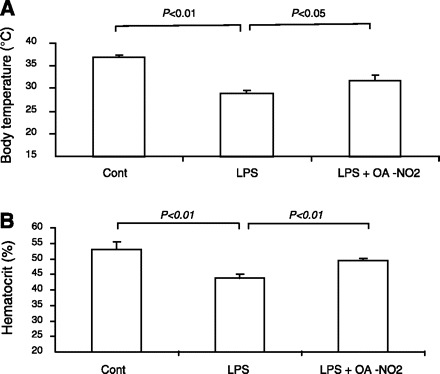
Effect of nitro-oleic acid (OA-NO2) on body temperature and hematocrit in LPS-induced endotoxemia in mice. Male C57/BL6 mice were pretreated with vehicle (LPS vehicle) or OA-NO2 (LPS OA-NO2) at 200 μg·kg−1·day−1 via osmotic minipumps, followed by LPS administration. LPS dissolved in saline was administered at 10 mg/kg via a single intraperitoneal (ip) injection. Body temperature and hematocrit was analyzed 18 h post-LPS. Control mice (Cont) received an ip injection of saline. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Renal function.
Renal function was reflected by plasma blood urea nitrogen (BUN) and creatinine (Fig. 2). LPS injection elevated plasma BUN from 27.3 ± 3.9 to 108.8 ± 3.4 mg/dl (P < 0.05) and plasma creatinine from 0.22 ± 0.03 to 0.34 ± 0.04 mg/dl (P < 0.05). Pretreatment with OA-NO2 attenuated the rise of plasma BUN (95 ± 1.4 vs. 108.8 ± 3.4 mg/dl, P < 0.05) and almost completely normalized plasma creatinine levels (0.23 ± 0.03 vs. 0.34 ± 0.04 mg/dl, P < 0.05). Renal histological changes in response to LPS were not evident as previously reported (13) and thus were not used as a parameter for evaluating the effect of OA-NO2 (data not shown).
Fig. 2.
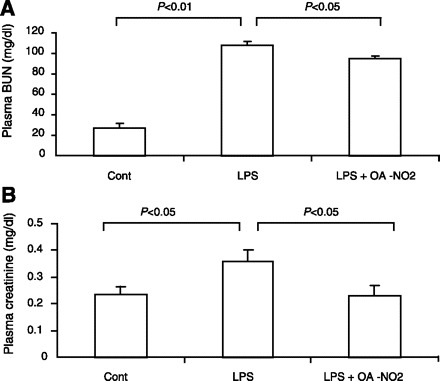
Plasma blood urea nitrogen (BUN; A) and creatinine (B) levels in control, LPS vehicle, and LPS OA-NO2 mice. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Proinflammatory cytokines, chemokines, and adhesion molecules.
TNF-α is a well-established pathogenic factor in the inflammatory responses in sepsis. Therefore, emphasis was placed on the analysis of renal TNF-α mRNA expression and circulating TNF-α. By real-time RT-PCR, renal TNF-α mRNA exhibited a 14-fold increase in LPS vehicle vs. control mice. Compared with LPS vehicle mice, this increase was reduced by 50% in the LPS OA-NO2 animals (Fig. 3A). Similar results were obtained concerning circulating TNF-α as assessed by enzyme immunoassay (Fig. 3B). Additionally, we examined renal mRNA expression of chemokines and adhesion molecules, including monocyte chemoattractant protein-1 (MCP-1), ICAM-1, and VCAM-1. The fold-inductions between LPS vehicle and LPS OA-NO2 groups were 27.1- vs. 9.7-fold for MCP-1, 12.0- vs. 4.8-fold for ICAM-1, and 4.2- vs. 1.8-fold for VCAM-1 (Fig. 4). Taken together, OA-NO2 pretreatment ameliorated the expression of proinflammatory cytokines, chemokines, and adhesion molecules in endotoxic shock.
Fig. 3.
