

Abstract
During infection, viruses cause global disruption to nuclear architecture in their attempt to take over the cell. In turn, the host responds with various defenses, which include chromatin-mediated silencing of the viral genome and activation of DNA damage signaling pathways. Dynamic exchanges at chromatin, and specific post-translational modifications on histones have recently emerged as master controllers of DNA damage signaling and repair. Studying viral control of chromatin modifications is identifying histones as important players in the battle between host and virus for control of cell cycle and gene expression. These studies are revealing new complexities of the virus-host interaction, uncovering the potential of chromatin as an anti-viral defense mechanism, and also providing unique insights into the role of chromatin in DNA repair.
2. Introduction
Chromatin is a complex and dynamic structure that must be successfully navigated by viruses in order to ensure an efficient infection cycle. There are two facets to the relationship between viruses and chromatin. On one side, the incoming viral genomes may be chromatinized by the host cell, while on the other side, the virus attempts to manipulate host chromatin proteins to promote its own replication. The effects of histone deposition on incoming viral genomes is emerging as an important aspect of the virus-host interaction, and is likely to have significant implications both for lytic replication and latent maintenance of viral genomes. These topics are covered elsewhere in this focus edition. The range of host proteins targeted by viruses in their attempt to gain control of the cell has recently been expanded to include histones and other chromatin modifying proteins. We will discuss the effects of viral infection on host chromatin, with a specific focus on targeting of proteins relevant to the DNA damage response.
2.1 Histone post-translational modifications in DNA repair
Repairing chromatinized DNA is a challenge, both in terms of accessibility of the break and maintenance of the histone code and chromatin organization. Histone post-translational modifications and chromatin remodeling proteins co-ordinate to allow access of repair molecules to the DNA, and to facilitate amplification and then termination of the damage signal [1, 2]. The cellular DNA damage response is a signal-transduction cascade. Lesions in DNA are detected by sensor proteins that activate kinases, which in turn lead to amplification of the signal through a series of downstream effector molecules. The major signaling kinases are ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK), which are members of the phosphoinositide 3 kinase-like kinase (PIKK) family. Upon sensing a break, one or more of these kinases is activated and phosphorylates the histone H2A variant, H2AX. H2AX is constitutively expressed and distributed throughout the genome, representing approximately 10% of the total cellular H2A. H2AX contains a conserved SQ(E/D) motif and this serine residue (S139) is rapidly phosphorylated in response to DNA damage (reviewed in [3]). The phosphorylated H2AX (now referred to as γH2AX) marks the site of damage, and recruits mediator proteins to form irradiation-induced foci (IRIF), which are visible by fluorescent microscopy. γH2AX extends up to 1 megabase in mammalian cells and this spreading is thought to have a role in amplification of the DNA damage signal (reviewed in [4]). Recent work has highlighted the importance of additional post-translational modifications on H2AX and other histones in the DNA damage response. These include a novel inhibitory tyrosine phosphorylation mark on H2AX (Y142) [5], ubiquitination sites on H2A and H2AX (reviewed in [6]), sumoylation sites on the H2A variant H2AZ [7], tyrosine phosphorylation on histone H3 [8], acetylation on H2AX [9], and methylation marks on histone H4 [10, 11]. The roles of these histone marks in DNA repair and the potential for their manipulation by viral infection are summarized in Figure 1 and discussed in the following sections.
Figure 1. Viral control of cellular histone modifications.
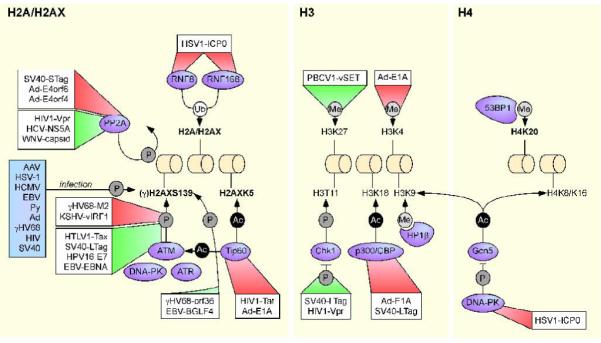
Chromatin remodeling proteins and histone post-translational modifications known to be targeted by viruses are depicted. Histone post-translational modificatons which have a known role in DNA repair are shown in bold. Effects of the viral proteins (boxed) on the chromatin-related substrates are indicated by the shaded trianges. Red triangles indicate interactions that are inhibitory to the pathways or cellular factors targeted and green triangles denote interactions that promote the activity of the pathways or cellular factors targeted. For simplicity, not all histone targets of the cellular enzymes are shown. The interactions depicted are limited to those covered in this review and many more examples of associations between viral and cellular proteins exist.
2.2 The role of chromatin remodeling in DNA repair
Although the significance of post-translational modifications on H2AX is becoming increasingly well understood, several questions remain. Key among these is how ubiquitin ligases, kinases and acetyltransferases gain access to the histones at the double strand break (DSB) site. Moreover, how γH2AX spreading is controlled, what facilitates its passage through compacted chromatin and what signals the conclusion of this spreading are unknown. It has been reported that DSBs trigger local recruitment of chromatin-remodeling proteins, such as SWI/SNF, RSC, SRCAP and INO80 enzyme complexes [12, 13]. These proteins co-ordinate histone eviction and decondensation of the chromatin in the region surrounding the break site and allow access of regulatory and repair enzymes to the DNA [14-17]. Recently, it was suggested that H2AX tyrosine de-phosphorylation in response to damage may adjust local chromatin structure to facilitate the access of kinases and other mediator proteins to damaged DNA [5]. In addition, the histone acetyltransferase (HAT) Tip60 acetylates H2AX and this event is required for the mobilization of γH2AX immediately after DSB break induction [9]. Further insight into the accessibility of H2AX has come from a study which demonstrated that the chromatin binding protein heterochromatin protein 1β (HP1β is phosphorylated by casein kinase II (CKII) in response to DNA damage [18]. HP1β is a chromodomain-containing adaptor molecule that binds chromatin at H3K9me3 and is implicated in gene silencing [19]. CKII-mediated phosphorylation of HP1β induces its transient dissociation from H3K9me and this is required for the phosphorylation signal to be deposited on H2AX. This model predicts that the dissociation of HP1β facilitates the formation of a break-specific open chromatin region that makes H2AX more accessible for kinases and ubiquitin ligases, and removes local repressive barriers to facilitate spreading of the γH2AX signal. This idea is attractive because targeting a histone binding protein ensures that the histone code itself remains unchanged after damage.
