

Abstract
In our analysis of a quantitative trait locus (QTL) for plasma triglyceride (TG) levels [logarithm of odds (LOD) = 3.7] on human chromosome 7q36, we examined 29 single nucleotide polymorphisms (SNPs) across INSIG1, a biological candidate gene in the region. Insulin-induced genes (INSIGs) are feedback mediators of cholesterol and fatty acid synthesis in animals, but their role in human lipid regulation is unclear. In our cohort, the INSIG1 promoter SNP rs2721 was associated with TG levels (P = 2 × 10−3 in 1,560 individuals of the original linkage cohort, P = 8 × 10−4 in 920 unrelated individuals of the replication cohort, combined P = 9.9 × 10−6). Individuals homozygous for the T allele had 9% higher TG levels and 2-fold lower expression of INSIG1 in surgical liver biopsy samples when compared with individuals homozygous for the G allele. Also, the T allele showed additional binding of nuclear proteins from HepG2 liver cells in gel shift assays. Finally, the variant rs7566605 in INSIG2, the only homolog of INSIG1, enhances the effect of rs2721 (P = 0.00117). The variant rs2721 alone explains 5.4% of the observed linkage in our cohort, suggesting that additional, yet-undiscovered genes and sequence variants in the QTL interval also contribute to alterations in TG levels in humans.
Keywords: triglyceride, INSIG2, SNP association, gene expression, EMSA
Increased plasma triglyceride (TG) levels are an important cardiovascular risk factor and are strongly associated with atherosclerotic heart disease (1, 2). Plasma TG levels vary widely between individuals, and both genetic and environmental factors have been shown to contribute to elevated plasma TG concentrations (3–7). Elevated plasma TG levels are often observed in obese and diabetic individuals and in individuals affected by the metabolic syndrome, a common chronic disorder associated with obesity, insulin resistance, hypertension, and alterations in plasma lipid profile such as elevated serum TG and low HDL levels. This characteristic pattern is similar to lipid abnormalities reported in familial combined hyperlipidemia (8).
The Metabolic Risk Complications of Obesity Genes (MRC-OB) project was established in 1994 to identify the genetic determinants of the metabolic syndrome and its metabolic abnormalities (9). As part of the project, basic anthropomorphic phenotypes, plasma lipid measures, and fasting glucose and insulin levels were ascertained for 2,209 individuals from 507 families. A genome-wide linkage scan of these families identified a quantitative trait locus (QTL) on human chromosome 7q36 linked to plasma TG levels (LOD = 3.7) (10). This region has also been implicated in numerous other studies (11–15) and represents one of the most replicated linkages for dyslipidemia and elevated TG levels. The genomic interval on chromosome 7q36 includes 22 known genes, including insulin-induced gene 1 (INSIG1), the only biological candidate.
INSIG1 was initially described as an insulin-induced transcript, designated CL-6 (16). It was later renamed INSIG1 following cloning and assignment to chromosome 7q36 (17). INSIG1 has been shown to affect the feedback regulation of cellular cholesterol (18, 19). Sterol regulatory element binding proteins (SREBPs), which are transcription factors located in the endoplasmic reticulum, control the synthesis of cholesterol and fatty acids. When cellular cholesterol levels fall, SREBPs bind to SREBP cleavage-activating proteins (SCAPs) to be transported to the Golgi, where they undergo proteolytic cleavage (20). The NH2-terminal fragment translocates to the nucleus to activate the transcription of target genes, including INSIG1 (21), that are involved in the synthesis of cholesterol and fatty acids. As cellular sterol levels rise, SCAP binds to INSIG1, thus preventing the transport of the SCAP/SREBP complex to the Golgi (18) and halting the synthesis of cholesterol and fatty acids [for detailed reviews, see (22, 23)]. INSIG1 and its only mammalian homolog, INSIG2, may also exert their feedback regulators of cholesterol synthesis by directly binding to 3-hydroxy-3-methylglutaryl CoA reductase, which triggers sterol-accelerated ubiquitination and degradation of 3-hydroxy-3-methylglutaryl CoA reductase, the rate-limiting enzyme of cholesterologenesis [for a detailed review, see (24)].
Overexpression of INSIG1 has been shown to reduce high levels of TG in both liver and plasma of Zucker diabetic fatty rats (25). It has been proposed that this effect is due to the mechanism outlined above (18). In addition, single knockout of either INSIG1 or INSIG2 in mice leads to increased total content of both cholesterol and TG in mouse livers, an effect exacerbated significantly when both INSIG genes are knocked out (26). These studies indicate that INSIG1 and INSIG2 interactively contribute to the control and regulation of lipid homeostasis in animals.
Based on this evidence, we hypothesized that sequence variation in INSIG1, which is located in the QTL region on human chromosome 7q36 linked to plasma TG levels, contributes to the observed genetic effect on plasma lipid levels. Here we present data on our comprehensive association analysis of sequence variants in INSIG1 in the initial MRC-OB cohort and a second replication cohort of unrelated individuals, and evidence for a synergistic effect of an associated sequence variant in INSIG1 with a variant in INSIG2 on plasma TG levels. Furthermore, our data suggest that a putative causal promoter variant in INSIG1 affects gene expression of INSIG1 in liver tissue, potentially by altering the binding of nuclear factors to the promoter sequence.
METHODS
Study design
We performed a family-based study on genetic contributions to plasma TG levels.
Study cohort.