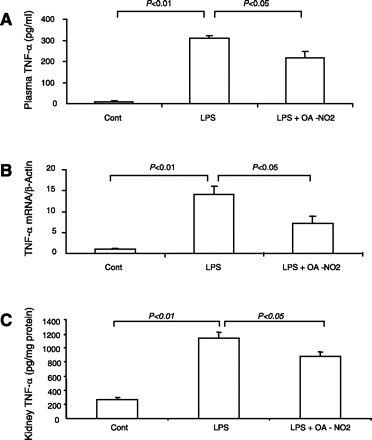
Renal TNF-α mRNA expression and circulating TNF-α levels in control, LPS vehicle, and LPS OA-NO2 mice. Renal TNF-α mRNA expression was determined by qRT-PCR and normalized by β-actin. Plasma TNF-α was measured by ELISA. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Fig. 4.
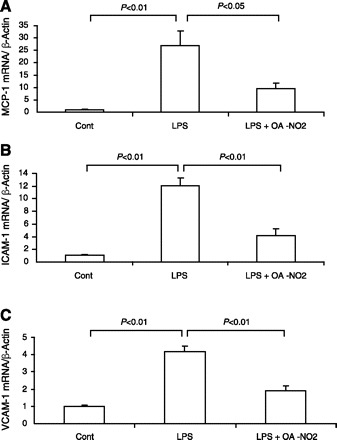
qRT-PCR analysis of renal mRNA expression of monocyte chemoattractant protein-1 (MCP-1; A), ICAM-1 (B), and VCAM-1 (C) in control, LPS vehicle, and LPS OA-NO2 mice. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Renal iNOS expression.
iNOS is highly inducible by proinflammatory stimuli and is a major source of enhanced NO generation in sepsis. Furthermore, iNOS-derived NO has been implicated in the pathogenesis of LPS-induced AKI (45). In the present study, the expression of iNOS was examined at both mRNA and protein levels. At 18 h post-LPS, renal iNOS mRNA exhibited a >1,000-fold increase that was reduced by 80% in LPS OA-NO2 mice (Fig. 5A). Similarly, LPS injection induced a 70-fold increase in renal iNOS protein expression that was reduced by 90% with OA-NO2 (Fig. 5B). Renal cGMP content exhibited a similar pattern of changes as renal iNOS expression (Fig. 5C).
Fig. 5.
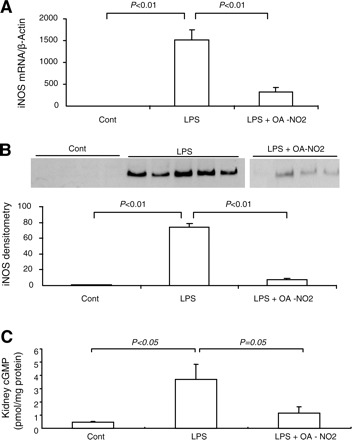
Renal inducible nitric oxide synthase (iNOS) expression in control, LPS vehicle, and LPS OA-NO2 mice. A: quantitative (q)RT-PCR. B: immunoblot analysis (top, immunoblots; bottom, densitometry). C: renal concentration of cGMP. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Renal COX-2 expression.
Like iNOS, COX-2 is highly inducible by proinflammatory stimuli and is another important marker of inflammatory responses in sepsis. In the present study, COX-2 expression was evaluated at mRNA and protein levels as well as at the level of the enzyme activity. Renal COX-2 mRNA and protein were 8.3- and 17.1-fold greater, respectively, in LPS vehicle vs. control mice; they were 2.1- and 8.9-fold greater in LPS OA-NO2 vs. control mice (Fig. 6, A and B). At 18 h post-LPS, renal content of PGE2, a major product of COX-2, exhibited a 1.5-fold increase that was almost normalized by pretreatment with OA-NO2 (Fig. 6C).
Fig. 6.