HP1β is also known to bind the cellular co-repressor, KAP1. In addition to HP1β, KAP1 interacts with other chromatin modifying proteins such as histone deacetylases (HDACs) [20], the H3K9 methyltransferase SETDB1 [21], and participates in gene expression and chromatin remodeling. Interestingly, KAP1 is phosphorylated by PIKKs following DNA damage, and KAP1 localizes to sites of DNA lesions [22]. The release of KAP1 from chromatin is dependent on ATM, raising the possibility that ATM may play a novel role in the phosphorylation and removal of chromatin proteins that are inhibitory to the repair process [23, 24]. The role of chromatin remodeling proteins in facilitating access of repair proteins to the break site is becoming increasingly well understood. However, how these chromatin modifications participate in the activation of ATM and concomitant induction of the signal transduction cascade has until recently remained obscure. Tip60 acetylates ATM in response to damage and this acetylation facilitates ATM autophosphorylation [25]. The recent identification of HMGN1 as a mediator of ATM activation provides another potential bridge between chromatin remodeling and signal transduction proteins. HMGN1 is required for the acetylation of H3K14 in response to IR, decreasing the retention of ATM at chromatin and increasing ATM autophosphorylation [26]. The fact that HMGN1 does not itself accumulate at DSBs suggests that it may affect the organization of ATM throughout the nucleus, raising the possibility that the activation dynamics of ATM are affected by chromatin interactions that occur before induction of a DNA break.
2.3 Viruses and the DNA damage response
As obligate intracellular parasites with small genomes, viruses have complex relationships with their hosts. Viruses have often co-evolved with the host and have developed a plethora of strategies to utilize cellular proteins to promote their own replication and facilitate maintenance of the viral genome. Many viruses are now known to interact with the DNA damage sensing and repair machinery [27]. These viruses have evolved tactics to eliminate, avoid or exploit the DNA damage machinery of the host cell. While some viruses activate DNA repair proteins, others target these cellular factors for degradation or mis-localization. The interface between viruses and the cellular DNA repair machinery is complex and it is becoming increasingly apparent that individual viruses interact with the cellular DNA repair machinery on multiple levels, inactivating certain aspects while exploiting others.
3. The effects of virally-induced DNA damage on nuclear architecture
Viral infection causes dramatic changes to the nuclear architecture as the virus attempts to take over the cell. In the initial stages of infection, host chromatin proteins can be mobilized and recruited to viral genomes. As the infection proceeds, cellular chromatin must be structurally re-organized to make room for small viral replication compartments that eventually coalesce and fill much of the nucleus. In the majority of examples of viral activation of γH2AX, no specific viral protein appears to be sufficient. In these cases, it is assumed that aberrant DNA structures on the incoming or replicating genomes may be the trigger, or that global changes to host chromatin may lead to ATM activation [26, 28] and subsequent damage-independent H2AX activation. In this section, we will review some examples of viral replication leading to changes in γH2AX.
3.1 Activation and nuclear localization of γH2AX
In the case of herpes simplex virus type 1 (HSV-1), infection induces activation of DNA damage signaling [29, 30], phosphorylation of H2AX and marginalization of this γH2AX to the periphery of viral replication centers [31]. In contrast, while infections with human cytomegalovirus (HCMV) also caused H2AX phosphorylation [32], the γH2AX co-localized with viral replication compartments, at least late in infection [32, 33]. In this case, activation of γH2AX was shown to be dependent on viral replication as it did not occur in p53 mutant cells, in which HCMV replication is abrogated [33]. Similarly, induction of Epstein Barr virus (EBV) lytic replication [34] or polyomavirus (Py) replication [35] induced phosphorylation of a number of DNA damage markers including γH2AX.
Infection with adenovirus (Ad) deleted of early region E4 resulted in activation of a cellular DNA damage response, with accumulation of damage mediator proteins at sites of viral replication [36]. Staining for γH2AX was observed around the edges of viral centers [37]. The viral trigger of this histone phosphorylation event is unknown but the accumulation of γH2AX was abrogated in the presence of caffeine, suggesting that the phosphorylation is mediated by cellular damage kinases. It is unclear whether the histone is modified on viral DNA or whether H2AX in cellular chromatin adjacent to viral replication centers becomes phosphorylated when damage kinases are activated in response to the virus. During infection with wild-type Ad, cellular DNA damage sensor proteins are degraded and the cellular DNA damage response is abrogated [37]. However, late during wild-type infection, H2AX is phosphorylated and the staining is distributed throughout the nucleus of infected cells, suggesting that the histone modification is on cellular DNA [38]. Control infections with a non-replicating Ad did not induce γH2AX staining, suggesting that viral replication is the trigger in this case.
Infection of cells with adeno-associated virus (AAV) also results in pan-nuclear activation of γH2AX [39, 40]. Wild-type and UV-treated AAV, but not recombinant AAV vectors, were able to elicit this response suggesting that the phosphorylation was due to a component of the wild-type AAV genome and not the viral capsid. The activation of γH2AX staining by wild-type AAV was narrowed down to a region of the genome containing the p5 promoter which may function as a potential origin of replication [40]. Although AAV infection or the viral Rep protein alone could induce some diffuse nuclear γH2AX staining, the response was much more robust during replication in the presence of Ad helper functions [39]. Together these studies suggest that γH2AX is activated in response to AAV due to specific viral sequences within the p5 region and by DNA structures generated during viral replication.
3.2 Virus-induced cellular re-organization
Viral infection usually leads to the formation of viral replication compartments that fill much of the interior of the nucleus. In the case of HSV-1, these replication compartments exclude histone H1 [41], heterochromatin marks H3K9Me3 and H4K20Me3 [42], and cause the marginalization of global host chromatin [43, 44], including γH2AX [31]. The exclusion of linker histones has been proposed to be due to a host cell-induced increase in H1 mobility in response to virus infection [45]. Interestingly, these histone exclusion effects were recently found to be dependent on nuclear lamin A/C. In the absence of lamin A/C, viral gene expression and replication were impaired, and this was a result of the presence of heterochromatin at viral replication compartments [42]. This led the authors to propose that lamin A/C is a molecular scaffold required both to control viral genome localization and to ensure that heterochromatin is not deposited on viral promoters during lytic infection.
In some examples, gross cellular changes induced by the virus are sufficient for phosphorylation of H2AX. For example, in the case of human immunodeficiency virus 1 (HIV-1) infection, virus-induced cell fusion was necessary and sufficient for ATM-dependent γH2AX activation [46]. This fusion is normally mediated by a gp120/CD4 interaction, and when this fusion event was prevented, H2AX phosphorylation was abolished. Surprisingly, H2AX phosphorylation could be restored by forcing cell fusion with polyethylene glycol, even in the absence of a gp120/CD4 interaction. The authors hypothesized that phosphorylation of H2AX may result from fusing of nuclei that are in different phases of the cell cycle.