Cohort recruitment and individual phenotyping have been described in detail previously (9, 10). In brief, families with at least two obese siblings [body mass index (BMI) >30], the availability of at least one parent, and one or more never obese siblings (BMI <27) were recruited from the Take Off Pounds Sensibly, Inc. membership in 10 Midwestern U.S. states. Health information of all participants was obtained by a questionnaire. Individuals with the following conditions were excluded from recruitment: pregnancy; type 1 diabetes; history of cancer, renal, or hepatic disease; severe coronary artery disease; substance abuse; corticosteroids or thyroid dosages above replacement dose; history of weight loss of >10% in the preceding 12 months; and individuals receiving lipid-lowering medications. Phenotypic measurements included height, weight, waist and hip circumferences, and fasting plasma levels of glucose, insulin, total cholesterol, LDL-cholesterol (LDL-C), HDL-cholesterol, and TG. A total of 2,209 individuals distributed over 507 families of Northern European descent qualified for the above-mentioned criteria and thus formed the initial study population, of which 1,560 individuals from 261 families that contributed to the linkage on chromosome 7q36 (family LOD >0) were selected for further studies reported here.
Confirmatory studies were performed from a cohort of unrelated individuals of Northern European descent recruited by the same criteria from the same geographic regions. Samples from 920 unrelated individuals were available for this study. All protocols were approved by the Institutional Review Board of the Medical College of Wisconsin, and all participants signed an informed consent. Details on the two cohorts used in this study are included in Table 1.
TABLE 1.
Summary statistics for the cohorts used in this study, the original 1,560 individuals of the MRC-OB family cohort and 920 unrelated individuals of the replication cohort
| MRC-OB Family Cohort |
Replication Cohort |
|||
|---|---|---|---|---|
| Average | Range | Average | Range | |
| n | 1560 | 920 | ||
| % female | 71 | 80 | ||
| Age (years) | 46 | 18–90 | 54 | 18–89 |
| BMI (kg/m2) | 31.9 | 17.1–75.3 | 32.6 | 15.9–86.1 |
| TGs (mg/dl) | 128.0 | 27–457 | 135.8 | 36–424 |
| LDL-C (mg/dl) | 106.8 | 13–245 | 131.0 | 33–251 |
Measurements
Genotyping.
Single nucleotide polymorphisms (SNPs) were genotyped using Invader technology as described for previous studies (27–30). Genotyping was carried out in a total volume of 6 μl, containing 0.5 μl of PCR product, 0.02 μl of each primary probe, 0.002 μl of Invader probe, 1.12 μl of 2.6 M betaine (Sigma, St. Louis, MO), 2.75 μl TE buffer, 0.35 μl Cleavase (TWT), and 1.24 μl FRET mix (TWT). Reactions were denatured at 95°C for 5 min and then heated at 63°C for 15–120 min. Additional SNPs were genotyped on an Affymetrix MegAllele custom-designed 3K array using molecular inversion probe technology, as described previously (28). The location of all SNPs included in the analysis is shown in Fig. 1.
Fig. 1.

Schematic illustration of the INSIG1 gene region. All SNPs investigated across the gene region (Chr7:154,696,933 bp–154,736,613 bp) are shown relative to the INSIG1 gene, depicted by the black line. Translated exons are shown by white boxes, untranslated exonic regions are indicated by gray boxes. The associated promoter SNP rs2721 is highlighted by the red box. The colored diagram depicts the pairwise linkage disequilibrium between SNPs (D'), as calculated for the family-based cohort of 1,560 individuals using the program Haploview.
Sequencing and SNP identification.
A 4,139 bp region was resequenced in 47 unrelated individuals selected from the MRC-OB cohort. The region was amplified by PCR using 21 overlapping amplicons 261–444 bp in size. PCR products were purified using MultiScreen-FB plates (Millipore) and eluted in sterile water for sequencing. Sequencing was carried out using BigDye v.3.1 chemistry (Applied Biosystems, Foster City, CA) with standard techniques and analyzed on an Applied Biosystems 3730xl DNA Analyzer. SNP loci were identified by aligning all sequences using POLYPHRED (31), and all putative sites were checked manually for confirmation. Results were independently confirmed by aligning all traces with the chromosome 7 reference sequence using the anchored alignment algorithm in POLYBAYES (32) and using POLYPHRED to screen for SNPs.
Expression profiling.
RNA isolation, anti-sense RNA generation, and Illumina BeadChip hybridization of the liver samples as well as transcript detection and normalization were performed as previously described (33). Briefly, a liver biopsy was obtained from each of 73 morbidly obese subjects of Northern European descent undergoing bariatric surgery. RNA was extracted and analyzed using Illumina Human-6 Expression BeadChips, which allow interrogation of almost 48,000 transcripts. Gene expression data for all liver samples were analyzed according to Goring et al. (33). To identify transcripts with detectable quantitative expression in the samples, distribution of expression values for a given transcript were compared with those of the controls imbedded in each chip. All significantly expressed transcripts were identified using an FDR of 0.05. To minimize the influence of overall signal levels, which may reflect RNA quantity and quality rather than a true biological difference between individuals, abundance values of all transcripts were standardized by z-scoring within individuals (using decile percentage bins of transcripts, grouped by average log-transformed raw signals across individuals), followed by linear regression against the individual-specific average log-transformed raw signal and its squared value, as discussed by Goring et al. (33).
For the analysis of allele-specific expression differences in INSIG1, individuals were grouped according to genotype (for rs2721), and their gene-specific normalized expression z-scores were compared for genotype-specific differences using ANOVA. Differences between individual genotypes were compared using t-tests.