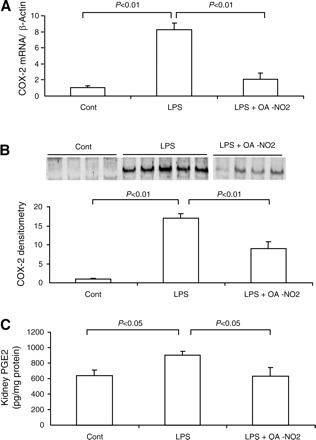
Renal cyclooxygenase 2 (COX-2) expression in control, LPS vehicle, and LPS OA-NO2 mice. A: qRT-PCR analysis of COX-2 mRNA, B: immunoblot analysis of COX-2 protein (top, immunoblots; bottom, densitometry). C: ELISA analysis of kidney PGE2 content. (top, immunoblots; bottom, densitometry). Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Hepatic injury.
LPS injection elevated plasma AST from 176.7 ± 43.3 to 264.8 ± 12.8 U/l (P < 0.05) (Fig. 7A) and plasma ALT from 81.1 ± 2.2. to 99.5 + 3.1 U/l (P < 0.05) (Fig. 7B). Pretreatment with OA-NO2 lowered plasma AST to 207.8 ± 15.9 U/l (P < 0.05) (Fig. 7A) and plasma ALT to 85.6 ± 5.6 U/l (P = 0.051) (Fig. 7B). ELISA and quantitative (q)RT-PCR were subsequently performed to evaluate markers of inflammatory response in the liver. By ELISA, the increase in hepatic TNF-α content was less in LPS OA-NO2 vs. LPS vehicle mice (Fig. 7C). LPS injection induced parallel increases in mRNA expression of hepatic TNF-α, IL-1, ICAM-1, MCP1, iNOS, and COX-2; these increases were all attenuated by OA-NO2 (Fig. 8).
Fig. 7.
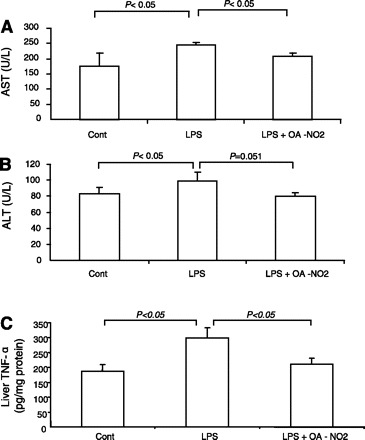
The activities of plasma aspartate aminotransferase (AST; A), alanine aminotransferase (ALT; B), and liver TNF-α (C) in control, LPS vehicle, and LPS OA-NO2 mice. Tissue TNF-α content was determined by ELISA. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Fig. 8.
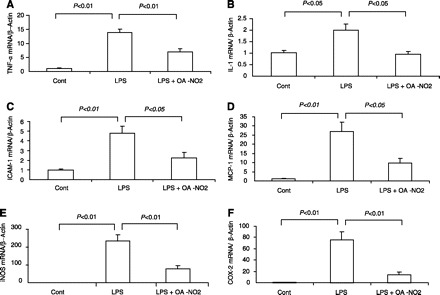
qRT-PCR analysis of hepatic mRNA expression of TNF-α (A), IL-1 (B), ICAM-1 (C), MCP-1 (D), iNOS (E), and COX-2 (F) in control, LPS vehicle, and LPS OA-NO2 mice. Control: n = 4; LPS: n = 8; LPS+OA-NO2: n = 9. Values are means ± SE.
Cardiac injury and hypotension.
Echocardiography revealed that LPS injection significantly reduced the ejection fraction, an index of left ventricular dysfunction, which was almost completely corrected by OA-NO2 (Fig. 9A). Plasma LDH was elevated by LPS injection (P < 0.05) that was attenuated by OA-NO2 (P < 0.05) (Fig. 9B). TNF-α content in the heart tended to show a similar pattern of changes as in the kidney and liver, but no statistical significance was detected between any groups (Fig. 9C). Hemodynamic parameters in LPS vehicle and LPS OA-NO2 mice were monitored using radiotelemetry. LPS-induced hypotension was evident in both groups. MAP in LPS OA-NO2 mice tended to be consistently ∼10 mmHg higher than in LPS vehicle animals (Fig. 10A). The statistical significance between the two groups can be detected by an unpaired t-test but not ANOVA, reflecting a small effect of OA-NO2. Similar results were obtained with heart rate (Fig. 10B).