4. Virus encoded proteins targeting chromatin
Viruses often inhibit or exploit specific cellular proteins to promote viral replication and facilitate maintenance of the viral genome. Recently, the repertoire of host proteins targeted directly or indirectly by viruses has been expanded to include histones. This section will consider several examples of specific viral proteins being necessary and sufficient to induce or remove post-translational modifications on histones.
4.1 Divorcing ubiquitin from histones - how a virus can remove the RING
It has been known for several years that HSV-1 and HSV-2 induce DNA damage signaling in the host cell, characterized by phosphorylation of many cellular proteins, including H2AX, and recruitment of several mediator proteins to viral replication centers [29, 30, 47, 48]. In addition to phosphorylation, ubiquitination of H2A and H2AX has recently emerged as a central co-ordinator of the DNA damage response. Herpesviruses need to gain control of the DNA repair pathway in order to promote their efficient replication, so it is therefore not surprising that they have evolved to target this key upstream regulatory step. Recent work from our laboratory has revealed that the HSV-1 encoded E3 ubiquitin ligase, ICP0, induces the proteasome-mediated degradation of cellular ubiquitin ligases normally serving to maintain the ubiquitinated mark on H2A and H2AX (CEL, MSC and MDW, unpublished observations). ICP0 is a strong transcriptional activator important for the efficient transition from latent to lytic infection. RNF8 and RNF168 are ubiquitin ligases for H2A and H2AX that mediate the accumulation of downstream repair proteins at the sites of double-strand breaks [49-54]. We propose that by targeting RNF8 and RNF168 for proteasome-mediated degradation, ICP0 dramatically reduces the levels of ubiquitinated H2A and H2AX in the cell and prevents the retention of downstream DNA repair proteins at sites of cellular damage. We hypothesize that one reason ICP0 may prevent this immobilization is to ensure that the cellular repair factors are available to be recruited to sites of viral replication. ICP0 has previously been reported to inhibit DNA repair since its expression led to the persistence of damage-induced γH2AX foci and impaired survival in a radiosensitivity assay [55]. This was attributed to the ICP0-mediated degradation of DNA-PKcs, but it is possible that impaired DNA damage signaling due to loss of RNF8, RNF168 and ubiquitinated H2A may also contribute to this effect.
4.2 Checking in on histone phosphorylation
DNA damage signaling initiated by ATM, ATR and DNA-PKcs leads to the phosphorylation of a number of downstream effector proteins. Among these is Chk1, which is a chromatin-associated protein phosphorylated primarily by ATR. Phosphorylated Chk1 has well-documented roles in cell-cycle arrest and apoptosis (reviewed in [56]). However, unphosphorylated Chk1 is still an active kinase and it has been proposed that it is the damage-induced dissociation of Chk1 from chromatin rather than its kinase activity per se which promotes the checkpoint response [57]. It has recently been shown that Chk1 phosphorylates histone H3 on T11 independently of DNA damage [8]. H3T11 is then dephosphorylated when Chk1 dissociates from chromatin after damage, and this coincides with a modest reduction in H3K9 acetylation, an event normally mediated by the HAT Gcn5. As viruses attempt to gain control of the cell cycle, they commonly target checkpoint kinases such as Chk1 and Chk2. It is likely that many viruses will induce Chk1 phosphorylation as part of a wider DNA damage response [27]. However, specific examples of viral proteins that are known to affect Chk1 phosphorylation include simian virus 40 (SV40) LTag [58] and HIV-1 Vpr [59].
: intended target or innocent bystander?
Phosphorylation of H2AX is considered a hallmark of the DNA damage response, but the specific trigger is unknown in most examples of viral induction of this event. As discussed in the previous section, it has often been assumed that this phosphorylation event may result from global disorganization of the nucleus or be caused by unusual DNA structures introduced by the virus. However, several recent papers suggest that specific virally-encoded proteins can also control the phosphorylation status of H2AX directly. Transfected Tax protein from human T-cell lymphotropic virus (HTLV-1) forms nuclear foci and is sufficient to induce phosphorylation of H2AX at these sites [60]. Concomitant with the activation of γH2AX by Tax, it was noted that the viral protein also induced phosphorylation of DNA-PKcs and activation of Chk2. IR-induced phosphorylation of H2AX is typically carried out by ATM, but in the absence of ATM, kinases such as DNA-PK can act, suggesting a possible mechanism for the effects of Tax on H2AX phosphorylation. Interestingly, when Tax-expressing cells were challenged with exogenous damage, the IR-induced increase in γH2AX was blunted and temporally delayed. These results suggest that Tax-expressing cells have an impaired capacity to respond to new damage, or that the damage recognition pathways are already “saturated” by the presence of Tax.
Previous reports indicated that infection with SV40 induced extensive DNA damage signaling with localization of many cellular repair and replication factors, including γH2AX, to large LTag-containing nuclear foci [61]. However, it was not clear whether LTag alone was sufficient to induce the response or whether viral replication structures were required. Recently, it was demonstrated that expression of LTag without a viral replication origin in normal human fibroblasts led to the induction of γH2AX foci and several other phosphorylation events indicative of a DNA damage response [58]. Interestingly, mutant LTag defective for Bub1 binding was unable to induce the formation of γH2AX foci, suggesting a role for this spindle assembly checkpoint kinase in H2AX activation.
Other viral proteins have been reported to lead to phosphorylation of γH2AX. Examples include human papillomavirus (HPV-16) E7 [62] and the EBV protein EBNA1 [63]. Interestingly, in the case of EBV, the intensity of γH2AX staining was decreased by treatment of EBNA1-expressing cells with ROS scavengers, suggesting that this viral protein may utilize a novel mechanism for viral induction of DNA damage signaling and histone modifications [64]. How these specific viral proteins lead to activation of H2AX is unclear. Possibilities include the induction of bone fide cellular double-strand breaks, deregulation of normal cellular DNA replication dynamics, global changes to host chromatin, or direct interaction with cellular PIKKs or other DNA repair proteins.