EMSA.
The human HepG2 cell line was maintained in DMEM, supplemented with 2 mM L-glutamine, 100 mg/ml each of streptomycin and penicillin, and 15% fetal bovine serum, at 37°C with 5% CO2. Complementary oligonucleotides representing both allelic forms of the INSIG1 promoter SNP (rs2721, G>T at −2253 to the start codon) were obtained commercially (Integrated DNA Technologies) and purified by HPLC. Electrophoretic mobility shift assay (EMSA) was performed as described previously (34). Briefly, nuclear extracts were prepared from approximately 8 × 107 cells. Extracts were frozen in liquid N2 and stored at −80°C. For EMSA, nuclear proteins (3 μg) were pre-incubated for 10 min on ice with 1 μg of poly (dI-dC) (Pharmacia) in a binding buffer [4% Ficoll, 20 mM HEPES (pH 7.9), 1 mM EDTA, 1 mM DTT, 50 mM KCl]. Nuclear proteins were then incubated with biotin-labeled double-stranded oligonucleotide probes (20 fmol) for 30 min on ice and then analyzed on 6% Novex® DNA retardation gels (Invitrogen, CA) and electro blotted onto a positively charged nylon membrane (Ambion, Austin, TX). Detection of protein/DNA complexes was achieved following incubation of the membrane with strepavidin-horseradish peroxidase and development with luminal substrate (Lightshift Chemiluminescent EMSA kit; Pierce, IL). Light emission was captured on X-ray film.
Statistical analysis
Initial association analysis discovered rs2721.
As TG levels are continuously distributed, the data were first examined for deviations from normality. Raw TG levels exhibited an increased number of high values, so the data were natural log (ln) transformed. Data were re-examined and observations exceeding 4 SD units were removed as outliers. In all models, TG were adjusted for age, sex, age by sex, age squared, age squared by sex, and type 2 diabetes status.
To test for statistical association with SNPs in INSIG1, we identified 11 tagSNPs in the INSIG1 gene region (∼40 kb) from the Hapmap database using the tagger function in Haploview with default settings (35, 36). After sequencing the promoter and the first exon of INSIG1 in 47 unrelated individuals, we identified an additional 17 SNPs. All 28 SNPs were genotyped in the full cohort (n = 1560) and were tested for association with ln(TG).
For each SNP, we used the measured genotype approach to test for association. Briefly, in this approach, the effects of the individual SNP genotypes will be modeled by assigning genotypic values such that the homozygotes will be assigned values of 1 and –1 and the heterozygotes will be assigned 0, providing an additive model (37). To account for the phenotypic correlation between family members, we used variance components analysis with the SNPs screened individually as covariates in the computer program SOLAR (38).
Confirmatory association analysis in unrelated individuals.
To confirm associations identified in the family-based analyses, we genotyped SNP rs2721 in a cohort of 920 unrelated individuals. Genotypes were coded as described above and were considered the independent variable. TG were ln transformed to improve normality of the distribution. As this was an unrelated cohort, we performed linear regression analysis using SAS v9.1 (SAS, Carey NC).
Meta-analysis of both cohorts.
To assess the combined effect of rs2721 in both cohorts, we combined the P-values using the Z-transformation. This approach is detailed by Whitlock et al. (39) and implemented in the Util program (http://www.genemapping.cn/util.htm).
Interaction analysis with INSIG2.
To determine whether there was an interaction between INSIG1 and INSIG2, as suggested from mouse knockout studies, we first examined the relationship between the family specific LOD scores for TG at the two loci (40). Second, we genotyped two SNPs in INSIG2 (rs7566605 and rs2422166) that had been reported to be associated with BMI and LDL-C in other studies (41–43) and tested for both main effects and interaction effects using the measured genotype analysis in SOLAR. Third, we examined the impact of INSIG1 variants conditional on INSIG2 variants. These analyses were performed by subsetting the data by INSIG2 genotype and then performing measured genotype analysis for the effect of INSIG1 genotype.
RESULTS
Association of INSIG1 sequence variants with TG in the MRC-OB cohort
The QTL on chromosome 7q35-q36 linked to plasma TG levels has been replicated in multiple cohorts (10–15). Among the 22 identified genes in this QTL, INSIG1 is the only candidate with known biological roles that are related to lipid metabolism and therefore has been the focus of this study.
Initially, we tested for INSIG1 association using 11 SNPs selected from the Hapmap database using the tagger function of Haploview (35, 36) (Fig. 1 and Table 2). These SNPs spanned the entire INSIG1 gene region (39.7 kb). One variant in the putative promoter region of INSIG1, rs2721, was significantly associated with plasma TG levels in 1,560 individuals from our cohort (P = 0.002). Two additional SNPs (rs9770068 and rs9769506) located in intron 6 of INSIG1 showed marginal association (P = 0.047 and P = 0.037, respectively), as well as one additional SNP in the promoter region (rs9690040, P = 0.045). As these three marginally associated SNPs are in almost complete linkage disequilibrium with rs2721 (Fig. 1), we speculate that their high degree of correlation may have led to the observed associations.
TABLE 2.