Fig. 9.
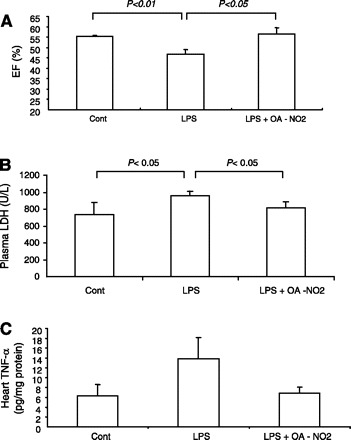
Evaluation of endotoxin-induced cardiac injury in control, LPS vehicle, and LPS OA-NO2 mice. A: echocardiography analysis of ejection fraction (EF). B: plasma LDH. C: qRT-PCR analysis of heart TNF-α. Control: n = 4–5; LPS: n = 8–11; LPS+OA-NO2: n = 9–12. Values are means ± SE.
Fig. 10.
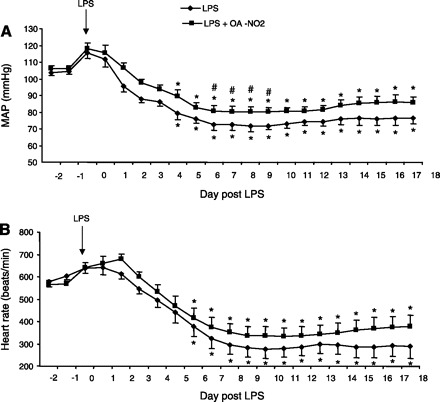
Radiotelemetric determination of mean arterial pressure (MAP; A) and heart rate (B) in LPS vehicle and LPS OA-NO2 mice. The arrow indicates the time of LPS injection. The immediate rise of MAP and heart rate following LPS injection reflects the influence from stress. LPS: n = 8; LPS+OA-NO2: n = 10. Values are means ± SE. *P < 0.05 vs. basal values in the same group. #P < 0.05 vs. LPS alone at the corresponding period.
Effectiveness of OA.
Separate experiments determined whether the protective effect of OA-NO2 in endotoxin-induced endotoxemia was specific to the nitrated (i.e., OA-NO2) or native form (i.e., OA). The indices of systemic inflammation (body temperature and hematocrit) and renal dysfunction (plasma BUN and creatinine) in the endotoxemic mice were significantly attenuated by OA-NO2 but were unaffected by OA (Fig. 11), documenting the lack of anti-inflammatory effect of OA. This finding suggests that the protective effect of OA-NO2 in endotoxic shock is attributable to the nitration of the fatty acid.
Fig. 11.
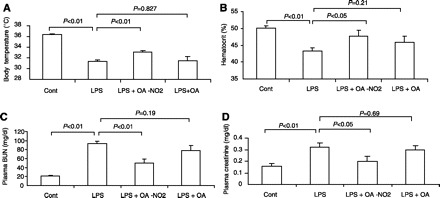
Comparison of the effects of OA-NO2 vs. oleic acid (OA) on body temperature (A), hematocrit (B), plasma BUN (C), and creatinine (D) in endotoxemic C57/BL6 mice; n = 5/group. Values are means ± SE.
DISCUSSION
NO2-FA, notably nitroalkene derivatives of LNO2 and OA-NO2, are naturally occurring products (11, 12). Abundant evidence demonstrates that nitroalkenes exert potent anti-inflammatory actions in vitro. The present study explored the in vivo anti-inflammatory effect of OA-NO2 in a mouse model of LPS administration. LPS is an endotoxin derived from the outer membrane of Gram-negative bacteria, and it is strongly associated with septic shock. Many of the pathophysiological abnormalities of human endotoxic shock can be reproduced in experimental animals by administration of LPS (18). In particular, murine models of LPS administration are frequently used to investigate the pathophysiology and therapies of endotoxin-induced ARF. Here, we report that preventative administration of OA-NO2 attenuated the systemic and local inflammatory responses and improved multiorgan dysfunction in the kidney, liver, and heart in septic C57/BL6 mice.