In the above examples, it is generally assumed that although the viral protein is the trigger, the actual phosphorylation event is performed by a cellular kinase. Recently, the first example of direct H2AX phosphorylation by a viral protein has been proposed. Tarakanova et al. reported that the murine gammaherpesvirus 68 (γHV68) protein orf36 and its EBV homolog, BGLF4, were necessary and sufficient to induce γH2AX phosphorylation [65]. The authors demonstrated that orf36 was necessary for the induction of γH2AX phosphorylation during γHV68 infection. They also showed that transient expression of plasmid-encoded orf36 was sufficient to cause H2AX phosphorylation and that, like other herpesviruses, the presence of ATM and H2AX are required for efficient γHV68 replication. Strikingly, immunopreciptated wild-type, but not mutant orf36, could phosphorylate recombinant H2AX in vitro, leading the authors to propose that the histone variant is a direct substrate of orf36. This attractive hypothesis would suggest that viral induction of the DNA damage signaling cascade can be an active event, rather than simply a “side-effect” of abnormal incoming DNA structures or intermediates generated during viral replication. Formal proof of this awaits further in vitro studies, since blunting of the orf36-induced phosphorylation event in ATM deficient cells raises the possibility that the viral protein may simply be stimulating ATM-mediated H2AX phosphorylation.
γHV68 has an unusual relationship with H2AX activation. In addition to encoding a potential histone kinase, γHV68 encodes another protein, M2, which increases endogenous ATM activity in an in vitro kinase assay. However, despite activation of this kinase, no γH2AX staining is detected in M2-expressing cells, even after treatment with DNA damaging agents [66]. This observation suggests that γHV68 M2 may specifically inhibit the ability of ATM to induce H2AX phosphorylation. γHV68 M2 is not the only viral protein known to inhibit H2AX activation. Expression of the vIRF1 protein from Kaposi’s sarcoma herpes virus (KSHV) was able to block the induction of both ATM and γH2AX by the topoisomerase inhibitor, etoposide [67].
Viral control of γH2AX induction could also be at the level of CKII-mediated phosphorylation of HP1β, the recently identified prerequisite step for H2AX phosphorylation [18]. The cellular co-repressor, KAP1, recruits HP1 family proteins to silence murine leukaemia virus (MLV) [68], and the HP1 isoform, HP1α, is known to be recruited to HIV-1 genomes [69]. Many herpesvirus genomes, including HSV-1 [70], human herpes virus 6 (HHV6) [71] and HCMV [72], are also known to co-localize with HP1 proteins in cells in which viral gene expression is repressed. This is likely an attempt to silence incoming viral genomes, and therefore may not directly affect host chromatin and γH2AX activation. However, whether these viruses have developed any counter-attack strategies against HP1 and whether these impact γH2AX activation remains to be seen. It is also interesting to note that KAP1 is phosphorylated on S824 in response to DNA damage [22] and phosphorylation of KAP1 on a different residue (S473) compromises the interaction of KAP1 with HP1β [73]. Whether viral induction of DNA damage signaling leads to phosphorylation of KAP1 on either site, and the potential implications of this modification for HP1β remain to be seen. Other viral proteins such as polyomavirus agnoprotein have been shown to bind HP1α, although the significance of this is unclear [74]. If HP1β is unable to be phosphorylated by CKII, it is constitutively bound to chromatin and prevents phosphorylation of H2AX in response to damage. Targeting of CKII might therefore be an attractive viral strategy to control H2AX phosphorylation and downstream signaling in response to damage.
4.4 A plethora of other post-translational modifications
Post-translational modifications of histones are a central theme in the recruitment of DNA repair proteins to sites of double strand breaks. Following phosphorylation of H2AX, mediator proteins are recruited and the H2AX surrounding the break site is ubiquitinated (reviewed in [6]). Poorly understood chromatin remodeling events then ensue, culminating in the exposure of methylated histone H4K20, the target of cellular SET domain methyltransferases and the binding site for the DNA damage protein 53BP1 [10, 11]. We have discussed viral strategies to control the phosphorylation and ubiquitination of histones involved in DNA repair, but viral targeting of this methylation step is as yet undiscovered. A recent report hints that examples of viral control at this level may emerge. Infection with paramecium bursaria chlorella virus 1 (PBCV-1) delivers a virion-encoded SET domain histone methyltransferase (vSET) that directly modifies chromatin by methylating histone H3K27 [75]. Although H3K27 is not known to be directly involved in DNA repair, this paper demonstrates that viruses can capture histone methyltransferase functions from their hosts. It will be interesting to see whether virally-encoded histone methytransferases are part of the weaponry used by viruses in their attempt to commandeer chromatin and control DNA repair pathways.
Acetylation of histones also provides an additional level of control in the damage signaling pathway. Tip60 is recruited to DSBs, where it acetylates histones [76]. This acetylation has a role in damage-induced histone exchange, since Tip60 is required for the mobilization of γH2AX immediately after break induction. Tip60 acetylates H2AX on K5, which promotes the UBC13-mediated polyubiquitination of γH2AX before releasing it from chromatin, and replacing it with unmodified H2AX [9, 77]. Tip60 forms a stable complex with ATM and acetylates it in response to damage, facilitating ATM autophosphorylation [25]. In a similar way, DNA-PKcs also binds to Tip60, an event required for the activation of this kinase [78]. These interactions imply that viral targeting of Tip60 would result in significant effects on DNA damage signaling and repair. So far, two viruses have been shown to interact directly with Tip60 [79, 80]. The most well studied example is HIV-1 Tat, for which Tip60 (Tat-interacting protein 60KDa) was named [79]. Interaction of Tat with Tip60 inhibits Tip60 HAT activity, presumably in an attempt to limit acetylation of histones on host transcriptional units [79, 81]. Adenovirus E1A binds TRRAP and Gcn5, components of the Tip60 HAT and nucleosome remodeling complex [82]. Given the recently appreciated significance of Tip60 and histone acetylation in the DNA damage response, it is likely that more examples of viral control at this level will emerge.
InadditiontoitseffectsonH2AX phosphorylation (discussed in section 4.3), γHV68 also impacts histone acetylation. The γHV68 protein M2 binds to a damaged DNA-binding protein 1 (DDB1)-containing complex that contains ATM and histone H3 and H4 [66]. Histone H3 and H4 acetylation was enhanced in M2-expressing cells, both in the presence and absence of DNA damaging agents. Since M2 expression also leads to ATM activation, the authors suggested that these histone acetylation events might induce chromatin remodeling, which in turn is known to induce autophosphorylation of ATM [28, 66].
There are undoubtedly many more examples of histone acetylation controlled at multiple levels, either directly or indirectly by viruses. For example, the ICP0 protein of HSV-1 inhibits deacetylation of histones by redistributing class II HDACs [83] and displacing HDAC1/2 from the CoREST/REST/LSD1 complex [84]. ICP0 also degrades DNA-PKcs [85]. These seemingly disparate observations may be linked by the fact that DNA-PKcs serves to phosphorylate and inactivate the HAT, Gcn5 [86]. Gcn5 is normally recruited to DSB sites where it contributes to the increase of H3 and H4 histone acetylation at sites flanking the break [87]. It therefore appears that ICP0 may have evolved to target acetylation from both directions: by indirectly activating acetyltransferases and by blocking the activity of HDACs.