Summary of SNP association results
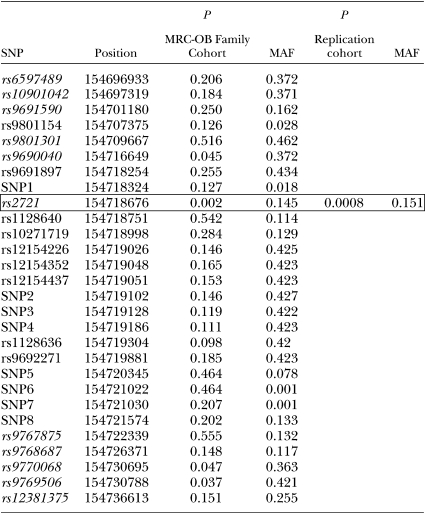 |
The promoter variant (rs2721) is located 2,253 bp upstream of the transcription start site of INSIG1. To include all other variants surrounding rs2721 in the association analysis, we resequenced the promoter region and exon 1 in 47 unrelated individuals from our cohort. SNPs in the remaining exons of the gene were in high linkage disequilibrium (LD) with each other and not in LD with rs2721; thus, we focused solely on the region surrounding rs2721. All selected individuals were parents from families contributing significantly to the initially observed linkage on chromosome7 in the MRC-OB cohort. The resequencing effort uncovered 17 additional SNPs, of which 8 were novel and not reported in dbSNP. The location of all SNPs is shown in Fig. 1.
All 28 SNPs were genotyped in the 1,560 individuals from the MRC-OB cohort. The results of the association analysis are summarized in Table 2. No additional SNPs were associated with plasma TG levels in this cohort, suggesting that the effect is primarily mediated by rs2721. The TG levels of individuals homozygous for the G allele (123.1 ± 2.3 mg/dl, n = 1073) were on average 9% lower than those of individuals homozygous for the T allele (135.9 ± 16.2 mg/dl, n = 31).
Next, we examined the effect of the SNP on the initial linkage. Overall, the QTL interval on chromosome 7q36 explains 31% of the variance in TG levels in our cohort. When including SNP rs2721 in INSIG1 as a covariate, the linkage drops 5.4% for our family-based cohort, suggesting a modest but significant effect on the initial linkage on chromosome 7q36 that explains 1.6% of the total observed variance in TG levels in the study cohort.
Replication of association analysis
To validate the association seen in the family-based MRC-OB cohort, we examined the effect of the associated variant rs2721 in a second study cohort. The details of the cohort are summarized in Table 1. A total of 920 individuals were genotyped and included in the analysis. The minor allele frequency (MAF) of rs2721 in this cohort is comparable to the MAF of rs2721 in the initial family-based cohort (15.1% vs. 14.5%). Again, rs2721 was strongly associated with plasma TG levels (P = 8 × 10−4), confirming the finding in our initial family-based cohort. In this replication cohort, individuals homozygous for the T allele of rs2721 (n = 21) had mean TG levels of 150.3 ± 18.8 mg/dl, compared with 135.9 ± 2.9 mg/dl for individuals homozygous for the G allele (n = 674).
When both datasets are combined using a Z-transformation (39), rs2721 is associated with altered TG levels with a P-value of 9.9 × 10−6.
Functional analysis of rs2721
To assess whether rs2721 may have a direct functional effect, we examined whether the sequence variant is correlated with INSIG1 expression levels, because the SNP is located in the promoter region of the gene. We examined mRNA expression levels of INSIG1 in 73 liver biopsy samples from morbidly obese patients (BMI > 36). All individuals were genotyped for rs2721, and average gene expression levels were compared within different genotype groups. The T allele reduced INSIG1 gene expression levels significantly. In liver, individuals with TT genotype (n = 2) had 2-fold lower levels of INSIG1 mRNA compared with those with TG (n = 23, P = 0.046) or GG types (n = 48, P = 0.030) (mean z-score difference of 0.52, P = 0.02). INSIG1 expression in heterozygous individuals was not significantly different from GG homozygous individuals (P = 0.40).
Based on this observed correlation between INSIG1 gene expression and rs2721 genotype, we investigated whether the two alleles may have differential affinities to nuclear proteins such as transcription factors, which may change the transcription levels that eventually lead to the altered lipid profiles. We performed EMSA using biotin-labeled, double-stranded DNA (41 bp) surrounding SNP rs2721 (20 nucleotides both upstream and downstream). As shown in Fig. 2, the presence of the T allele in the promoter DNA leads to a shift in observed bands after binding of nuclear extracts from HepG2 cells compared with the G allele. This shift is not seen in control experiments without nuclear extract or biotinylated probes. These results show that the two alleles of rs2721 have different affinities to nuclear proteins. While the protein(s) binding to the sequence surrounding rs2721 are currently unknown, it is possible that this differential binding mediates the observed difference in INSIG1 gene expression and thereby affects plasma TG levels in individuals homozygous for the T allele.
Fig. 2.

EMSA of rs2721 using nuclear extract of liver HepG2 cells. Oligos of DNA sequences from INSIG1 promoter region centered with either allele of rs2721 (T or G) were incubated with total nuclear proteins from cultured liver cells. Arrows point at mobility shift of oligonucleotides with the T allele caused by differential protein binding. Results are representative of at least two independent experiments.
Interaction between INSIG1 and INSIG2
In humans, the INSIG1 protein is 78% similar to its only homolog, INSIG2. Interestingly, while INSIG1 is involved in the feedback regulation of lipid synthesis, INSIG2 has been shown to be associated with obesity and adipocyte metabolism in a number of recent studies (41–43). However, no study to date has addressed whether these two genes work cooperatively in humans to affect lipid levels, as has been shown in mice (26).