Endotoxic shock represents a systemic inflammatory response arising from a complicated interplay of mediators of cellular function and inflammation, caused by the release of bacterial constituents, such as LPS. The binding of LPS to Toll-like receptor 4 (TLR4) initiates the release of inflammatory cytokines (14). In patients with ARF, plasma cytokine levels are positively correlated with mortality (42). Among various proinflammatory cytokines induced by LPS, TNF-α is of major importance (31). The pathogenic role of TNF-α in endotoxic renal failure is demonstrated by the observations that administration of TNF-α to intact animals or isolated, perfused kidneys reduced the glomerular filtration rate (5, 44). Along this line, TNF-α neutralization with a TNF-α antibody or a TNF-α-soluble receptor (TNFsRp55) protects against septic shock in baboons (43) and mice (6, 25). More importantly, mice deficient in TNFR1, the major receptor subtype for the inflammatory action of TNF-α, are resistant to LPS-induced ARF (12) despite the lack of improvement of mortality (33, 35); the cross-kidney transplantation between TNFR +/+ and −/− mice further demonstrates that LPS-induced ARF is caused by the direct renal action of TNF-α (12). In the present study, we found that pretreatment with OA-NO2 effectively reduced renal and hepatic TNF-α mRNA expression as well as circulating TNF-α level in septic mice. We suspect that suppression of TNF-α expression may be a primary mechanism responsible for the protective action of OA-NO2 in endotoxic shock. The inhibition of TNF-α appears to be direct since exposure of cultured RAW264.7 cells to OA-NO2 as well as LNO2 significantly reduced the release of TNF-α in response to LPS (11).
LPS administration is well known to trigger an influx of inflammatory cells, namely, neutrophils and macrophages, to various organs, including the liver, lung, and kidney (1, 22, 47). This process is directed by local expression of adhesion molecules and chemokines. For example, the adhesion molecule ICAM-1 is mainly expressed on the endothelial cell surfaces, interacting with ligands present on circulating leukocytes, thereby allowing attachment and adhesion of the leukocytes to the endothelium, a key event of the inflammatory response. The importance of ICAM-1 in sepsis is demonstrated by the observation that mice deficient in ICAM-1 are resistant to LPS-induced ARF (47), with improved mortality (48). We found that pretreatment with OA-NO2 attenuated LPS-induced expression of ICAM-1 as well as VCAM-1 in the kidney or/and liver. We suspect that OA-NO2 may directly target vascular endothelial cells, where it attenuates attachment of circulating leukocytes via suppressing local expression of adhesion molecules. Since the expression of MCP-1 is similarly suppressed by OA-NO2, it is possible that the nitrated lipid may also attenuate macrophage recruitment via suppressing chemokine expression.
Other inflammatory mediators including iNOS and COX-2 are well known to be responsive to proinflammatory stimuli such as LPS. NO is generated by three NOS isoenzymes: endothelial (eNOS), neuronal (nNOS), and iNOS. The involvement of NO in endotoxemic organ failure has been extensively studied, revealing distinct actions of NO derived from different enzymatic sources. eNOS-derived NO protects the cardiovascular system (41), while iNOS-derived NO mediates kidney injury in endotoxic shock (25). In particular, the iNOS inhibitor l-N6-(1-iminoethyl)-lysine and iNOS deficiency protect against entotoxemic renal failure (25, 46). COX exists in two isoforms: constitutive (COX-1) and inducible (COX-2). Like iNOS, COX-2 is highly inducible by proinflammatory stimuli. COX-2 −/− mice are resistant to LPS-induced inflammation and mortality, accompanied by blunted induction of renal iNOS expression (15, 16). The finding suggests that LPS-induced iNOS expression may be mediated by COX-2-derived products. Along this line, we observed a robust induction of iNOS and COX-2 in response to LPS administration in the kidney, which was suppressed in parallel by pretreatment with OA-NO2. To date, there are no reports as to whether nitraolkenes are capable of directly suppressing COX-2 expression in the context of inflammation. Of note, the changes in the cGMP concentration in the kidney correspond to the expression level of iNOS in control, LPS, and LPS + OA-NO2 mice, suggesting that the increased renal cGMP production after LPS administration may be primarily due to iNOS-derived NO and therefore may be detrimental. However, cGMP is renoprotective in other experimental disease settings such as renal ischemia-reperfusion injury and nephrectomy (7, 34). Likely, cGMP can exert distinct roles in renal pathophysiology depending on the coupling to a specific NOS isoform.