4.5 Prolonging the signal by targeting the γH2AX phosphatases
Although H2AX phosphorylation has long been known to be a key event in the induction of the DNA damage signaling cascade, the cellular players controlling the resolution of this signal were until recently not characterized. However, it has now been shown that protein phosphatase 2A (PP2A) dephosphorylates γH2AX. When PP2A was silenced by RNA interference, γH2AX foci persisted, DNA repair was inefficient, and cells were hypersensitive to DNA damaging agents [88]. PP2A may also play a role in the regulation of ATM autophosphorylation. ATM interacts with PP2A in undamaged cells but exposure to IR induces the phosphorylation-dependent dissociation of PP2A from ATM [89]. These data suggest that PP2A is required to resolve γH2AX foci and has an important role in both activation of ATM and the completion of repair after damage.
There are a number of examples of viruses targeting PP2A. The best characterized of these is SV40 small T antigen (STag) which interacts with PP2A and inhibits its activity. The interaction between STag and PP2A is required for SV40-mediated transformation of human cells but the exact mechanism by which this transformation is afforded is unclear. SV40 is known to induce DNA damage signaling [61, 90] including LTag-mediated induction of γH2AX foci [58]. Therefore, it seems possible that large and small T antigens co-operate during a SV40 infection to prolong the phosphorylation mark on H2AX. This could be favorable for the virus, since it has been demonstrated that SV40 replication is inhibited in situations where H2AX phosphorylation might be compromised, for example in the presence of an ATM inhibitor [90]. The SV40-related viruses, polyomavirus and John Cunningham virus (JCV), also have proteins that interact with PP2A. PP2A associates with and dephosphorylates JCV agnoprotein and downregulation of PP2A by siRNA inhibited JCV replication [91].
Adenovirus also has a multifaceted relationship with PP2A and H2AX phosphorylation. Ad E4orf4 binds and relocalizes PP2A, and this interaction mediates all the known functions of E4orf4 [92] including the downregulation of Myc, possibly to counteract negative effects of Myc on Ad replication [93]. Ad is not alone in using PP2A to control myc expression; myc is upregulated in a PP2A-dependent manner by both Py and SV40 small T antigen [94, 95]. Other Ad proteins also affect DNA damage signaling and PP2A. E4orf6 interacts with E1B55K and together these form an ubiquitin ligase complex that targets several DNA repair proteins for proteasome-mediated degradation (reviewed in [96]). However, it was subsequently found that E4orf6 alone is necessary and sufficient to radiosensitize human tumor cells, suggesting that E4orf6 has additional functions in DNA repair that are independent of E1B55K [97]. Cells expressing E4orf6 displayed phosphorylation of H2AX and DNA-PKcs that extended beyond time points when DNA repair should be completed and these signaling proteins would normally be returned to their dephosphorylated state. Expression of E4orf6 did not appear to inhibit directly the re-ligation of damaged DNA ends, but the viral protein could inhibit the activation of PP2A normally observed in response to irradiation [98]. The authors suggest that the resultant prolonged signaling of DNA damage in the presence of E4orf6 falsely alerts the cell that significant levels of unrepaired damage remain and this initiates caspase-dependent and independent cell death.
While viruses such as SV40 and Ad inhibit PP2A, other viruses or viral proteins induce the activity of the cellular phosphatase. These include west nile virus (WNV) capsid protein, HIV-1 Vpr and hepatitis C virus (HCV) NS5A [99-103]. In the case of the WNV capsid protein, this PP2A activation is manifested by viral targeting of I(2)(PP2A), a PP2A inhibitor [100]. The consequences of these interactions on the γH2AX signal and viral manipulation of the DNA damage signal and cellular chromatin remain to be determined.
A second phosphatase acting on γH2AX was recently identified. Like PP2A, protein phosphatase 4 (PP4) can dephosphorylate γH2AX in vitro and in cells. Depletion of PP4 in cells resulted in a sustained presence of Mdc1 and Mre11 on damaged DNA, prolonged checkpoint arrest, defective repair, and hypersensitivity to DNA replication inhibitors [104, 105]. PP4 appears to have a closely related but distinct role from PP2A in mediating the dephosphorylation of H2AX. While PP2A acted on γH2AX formed by various types of exogenous damage, PP4 mainly dephosphorylated γH2AX generated during DNA replication stress [105]. This lack of redundancy led the authors to suggest that distinct phosphatases may differentially regulate γH2AX originating from different types or levels of DNA damage [105]. If this is the case, one might expect viruses would have evolved to target both phosphatases in order to control H2AX phosphorylation throughout the cell cycle. The fact that PP4 negatively regulates HDAC3 activity, combined with the observation that phosphorylation by CKII is required for HDAC3’s deacetylase activity [106], suggests that PP4 may have a more widespread role in regulating DNA damage and chromatin remodeling than is currently appreciated. Future work will reveal if viruses have indeed also evolved to target PP4.
5. Cellular implications of viral control of cellular histone modifications
The widespread or dysregulated H2AX phosphorylation seen during viral infections may have severe consequences for the cell. This section will discuss how viral control of histone post-translational modifications could impact DNA repair and transformation.
5.1 Implications for DNA repair
In yeast, H2A phosphorylation-mutant strains exhibit a decreased ability to perform end-joining reactions, suggesting that this histone phosphorylation event is important in DNA repair [107]. γH2AX foci do not form after irradiation in mice in which the H2AX cannot be phosphorylated or in which a glutamic acid substitution mimics a constitutively phosphorylated H2AX. Moreover, although both of these H2AX mutants allowed initial recruitment of downstream repair factors such as Nbs1 to the damaged DNA, these proteins were not maintained at the break site [108]. Metaphase spreads and spectral karyotype analysis of fibroblasts from H2AX-/- mice showed a marked increase in chromosomal abnormalities, including chromatid breaks, dicentric chromosomes, random translocations and complex rearrangements [109]. H2AX null cells were extremely sensitive to IR and demonstrated a defective capacity for DNA repair, as measured by DNA fragmentation assays, class switch recombination assays, gene targeting efficiencies, and homologous recombination experiments [109, 110]. Thus, improperly regulated H2AX phosphorylation, as seen during the viral infections discussed above, may result in aberrant recruitment of DNA damage responders and lead to faulty DNA repair. This could result in repair proteins being recruited to undamaged cellular DNA, accumulation of cellular mutations, “repair” of viral DNA, or prevention of maintenance of repair factors at sites of cellular damage. An example of a viral protein changing histone phosphorylation status and affecting the cellular capacity for repair comes from HCV. Here the NS3/4A protein interacts with and mislocalizes ATM, causes delayed dephosphorylation of ATM and γH2AX following ionizing irradiation, and impairs the ability of the cell to repair exogenous damage as assessed by a comet assay [111].