Correlation analysis based on family-specific LOD scores of the locus of INSIG1 and the locus of INSIG2 suggested a nonadditive interaction between the two genes (ρ = −0.239, P < 0.0001). Based on these results, we selected two SNPs in INSIG2 (rs2422166 and rs7566605) that have been repeatedly shown to be associated with BMI and LDL-C in other studies (41–43). Gene-gene interaction analysis was performed on the two INSIG2 SNPs and rs2721 of INSIG1. In this analysis, SNP rs2422166 did not show any effect on TG levels directly or through interaction with rs2721. In contrast, SNP rs7566605 showed marginal interaction with rs2721 (P = 0.0473) although it was not directly associated with TG levels. To measure the impact of genotype at rs7566605 on the association of rs2721 with lipid traits, we grouped the data based on genotype at rs7566605. Measured genotype analysis was then performed in these subsets (Table 3). In the GG subgroup, the associations with TG levels disappeared. In contrast, in the CC-CG subgroup, the association was significantly strengthened (β = 0.13 ± 0.04, P = 0.00117). Here, individuals homozygous for the T allele of rs2721 had 31% higher TG levels than individuals homozygous for the G allele (138.4 ± 1.9 mg/dl vs. 105.6 ± 1.7 mg/dl). Before we stratified the study cohort based on genotypes of INSIG2 rs7566605, the difference between the INSIG1 rs2721 homozygous groups was only 8%.
TABLE 3.
Interaction analysis of rs7566605 in INSIG2 and rs2721 in INSIG1. The C-allele of rs7566605 significantly enhances the association of rs2721 with plasma TG levels
| rs7566605 | rs2721 | TG (mg/dl) |
|---|---|---|
| CC and CG (n = 763) | GG | 105.6 ± 1.7 |
| GT | 117.9 ± 1.8 | |
| TT | 138.4 ± 1.9* | |
| GG (n = 768) | GG | 107.8 ± 1.7 |
| GT | 108.9 ± 1.8 | |
| TT | 98.5 ± 1.6 |
* = significant difference when compared with GG of rs2721 (P = 0.00117).
DISCUSSION
Studies conducted in animals have revealed intriguing and critical roles of INSIG genes in lipid homeostasis (26). By anchoring a sterol-sensing retainer protein that is able to block the access to the nucleus of a number of transcription factors, INSIG proteins have been recognized as key regulators of both cholesterol and fatty acid synthesis (18, 19, 21). No study, however, has shown evidence of their proposed roles in lipid homeostasis in humans. Based on the fact that the gene for INSIG1 is located in a QTL region linked to plasma TG levels in our MRC-OB cohort, we examined the role of sequence variants in the gene in the modulation of plasma TG levels.
Our initial association analysis using 11 tagSNPs revealed four SNPs associated with TG levels. Further analysis of additional sequence variants uncovered by resequencing suggested the promoter SNP rs2721 as the likely causal variant for the observed hyperlipidemia. Genotype effect assessment showed that people homozygous for the T allele at rs2721 had significantly elevated plasma TG levels. The association was replicated in a second independent cohort, detecting a similar effect in individuals homozygous for the T allele. These data suggest that rs2721 in INSIG1 may be causally associated with TG levels in our linkage region. However, our analysis also suggests that this variant accounts for only a portion of the overall linkage (∼5%), suggesting that other variants and genes in the QTL interval also affect plasma TG levels. Overall, variance at rs2721 explains 1.6% of the observed variance in TG levels in our family cohort. This contribution of this polymorphism, however, may be underestimated, because the interaction with other genes and beyond INSIG2 locus have not been considered in this study.
The SNP rs2721 resides 2,253 bp upstream of the start codon of INSIG1 in the putative promoter region. To test the genotype-specific effect on the transcription of INSIG1 in humans, expression profiling was conducted in liver tissue, the putative target tissue for INSIG1 action on lipid homeostasis in mice. Our analysis revealed that individuals homozygous for the T allele had significantly lower INSIG1 expression levels, suggesting that reduced expression of INSIG1 increases circulating plasma TG levels. While the expression analysis revealed only two individuals homozygous for the T allele in our sample set of 73 individuals, the data nonetheless provide suggestive evidence for an effect of this promoter variant on gene repression in vivo. Additional samples and tissues will need to be investigated to further substantiate this effect of rs2721.
To further validate this potential effect of SNP rs2721 on the promoter function of INSIG1, we examined the binding of nuclear proteins to the sequence surrounding rs2721 by EMSA. Separate custom oligonucleotides specific for the alleles of rs2721 and comprising the immediate sequence surrounding the SNP location were designed. Oligonucleotides specific for the T allele were able to bind nuclear proteins extracted from HepG2 cells, in contrast to the G allele (Fig. 2). These data demonstrate that the T allele of rs2721 shows differential binding of nuclear proteins in vitro, further supporting the hypothesis that this sequence variant affects promoter function of INSIG1. Analysis of the sequence using TFBIND (http://tfbind.ims.u-tokyo.ac.jp/) reveals that rs2721 is located in a putative binding site for the transcription factor PreB-cell leukemia homeobox 1 (PBX1), which is expressed in the liver and a wide variety of other tissues. The binding site is altered in the G allele, reducing the probability of PBX1 binding more than 6-fold over the T allele. Additional transcription factors with differential binding affinity to the two alleles of rs2721 include Forkhead transcription factor and Caudal type homeobox A. This differential binding affinity may explain the observed EMSA results and may be related to the observed allele-specific expression difference, although none of the transcription factors have been reported to modulate plasma TG levels. Nonetheless, PBX1 has been reported to affect glucose tolerance and steroidogenesis, and it has been suggested that mutations in PBX1 may promote susceptibility to diabetes (44). However, at this point, the specific proteins mediating this effect (binding to the T allele, reducing expression of INSIG1, and thereby elevating plasma TG levels) remain to be identified. While the EMSA results, in conjunction with the expression analysis of the liver biopsy samples, suggest a functional mechanism for rs2721, it is clear that EMSA results do not prove functional interactions, and additional molecular studies using transfection assays may be required to confirm the initial findings.