Overall, our data suggest that the anti-inflammatory action of nitroalkenes may be attributable to their ability to suppress diverse inflammatory pathways mediated by TNF-α, MCP-1, ICAM-1/VCAM-1, iNOS, and COX-2. Although TNF-α is reported to induce expression of most of these inflammatory mediators, evidence exists to suggest dissociation of TNF-α with iNOS in endotoxemic renal failure in mice (25). In particular, this study showed that TNF neutralization produces a similar degree of renoprotection in endotoxemic iNOS +/+ and −/− mice (25). Despite favorable preclinical elevations, clinical trials attempting to inhibit TNF-α by using either anybodies (8) or fusion proteins composed of the extracellular domain of TNFR1 or TNFR2 have failed to influence sepsis-related mortality (2, 17). The negative clinical trial data may stimulate interest in targeting an inflammatory mediator (s) upstream of TNF-α, such as NF-κB. Indeed, nitroalkenes are reported to inhibit LPS-induced NF-κB activation via nitroalkylation of p65 (11). Moreover, nitroalkenes activate the mitogen-activated protein kinase phosphatase-1, a negative regulator of LPS-induced STAT signaling, resulting in suppression of iNOS and MCP-1 (21). Most likely, the effective anti-inflammatory effect of nitroalkenes may lie in their ability to intervene in the early event of inflammatory signaling.
The present study is associated with a number of limitations. For example, OA-NO2 was administered in a preventative rather than therapeutic way. The latter was not performed due to the consideration that with the slow infusion rate of minipumps, the effective and stable plasma concentrations of OA-NO2 might not be achieved within the short period post-LPS administration. Second, the molecular mechanisms responsible for the anti-inflammatory action of OA-NO2 were not tested. The mechanisms appear to be complex, involving activation of PPARγ (3, 39), release of NO (26, 38), inhibition of NF-κB (11), and regulation of phosphorylation of STAT (21), etc. Dissection of each of the signaling pathways needs to be performed in future studies.
In summary, the present study is the first to evaluate the in vivo anti-inflammatory action of OA-NO2 in a mouse model of LPS administration. Preventative treatment with OA-NO2 attenuated the systemic and local inflammatory responses and improved multiorgan dysfunction in endotoxic mice. The effective anti-inflammatory effect of OA-NO2 appears to be attributable to the suppression of diverse proinflammatory mediators, including cytokines, chemokines, adhesion molecules, iNOS, and COX-2. Together, nitroalkenes may hold promise for the prevention of and possibly therapy for endotoxic shock.
GRANTS
This work was supported by National Institutes of Health Grants HL079453 and DK079162 and a grant from the American Heart Association (to T. Yang). H. Wang was an exchange graduate student and was partially supported by Shandong University. T. Yang is an Established Investigator from the American Heart Association.
DISCLOSURES
No conflicts of interest are declared by the authors.