5.2 Implications for transformation
Recent studies have suggested that aberrant, pre-cancerous cell division can trigger H2AX phosphorylation [112, 113]. This is thought to be a protective mechanism since the downstream signaling events arrest cell cycle and direct the cell towards apoptosis, thereby limiting malignant progression. The authors predicted that progression from pre-cancerous lesions to mature tumors occurs in cells defective in their DNA damage response components. These cells are therefore able to evade checkpoint control, suggesting that inactivation of the DNA damage response may be a contributing factor in the switch between early lesions and cancer. In support of this prediction, there is evidence that both H2AX alleles are required for optimal protection against tumorigenesis in mice [114] and that improper levels of H2AX expression in humans may also contribute to the malignant progression (reviewed in [115]). Viral inhibition of H2AX phosphorylation or viral induction of premature γH2AX dephosphorylation might therefore be a novel mechanism by which these viruses contribute to cellular transformation.
There is also evidence of more direct links between chromatin modifications and viral transformation. Ad E1A induces a genome-wide relocalization of p300/CBP to ensure that these HATs are concentrated at genes the virus needs to be acetylated/expressed, and sequestered from genes that the virus requires to be hypoacetylated/silenced [116]. This dramatic effect of a single viral protein highlights the importance of chromatin modifications to successful infection and suggests that many more examples of virally-induced global epigenetic reprogramming will be uncovered. The overall effect of Ad E1A was global hypoacetylation of H3K18 and hypomethylation of H3K4 [117]. The acetylation changes were found to be directly attributable to the interaction of E1A with p300/CBP, since viral mutants unable to bind this HAT had no effect on the H3K18 acetylation profile. Interestingly, SV40 LTag binds and is acetylated by p300/CBP [118] and expression of SV40 LTag also leads to hypoacetylation of H3K18 [117]. This interesting parallel led the authors to propose that hypoacetylation of histones may be a general theme used by tumor virus oncoproteins in their quest to transform cells. In support of this hypothesis, the pattern of global chromatin changes seen with E1A expression (hypoacetylation of H3K18 and hypomethylation of H3K4) was strikingly similar to those seen in primary prostate cancers with poor prognosis [119].
6. Chromatin as an anti-viral defense mechanism
There are many examples of control of viral gene expression by assembly of chromatin on incoming or latent viral genomes. These have been previously discussed [120, 121] and are extensively addressed elsewhere in this focus edition. In addition to traditional anti-viral defenses such as the interferon response and apoptosis, it is becoming clear that chromatinization of viral genomes should be considered part of the host’s defense to invading pathogens. For example, transcription from many viral promoters, including the HCMV IE promoter, is known to be stimulated by treatment with HDAC inhibitors [72]. More recently, chromatin control of viral latency has also emerged as an exciting area of study. Reactivation of HSV-1, EBV, and KSHV from latency is stimulated by treatment of cells with HDAC inhibitors, suggesting a role for HDACs in the silencing of viral genomes [120, 122-125]. HSV-1 ICP0 is a strong transactivator of viral and cellular promoters, is required for lytic replication at low multiplicities of infection, and is necessary for the efficient reactivation of quiescent viral genomes (reviewed in [126]). We have observed that ICP0 leads to degradation of histone ubiquitin ligases RNF8 and RNF168 and a loss of ubiquitinated forms of H2A (CEL, MSC and MDW unpublished observations). Ubiquitinated H2A is strongly linked to transcriptional repression, with roles in silencing the inactive X chromosome (Xi) in mice and the promoters of polycomb-target genes such as Hox (reviewed in [127]). During lytic infection, incoming HSV-1 genomes are quickly marked by cellular histones, presumably in an attempt to silence expression of viral genes, and the latent viral genome is also nucleosome bound (reviewed in [120]). We propose that histone ubiquitination may be a novel means to silence pathogens and that removing this repressive mark on viral genomes may be one way in which ICP0 overcomes silencing during the lytic cycle and during reactivation from latency.
7. Summary and future directions
Cellular chromatin presents a challenge to viral transcription and viral manipulation of the DNA damage response. Chromatin plays an important role in the lifecycle of a diverse set of viruses, and we have reviewed some of the counter-strategies that these viruses have evolved to navigate, exploit or circumvent this potential obstacle. Viral control of histone methylation, phosphorylation and acetylation and the roles of these modifications in DNA repair are being increasingly well understood. Examples of viral interactions with cellular chromatin modifications covered in this review are summarized in Figure 1. As more post-translational modifications are uncovered, it is likely that this list will continue to expand. For example, recent work in yeast is beginning to uncover a role for histone sumoylation in DNA damage responses [7] and it has been suggested that sumoylation may serve as a block to other histone modifications [128]. It remains to be seen whether sumoylation is inversely correlated with any of the repair-associated post-translational modifications we have discussed, and whether viruses have evolved to exploit this potential blocking function of histone sumoylation. The reversible nature of histone modifications provides an extra level of control in the dynamics of chromatin remodeling.
There are already examples of virally-encoded histone kinases and viral interactions with histone phosphatases. Future work will also reveal whether viruses have evolved to encode histone phosphatases, deubiquitinases and demethylases. Investigation of the interplay between viruses, DNA repair, and the multitude of histone post-translational modifications is a productive area of research. Studying viral control of cellular epigenetics not only provides insights into the role of chromatin in DNA repair but may also lead to new opportunities for therapeutic intervention [120, 129, 130].
Table 1.