It has been shown in animal models that double knockout animals for INSIG1 and its only homolog INSIG2 have more significant lipid abnormalities compared with knockout animals for either gene individually (26). We therefore explored the putative interactive effect of sequence variants in both human INSIG1 and INSIG2 on plasma lipids. By focusing on the functional SNP rs2721 of INSIG1, we tested the gene-gene interaction with two INSIG2 variants that have shown repeatedly to be associated with BMI and lipid abnormalities in obesity studies. Based on our analysis, INSIG1 does interact with INSIG2 in relation to lipid homeostasis. The two INSIG2 SNPs have no independent effect on plasma TG levels in our cohort. However, rs7566605 significantly enhances the effect seen with rs2721 of INSIG1, and their interaction strengthened the association with plasma TG levels (Table 3). Specifically, people possessing the GG genotype at rs7566605 of INSIG2 no longer showed a detectable effect of the TT genotype of rs2721 in INSIG1 on TG levels. In contrast, individuals with at least one C allele at rs7566605 of INSIG2 showed an enhanced effect of the T allele of rs2721 on TG. These data suggest that INSIG2 (or at least the G allele of rs7566605) may be able to compensate for the effect of rs2721 in INSIG1. While the exact mechanism remains to be elucidated, our analysis overall clearly suggests that rs2721 affects plasma TG levels by lowering INSIG1 gene expression in the liver. This effect can be reduced or enhanced by the different alleles of rs7566605 in INSIG2.
Clinically, a reduction of 10 mg/dl in plasma TG levels, independently of the impact of treatment on LDL-C levels, resulted in a 1.4% reduction in risk of developing coronary heart disease in a report by Miller M et al. (45). In our study, the elevation in TG levels in individuals with the TT genotype of rs2721 was 12.8 mg/dl and 14.4 mg/dl for families and unrelated individuals, respectively, when the population was not stratified by genotypes of INSIG2 at rs7566605. This effect is further exacerbated by the cooccurrence of the C allele of rs7566605 in INSIG2 and the TT allele of rs2721, resulting in a 32.8 mg/dl elevation in TG levels.
Despite the replication of the linkage finding on chromosome 7q36, all genome-wide association studies (GWAS) to date exploring lipid traits have failed to detect any significant association in this genomic interval. Neither INSIG1 nor any other gene in the QTL region has been implicated in any of the lipid trait-related GWAS efforts. Given the low linkage disequilibrium between rs2721 and any of the adjacent SNPs on common GWAS platforms, it is not surprising why the association of rs2721 with plasma TG levels detected in our analysis was not uncovered by GWAS. However, the complete lack of association evidence for the entire genomic interval, despite repeated linkage evidence, needs to be investigated further. This effect has been reported before and is not specific to this QTL region or lipid traits (46).
The lack of comparable findings in other studies may also be due to differences in the recruitment of the study cohorts. In our study design, we excluded individuals with hepatic disease, severe coronary artery disease, dramatic weight loss in the preceding 12 months, or individuals taking lipid-lowering medications. Given the suggested function of the INSIG genes, these criteria may themselves be associated with INSIG function and thus may have a potential influence on the results. Likewise, it remains to be seen whether the expression profiles obtained from liver samples collected from bariatric surgery patients reflect the levels of INSIG1 expression in the general population.
While INISG1 is not the only gene and rs2721 not the only sequence variant responsible for the initially observed linkage on human chromosome 7q36, our analysis, for the first time, demonstrates a significant role for this gene in the control and regulation of plasma TG levels and suggests a functional mechanism by which the causal variant rs2721 may exert its effect. Further QTL-wide analyses will be required to elucidate the other remaining genetic factors affecting plasma TG levels in our cohort and validate the effect on this important cardiovascular disease risk factor in other patient cohorts.
Footnotes
Abbreviations:
- BMI
- body mass index
- EMSA
- electrophoretic mobility shift assay
- GWAS
- genome-wide association studies
- INSIGs
- insulin-induced genes
- LD
- linkage disequilibrium
- LDL-C
- LDL-cholesterol
- ln
- natural log
- LOD
- logarithm of odds
- MAF
- minor allele frequency
- MRC-OB
- Metabolic Risk Complications of Obesity Genes project
- PBX1
- PreB-cell leukemia homeobox 1
- QTL
- quantitative trait locus
- SCAPs
- SREBP cleavage-activating proteins
- SNP
- single nucleotide polymorphism
- SREBP
- sterol regulatory element binding protein
- TG
- tri-glyceride
This work was supported by grant HL74168 from the Heart, Lung, and Blood Institute of the National Institutes of Health (M.O.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies.