REFERENCES
- 1. Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 279: L1137–L1145, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Abraham E, Glauser MP, Butler T, Garbino J, Gelmont D, Laterre PF, Kudsk K, Bruining HA, Otto C, Tobin E, Zwingelstein C, Lesslauer W, Leighton A. p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial Ro 45–2081 Study Group. JAMA 277: 1531–1538, 1997 [PubMed] [Google Scholar]
- 3. Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem 280: 42464–42475, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem 281: 20450–20463, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, Remuzzi G. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol 134: 419–430, 1989 [PMC free article] [PubMed] [Google Scholar]
- 6. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229: 869–871, 1985 [DOI] [PubMed] [Google Scholar]
- 7. Choi DE, Jeong JY, Lim BJ, Chung S, Chang YK, Lee SJ, Na KR, Kim SY, Shin YT, Lee KW. Pretreatment of sildenafil attenuates ischemia-reperfusion renal injury in rats. Am J Physiol Renal Physiol 297: F362–F370, 2009 [DOI] [PubMed] [Google Scholar]
- 8. Cohen J, Carlet J. INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit Care Med 24: 1431–1440, 1996 [DOI] [PubMed] [Google Scholar]
- 9. Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, O'Donnell VB. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circ Res 91: 375–381, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O'Donnell VB. Nitrolinoleate inhibits platelet activation by attenuating calcium mobilization and inducing phosphorylation of vasodilator-stimulated phosphoprotein through elevation of cAMP. J Biol Chem 277: 5832–5840, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem 281: 35686–35698, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cunningham PN, Dyanov HM, Park P, Wang J, Newell KA, Quigg RJ. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol 168: 5817–5823, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Cunningham PN, Holers VM, Alexander JJ, Guthridge JM, Carroll MC, Quigg RJ. Complement is activated in kidney by endotoxin but does not cause the ensuing acute renal failure. Kidney Int 58: 1580–1587, 2000 [DOI] [PubMed] [Google Scholar]
- 14. Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol 172: 2629–2635, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Ejima K, Layne MD, Carvajal IM, Kritek PA, Baron RM, Chen YH, Vom Saal J, Levy BD, Yet SF, Perrella MA. Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J 17: 1325–1327, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Ejima K, Perrella MA. Alteration in heme oxygenase-1 and nitric oxide synthase-2 gene expression during endotoxemia in cyclooxygenase-2-deficient mice. Antioxid Redox Signal 6: 850–857, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Fisher CJ, Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, Abraham E, Schein RM, Benjamin E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med 334: 1697–1702, 1996 [DOI] [PubMed] [Google Scholar]
- 18. Haeffner-Cavaillon N, Carreno MP, Aussel L, Caroff M. Molecular aspects of endotoxins relevant to their biological functions. Nephrol Dial Transplant 14: 853–860, 1999 [DOI] [PubMed] [Google Scholar]
- 19. Hoyert D, Arias E, Smith B, Murphy S, Kochanek K. Deaths: Final Data for 1999. Hyattsville, MD: National Center for Health Statistics, National Vital Statistics Reports 49, 2001 [DHHS publication n. (PHS) 2001–1120 PRS 2001–0573, 1999] [PubMed] [Google Scholar]
- 20. Hsu BG, Yang FL, Lee RP, Peng TC, Chen HI. Effects of post-treatment with low-dose propofol on inflammatory responses to lipopolysaccharide-induced shock in conscious rats. Clin Exp Pharmacol Physiol 32: 24–29, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Ichikawa T, Zhang J, Chen K, Liu Y, Schopfer FJ, Baker PR, Freeman BA, Chen YE, Cui T. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology 149: 4086–4094, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamochi M, Kamochi F, Kim YB, Sawh S, Sanders JM, Sarembock I, Green S, Young JS, Ley K, Fu SM, Rose CE., Jr P-selectin and ICAM-1 mediate endotoxin-induced neutrophil recruitment and injury to the lung and liver. Am J Physiol Lung Cell Mol Physiol 277: L310–L319, 1999 [DOI] [PubMed] [Google Scholar]