Virus Names and Abbreviations
| VIRUS NAME | ABBREVIATION |
|---|---|
| Herpes Simplex Virus | HSV |
| Human Cytomegalovirus | HCMV |
| Epstein Barr Virus | EBV |
| Kaposi’s Sarcoma Herpes Virus | KSHV |
| Simian Virus 40 | SV40 |
| Adeno-associated Virus | AAV |
| Adenovirus | Ad |
| Human Immunodeficiency Virus | HIV |
| Human T-cell Lymphotropic Virus | HTLV |
| John Cunningham Virus | JCV |
| Murine Gamma Herpes Virus 68 | γHV68 |
| Murine Leukaemia Virus | MLV |
| Hepatitis C Virus | HCV |
| West Nile Virus | WNV |
| Human Herpes Virus 6 | HHV6 |
| Polyoma Virus | Py |
| Human Papillomavirus | HPV |
8. Acknowledgements
We apologize to the many groups whose primary research papers could not be cited due to space constraints. We thank our colleagues in the fields of virology and DNA repair for helpful discussions and members of the Weitzman lab for comments on the manuscript. CEL received support from a Wellcome Trust International Research Fellowship. Work on viruses and DNA repair in the Weitzman lab has been supported by grants from the National Institutes of Health (AI067952, CA097093 and AI051686) and a Pioneer Developmental Chair from the Salk Institute. MSC is supported by a predoctoral Ruth L. Kirschstein National Research Service Award (NIH/NCI T32 CA009523).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol. 2009 doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- [3].Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- [4].Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- [5].Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, Patel DJ, Elledge SJ, Allis CD. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst) 2009 doi: 10.1016/j.dnarep.2009.01.013. [DOI] [PubMed] [Google Scholar]
- [7].Kalocsay M, Hiller NJ, Jentsch S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell. 2009;33:335–343. doi: 10.1016/j.molcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- [8].Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- [9].Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, Kimura H, Ikura M, Nishikubo S, Ito T, Muto A, Miyagawa K, Takeda S, Fishel R, Igarashi K, Kamiya K. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol. 2007;27:7028–7040. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huyen Y, Zgheib O, Ditullio RA, Jr., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- [11].Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].van Attikum H, Fritsch O, Gasser SM. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007;26:4113–4125. doi: 10.1038/sj.emboj.7601835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Osley MA, Tsukuda T, Nickoloff JA. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res. 2007;618:65–80. doi: 10.1016/j.mrfmmm.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Berkovich E, Monnat RJ, Jr., Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- [16].Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005;6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- [18].Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- [19].Eissenberg JC, Elgin SC. The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev. 2000;10:204–210. doi: 10.1016/s0959-437x(00)00058-7. [DOI] [PubMed] [Google Scholar]
- [20].Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].White DE, Negorev D, Peng H, Ivanov AV, Maul GG, Rauscher FJ., 3rd KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006;66:11594–11599. doi: 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- [23].Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- [24].Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- [25].Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim YC, Gerlitz G, Furusawa T, Catez F, Nussenzweig A, Oh KS, Kraemer KH, Shiloh Y, Bustin M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat Cell Biol. 2009;11:92–96. doi: 10.1038/ncb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15:119–126. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- [28].Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- [29].Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wilkinson DE, Weller SK. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J Cell Sci. 2006;119:2695–2703. doi: 10.1242/jcs.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gaspar M, Shenk T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A. 2006;103:2821–2826. doi: 10.1073/pnas.0511148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol. 2007;81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem. 2005;280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- [35].Dahl J, You J, Benjamin TL. Induction and utilization of an ATM signaling pathway by polyomavirus. J Virol. 2005;79:13007–13017. doi: 10.1128/JVI.79.20.13007-13017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- [37].Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nichols GJ, Schaack J, Ornelles DA. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J Virol. 2009;83:5987–5998. doi: 10.1128/JVI.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schwartz RA, Carson CT, Schuberth C, Weitzman MD. Adeno-associated virus replication induces a DNA damage response coordinated by DNA-dependent protein kinase. J Virol. 2009;83:6269–6278. doi: 10.1128/JVI.00318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fragkos M, Breuleux M, Clement N, Beard P. Recombinant adeno-associated viral vectors are deficient in provoking a DNA damage response. J Virol. 2008;82:7379–7387. doi: 10.1128/JVI.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Simpson-Holley M, Colgrove RC, Nalepa G, Harper JW, Knipe DM. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J Virol. 2005;79:12840–12851. doi: 10.1128/JVI.79.20.12840-12851.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Silva L, Cliffe A, Chang L, Knipe DM. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008;4:e1000071. doi: 10.1371/journal.ppat.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Monier K, Armas JC, Etteldorf S, Ghazal P, Sullivan KF. Annexation of the interchromosomal space during viral infection. Nat Cell Biol. 2000;2:661–665. doi: 10.1038/35023615. [DOI] [PubMed] [Google Scholar]
- [44].Simpson-Holley M, Baines J, Roller R, Knipe DM. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J Virol. 2004;78:5591–5600. doi: 10.1128/JVI.78.11.5591-5600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Conn KL, Hendzel MJ, Schang LM. Linker histones are mobilized during infection with herpes simplex virus type 1. J Virol. 2008;82:8629–8646. doi: 10.1128/JVI.00616-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Perfettini JL, Nardacci R, Bourouba M, Subra F, Gros L, Seror C, Manic G, Rosselli F, Amendola A, Masdehors P, Chessa L, Novelli G, Ojcius DM, Siwicki JK, Chechlinska M, Auclair C, Regueiro JR, de The H, Gougeon ML, Piacentini M, Kroemer G. Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia. PLoS ONE. 2008;3:e2458. doi: 10.1371/journal.pone.0002458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem. 2005;280:30336–30341. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- [49].Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- [52].Sakasai R, Tibbetts R. RNF8-dependent and RNF8-independent regulation of 53BP1 in response to DNA damage. J Biol Chem. 2008;283:13549–13555. doi: 10.1074/jbc.M710197200. [DOI] [PubMed] [Google Scholar]
- [53].Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- [54].Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, Oldreive C, Wildenhain J, Tagliaferro A, Pelletier L, Taubenheim N, Durandy A, Byrd PJ, Stankovic T, Taylor AM, Durocher D. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- [55].Hadjipanayis CG, DeLuca NA. Inhibition of DNA repair by a herpes simplex virus vector enhances the radiosensitivity of human glioblastoma cells. Cancer Res. 2005;65:5310–5316. doi: 10.1158/0008-5472.CAN-04-3793. [DOI] [PubMed] [Google Scholar]