REFERENCES
- 1.Forrester J. S.2001. Triglycerides: risk factor or fellow traveler? Curr. Opin. Cardiol. 16: 261–264 [DOI] [PubMed] [Google Scholar]
- 2.Malloy M. J., Kane J. P. 2001. A risk factor for atherosclerosis: triglyceride-rich lipoproteins. Adv. Intern. Med. 47: 111–136 [PubMed] [Google Scholar]
- 3.Connelly P. W., Petrasovits A., Stachenko S., MacLean D. R., Little J. A., Chockalingam A. 1999. Prevalence of high plasma triglyceride combined with low HDL-C levels and its association with smoking, hypertension, obesity, diabetes, sedentariness and LDL-C levels in the Canadian population. Canadian Heart Health Surveys Research Group. Can. J. Cardiol. 15: 428–433 [PubMed] [Google Scholar]
- 4.Tai E. S., Emmanuel S. C., Chew S. K., Tan B. Y., Tan C. E. 1999. Isolated low HDL cholesterol: an insulin-resistant state only in the presence of fasting hypertriglyceridemia. Diabetes. 48: 1088–1092 [DOI] [PubMed] [Google Scholar]
- 5.Austin M. A., King M. C., Bawol R. D., Hulley S. B., Friedman G. D. 1987. Risk factors for coronary heart disease in adult female twins. Genetic heritability and shared environmental influences. Am. J. Epidemiol. 125: 308–318 [DOI] [PubMed] [Google Scholar]
- 6.Heller D. A., de Faire U., Pedersen N. L., Dahlen G., McClearn G. E. 1993. Genetic and environmental influences on serum lipid levels in twins. N. Engl. J. Med. 328: 1150–1156 [DOI] [PubMed] [Google Scholar]
- 7.Perusse L., Rice T., Despres J. P., Bergeron J., Province M. A., Gagnon J., Leon A. S., Rao D. C., Skinner J. S., Wilmore J. H., et al. 1997. Familial resemblance of plasma lipids, lipoproteins and postheparin lipoprotein and hepatic lipases in the HERITAGE Family Study. Arterioscler. Thromb. Vasc. Biol. 17: 3263–3269 [DOI] [PubMed] [Google Scholar]
- 8.Kissebah A. H., Alfarsi S., Adams P. W. 1981. Integrated regulation of very low density lipoprotein triglyceride and apolipoprotein-B kinetics in man: normolipemic subjects, familial hypertriglyceridemia and familial combined hyperlipidemia. Metabolism. 30: 856–868 [DOI] [PubMed] [Google Scholar]
- 9.Kissebah A. H., Sonnenberg G. E., Myklebust J., Goldstein M., Broman K., James R. G., Marks J. A., Krakower G. R., Jacob H. J., Weber J., et al. 2000. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc. Natl. Acad. Sci. USA. 97: 14478–14483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonnenberg G. E., Krakower G. R., Martin L. J., Olivier M., Kwitek A. E., Comuzzie A. G., Blangero J., Kissebah A. H. 2004. Genetic determinants of obesity-related lipid traits. J. Lipid Res. 45: 610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W. D., Dong C., Li D., Garrigan C., Price R. A. 2005. A genome scan for serum triglyceride in obese nuclear families. J. Lipid Res. 46: 432–438 [DOI] [PubMed] [Google Scholar]
- 12.Duggirala R., Blangero J., Almasy L., Dyer T. D., Williams K. L., Leach R. J., O'Connell P., Stern M. P. 2000. A major susceptibility locus influencing plasma triglyceride concentrations is located on chromosome 15q in Mexican Americans. Am. J. Hum. Genet. 66: 1237–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horne B. D., Malhotra A., Camp N. J. 2003. Comparison of linkage analysis methods for genome-wide scanning of extended pedigrees, with application to the TG/HDL-C ratio in the Framingham Heart Study. BMC Genet. 4(Suppl. 1): S93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin J. P.2003. Genome-wide scan on plasma triglyceride and high density lipoprotein cholesterol levels, accounting for the effects of correlated quantitative phenotypes. BMC Genet. 4(Suppl. 1): S47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shearman A. M., Ordovas J. M., Cupples L. A., Schaefer E. J., Harmon M. D., Shao Y., Keen J. D., DeStefano A. L., Joost O., Wilson P. W., et al. 2000. Evidence for a gene influencing the TG/HDL-C ratio on chromosome 7q32.3-qter: a genome-wide scan in the Framingham study. Hum. Mol. Genet. 9: 1315–1320 [DOI] [PubMed] [Google Scholar]
- 16.Diamond R. H., Du K., Lee V. M., Mohn K. L., Haber B. A., Tewari D. S., Taub R. 1993. Novel delayed-early and highly insulin-induced growth response genes. Identification of HRS, a potential regulator of alternative pre-mRNA splicing. J. Biol. Chem. 268: 15185–15192 [PubMed] [Google Scholar]
- 17.Peng Y., Schwarz E. J., Lazar M. A., Genin A., Spinner N. B., Taub R. 1997. Cloning, human chromosomal assignment, and adipose and hepatic expression of the CL-6/INSIG1 gene. Genomics. 43: 278–284 [DOI] [PubMed] [Google Scholar]
- 18.Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., Brown M. S. 2002. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 110: 489–500 [DOI] [PubMed] [Google Scholar]
- 19.Radhakrishnan A., Ikeda Y., Kwon H. J., Brown M. S., Goldstein J. L. 2007. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA. 104: 6511–6518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown M. S., Goldstein J. L. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA. 96: 11041–11048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horton J. D., Shimomura I., Ikemoto S., Bashmakov Y., Hammer R. E. 2003. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J. Biol. Chem. 278: 36652–36660 [DOI] [PubMed] [Google Scholar]