- 23. Kao SJ, Wang D, Lin HI, Chen HI. N-acetylcysteine abrogates acute lung injury induced by endotoxin. Clin Exp Pharmacol Physiol 33: 33–40, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Klenzak J, Himmelfarb J. Sepsis and the kidney. Crit Care Clin 21: 211–222, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Knotek M, Rogachev B, Wang W, Ecder T, Melnikov V, Gengaro PE, Esson M, Edelstein CL, Dinarello CA, Schrier RW. Endotoxemic renal failure in mice: role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int 59: 2243–2249, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O'Donnell VB, Eiserich JP, Freeman BA. Nitrolinoleate, a nitric oxide-derived mediator of cell function: synthesis, characterization, and vasomotor activity. Proc Natl Acad Sci USA 99: 15941–15946, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H, Jia Z, Soodvilai S, Guan G, Wang MH, Dong Z, Symons JD, Yang T. Nitro-oleic acid protects the mouse kidney from ischemia and reperfusion injury. Am J Physiol Renal Physiol 295: F942–F949, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348: 1546–1554, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Neveu H, Kleinknecht D, Brivet F, Loirat P, Landais P. Prognostic factors in acute renal failure due to sepsis. Results of a prospective multicentre study The French Study Group on Acute Renal Failure. Nephrol Dial Transplant 11: 293–299, 1996 [DOI] [PubMed] [Google Scholar]
- 30. O'Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol 12: 83–92, 1999 [DOI] [PubMed] [Google Scholar]
- 31. Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med 328: 1471–1477, 1993 [DOI] [PubMed] [Google Scholar]
- 32. Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med 113: 227–242, 1990 [DOI] [PubMed] [Google Scholar]
- 33. Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 160: 943–952, 1998 [PubMed] [Google Scholar]
- 34. Rodriguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-kappaB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther 315: 51–57, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364: 798–802, 1993 [DOI] [PubMed] [Google Scholar]
- 36. Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem 269: 26066–26075, 1994 [PubMed] [Google Scholar]
- 37. Russell JA. Management of sepsis. N Engl J Med 355: 1699–1713, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, Patel RP, Hogg N, Branchaud BP, Lancaster JR, Jr, Freeman BA. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem 280: 19289–19297, 2005 [DOI] [PubMed] [Google Scholar]
- 39. Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci USA 102: 2340–2345, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med 351: 159–169, 2004 [DOI] [PubMed] [Google Scholar]
- 41. Schwartz D, Mendonca M, Schwartz I, Xia Y, Satriano J, Wilson CB, Blantz RC. Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats. J Clin Invest 100: 439–448, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simmons EM, Himmelfarb J, Sezer MT, Chertow GM, Mehta RL, Paganini EP, Soroko S, Freedman S, Becker K, Spratt D, Shyr Y, Ikizler TA. Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int 65: 1357–1365, 2004 [DOI] [PubMed] [Google Scholar]
- 43. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 330: 662–664, 1987 [DOI] [PubMed] [Google Scholar]
- 44. van der Veen AH, Seynhaeve AL, Breurs J, Nooijen PT, Marquet RL, Eggermont AM. In vivo isolated kidney perfusion with tumour necrosis factor alpha (TNF-alpha) in tumour-bearing rats. Br J Cancer 79: 433–439, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang W, Jittikanont S, Falk SA, Li P, Feng L, Gengaro PE, Poole BD, Bowler RP, Day BJ, Crapo JD, Schrier RW. Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am J Physiol Renal Physiol 284: F532–F537, 2003 [DOI] [PubMed] [Google Scholar]
- 46. Wu L, Mayeux PR. Effects of the inducible nitric-oxide synthase inhibitor l-N6-(1-iminoethyl)-lysine on microcirculation and reactive nitrogen species generation in the kidney following lipopolysaccharide administration in mice. J Pharmacol Exp Ther 320: 1061–1067, 2007 [DOI] [PubMed] [Google Scholar]
- 47. Wu X, Guo R, Wang Y, Cunningham PN. The role of ICAM-1 in endotoxin-induced acute renal failure. Am J Physiol Renal Physiol 293: F1262–F1271, 2007 [DOI] [PubMed] [Google Scholar]
- 48. Xu H, Gonzalo JA, St Pierre Y, Williams IR, Kupper TS, Cotran RS, Springer TA, Gutierrez-Ramos JC. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med 180: 95–109, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci USA 102: 9406–9411, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]