- 22.Horton J. D., Goldstein J. L., Brown M. S. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109: 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein J. L., DeBose-Boyd R. A., Brown M. S. 2006. Protein sensors for membrane sterols. Cell. 124: 35–46 [DOI] [PubMed] [Google Scholar]
- 24.DeBose-Boyd R. A.2008. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 18: 609–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takaishi K., Duplomb L., Wang M. Y., Li J., Unger R. H. 2004. Hepatic insig-1 or -2 overexpression reduces lipogenesis in obese Zucker diabetic fatty rats and in fasted/refed normal rats. Proc. Natl. Acad. Sci. USA. 101: 7106–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engelking L. J., Liang G., Hammer R. E., Takaishi K., Kuriyama H., Evers B. M., Li W. P., Horton J. D., Goldstein J. L., Brown M. S. 2005. Schoenheimer effect explained–feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J. Clin. Invest. 115: 2489–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivier M.2005. The Invader assay for SNP genotyping. Mutat. Res. 573: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivier M., Chuang L. M., Chang M. S., Chen Y. T., Pei D., Ranade K., de Witte A., Allen J., Tran N., Curb D., et al. 2002. High-throughput genotyping of single nucleotide polymorphisms using new biplex invader technology. Nucleic Acids Res. 30: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pennacchio L. A., Olivier M., Hubacek J. A., Cohen J. C., Cox D. R., Fruchart J. C., Krauss R. M., Rubin E. M. 2001. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 294: 169–173 [DOI] [PubMed] [Google Scholar]
- 30.Pennacchio L. A., Olivier M., Hubacek J. A., Krauss R. M., Rubin E. M., Cohen J. C. 2002. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum. Mol. Genet. 11: 3031–3038 [DOI] [PubMed] [Google Scholar]
- 31.Nickerson D. A., Tobe V. O., Taylor S. L. 1997. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 25: 2745–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marth G. T., Korf I., Yandell M. D., Yeh R. T., Gu Z., Zakeri H., Stitziel N. O., Hillier L., Kwok P. Y., Gish W. R. 1999. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 23: 452–456 [DOI] [PubMed] [Google Scholar]
- 33.Goring H. H., Curran J. E., Johnson M. P., Dyer T. D., Charlesworth J., Cole S. A., Jowett J. B., Abraham L. J., Rainwater D. L., Comuzzie A. G., et al. 2007. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 39: 1208–1216 [DOI] [PubMed] [Google Scholar]
- 34.Li Y. C., Ross J., Scheppler J. A., Jr. Franza B. R. 1991. An in vitro transcription analysis of early responses of the human immunodeficiency virus type 1 long terminal repeat to different transcriptional activators. Mol. Cell. Biol. 11: 1883–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett J. C., Fry B., Maller J., Daly M. J. 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 21: 263–265 [DOI] [PubMed] [Google Scholar]
- 36.de Bakker P. I., Yelensky R., Pe'er I., Gabriel S. B., Daly M. J., Altshuler D. 2005. Efficiency and power in genetic association studies. Nat. Genet. 37: 1217–1223 [DOI] [PubMed] [Google Scholar]
- 37.Falconer D. S.1989. Introduction to quantitative genetics. 3rd edition Longman Scientific and Technical, New York, NY [Google Scholar]
- 38.Almasy L., Blangero J. 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62: 1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitlock M. C.2005. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J. Evol. Biol. 18: 1368–1373 [DOI] [PubMed] [Google Scholar]
- 40.Cox N. J., Frigge M., Nicolae D. L., Concannon P., Hanis C. L., Bell G. I., Kong A. 1999. Loci on chromosome 2 (NIDDM1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat. Genet. 21: 213–215 [DOI] [PubMed] [Google Scholar]
- 41.Herbert A., Gerry N. P., McQueen M. B., Heid I. M., Pfeufer A., Illig T., Wichmann H. E., Meitinger T., Hunter D., Hu F. B., et al. 2006. A common genetic variant is associated with adult and childhood obesity. Science. 312: 279–283 [DOI] [PubMed] [Google Scholar]
- 42.Hotta K., Nakamura M., Nakata Y., Matsuo T., Kamohara S., Kotani K., Komatsu R., Itoh N., Mineo I., Wada J., et al. 2008. INSIG2 gene rs7566605 polymorphism is associated with severe obesity in Japanese. J. Hum. Genet. 53: 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oki K., Yamane K., Kamei N., Asao T., Awaya T., Kohno N. 2009. The single nucleotide polymorphism upstream of insulin-induced gene 2 (INSIG2) is associated with the prevalence of hypercholesterolaemia, but not with obesity, in Japanese American women. Br. J. Nutr. 101: 322–327 [DOI] [PubMed] [Google Scholar]
- 44.Kim S. K., Selleri L., Lee J. S., Zhang A. Y., Gu X., Jacobs Y., Cleary M. L. 2002. Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 30: 430–435 [DOI] [PubMed] [Google Scholar]
- 45.Miller M., Cannon C. P., Murphy S. A., Qin J., Ray K. K., Braunwald E. 2008. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 Trial. J. Am. Coll. Cardiol. 51: 724–730 [DOI] [PubMed] [Google Scholar]
- 46.Kitsios G. D., Zintzaras E. 2009. Genomic convergence of genome-wide investigations for complex traits. Ann. Hum. Genet. 73: 514–519 [DOI] [PMC free article] [PubMed] [Google Scholar]