- [56].Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- [57].Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- [58].Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol. 2009;83:117–127. doi: 10.1128/JVI.01515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li G, Elder RT, Qin K, Park HU, Liang D, Zhao RY. Phosphatase type 2A-dependent and -independent pathways for ATR phosphorylation of Chk1. J Biol Chem. 2007;282:7287–7298. doi: 10.1074/jbc.M607951200. [DOI] [PubMed] [Google Scholar]
- [60].Durkin SS, Guo X, Fryrear KA, Mihaylova VT, Gupta SK, Belgnaoui SM, Haoudi A, Kupfer GM, Semmes OJ. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem. 2008;283:36311–36320. doi: 10.1074/jbc.M804931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–5328. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- [63].Kamranvar SA, Gruhne B, Szeles A, Masucci MG. Epstein-Barr virus promotes genomic instability in Burkitt’s lymphoma. Oncogene. 2007;26:5115–5123. doi: 10.1038/sj.onc.1210324. [DOI] [PubMed] [Google Scholar]
- [64].Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A. 2009;106:2313–2318. doi: 10.1073/pnas.0810619106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin H.W.t. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe. 2007;1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liang X, Pickering MT, Cho NH, Chang H, Volkert MR, Kowalik TF, Jung JU. Deregulation of DNA damage signal transduction by herpesvirus latency-associated M2. J Virol. 2006;80:5862–5874. doi: 10.1128/JVI.02732-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, Jung JU. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol. 2006;80:2257–2266. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wolf D, Cammas F, Losson R, Goff SP. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J Virol. 2008;82:4675–4679. doi: 10.1128/JVI.02445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, Leid M, Schaeffer E. Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol. 2003;77:5415–5427. doi: 10.1128/JVI.77.9.5415-5427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Everett RD, Murray J, Orr A, Preston CM. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol. 2007;81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kofod-Olsen E, Ross-Hansen K, Mikkelsen JG, Hollsberg P. Human herpesvirus 6B U19 protein is a PML-regulated transcriptional activator that localizes to nuclear foci in a PML-independent manner. J Gen Virol. 2008;89:106–116. doi: 10.1099/vir.0.83224-0. [DOI] [PubMed] [Google Scholar]
- [72].Murphy JC, Fischle W, Verdin E, Sinclair JH. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002;21:1112–1120. doi: 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chang CW, Chou HY, Lin YS, Huang KH, Chang CJ, Hsu TC, Lee SC. Phosphorylation at Ser473 regulates heterochromatin protein 1 binding and corepressor function of TIF1beta/KAP1. BMC Mol Biol. 2008;9:61. doi: 10.1186/1471-2199-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 2005;6:452–457. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Manzur KL, Farooq A, Zeng L, Plotnikova O, Koch AW, Sachchidanand, Zhou MM. A dimeric viral SET domain methyltransferase specific to Lys27 of histone H3. Nat Struct Biol. 2003;10:187–196. doi: 10.1038/nsb898. [DOI] [PubMed] [Google Scholar]
- [76].Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- [77].Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- [78].Jiang X, Sun Y, Chen S, Roy K, Price BD. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem. 2006;281:15741–15746. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- [79].Kamine J, Elangovan B, Subramanian T, Coleman D, Chinnadurai G. Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology. 1996;216:357–366. doi: 10.1006/viro.1996.0071. [DOI] [PubMed] [Google Scholar]
- [80].Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, Motloch P, Takemoto S, Taguchi H, Cole MD, Luscher B, Dittrich O, Tagami H, Nakatani Y, McGee M, Girard AM, Gaughan L, Robson CN, Monnat RJ, Jr., Harrod R. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol. 2005;25:6178–6198. doi: 10.1128/MCB.25.14.6178-6198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Creaven M, Hans F, Mutskov V, Col E, Caron C, Dimitrov S, Khochbin S. Control of the histone-acetyltransferase activity of Tip60 by the HIV-1 transactivator protein, Tat. Biochemistry. 1999;38:8826–8830. doi: 10.1021/bi9907274. [DOI] [PubMed] [Google Scholar]
- [82].Lang SE, Hearing P. The adenovirus E1A oncoprotein recruits the cellular TRRAP/GCN5 histone acetyltransferase complex. Oncogene. 2003;22:2836–2841. doi: 10.1038/sj.onc.1206376. [DOI] [PubMed] [Google Scholar]
- [83].Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol. 2004;78:6744–6757. doi: 10.1128/JVI.78.13.6744-6757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73:650–657. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Barlev NA, Poltoratsky V, Owen-Hughes T, Ying C, Liu L, Workman JL, Berger SL. Repression of GCN5 histone acetyltransferase activity via bromodomain-mediated binding and phosphorylation by the Ku-DNA-dependent protein kinase complex. Mol Cell Biol. 1998;18:1349–1358. doi: 10.1128/mcb.18.3.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005;25:4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- [89].Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J Biol Chem. 2005;280:40195–40200. doi: 10.1074/jbc.C500400200. [DOI] [PubMed] [Google Scholar]
- [91].Sariyer IK, Khalili K, Safak M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: inhibition by small t antigen. Virology. 2008;375:464–479. doi: 10.1016/j.virol.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].O’Shea CC, Choi S, McCormick F, Stokoe D. Adenovirus overrides cellular checkpoints for protein translation. Cell Cycle. 2005;4:883–888. doi: 10.4161/cc.4.7.1791. [DOI] [PubMed] [Google Scholar]
- [93].Ben-Israel H, Sharf R, Rechavi G, Kleinberger T. Adenovirus E4orf4 protein downregulates MYC expression through interaction with the PP2A-B55 subunit. J Virol. 2008;82:9381–9388. doi: 10.1128/JVI.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- [95].Klucky B, Koch B, Radolf M, Steinlein P, Wintersberger E. Polyomavirus tumorantigens have a profound effect on gene expression in mouse fibroblasts. Oncogene. 2004;23:4707–4721. doi: 10.1038/sj.onc.1207640. [DOI] [PubMed] [Google Scholar]
- [96].Weitzman MD, Carson CT, Schwartz RA, Lilley CE. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 2004;3:1165–1173. doi: 10.1016/j.dnarep.2004.03.018. [DOI] [PubMed] [Google Scholar]
- [97].Hart LS, Yannone SM, Naczki C, Orlando JS, Waters SB, Akman SA, Chen DJ, Ornelles D, Koumenis C. The adenovirus E4orf6 protein inhibits DNA double strand break repair and radiosensitizes human tumor cells in an E1B-55K-independent manner. J Biol Chem. 2005;280:1474–1481. doi: 10.1074/jbc.M409934200. [DOI] [PubMed] [Google Scholar]
- [98].Hart LS, Ornelles D, Koumenis C. The adenoviral E4orf6 protein induces atypical apoptosis in response to DNA damage. J Biol Chem. 2007;282:6061–6067. doi: 10.1074/jbc.M610405200. [DOI] [PubMed] [Google Scholar]
- [99].Georgopoulou U, Tsitoura P, Kalamvoki M, Mavromara P. The protein phosphatase 2A represents a novel cellular target for hepatitis C virus NS5A protein. Biochimie. 2006;88:651–662. doi: 10.1016/j.biochi.2005.12.003. [DOI] [PubMed] [Google Scholar]
- [100].Hunt TA, Urbanowski MD, Kakani K, Law LM, Brinton MA, Hobman TC. Interactions between the West Nile virus capsid protein and the host cell-encoded phosphatase inhibitor, I2PP2A. Cell Microbiol. 2007;9:2756–2766. doi: 10.1111/j.1462-5822.2007.01046.x. [DOI] [PubMed] [Google Scholar]
- [101].Hrimech M, Yao XJ, Branton PE, Cohen EA. Human immunodeficiency virus type 1 Vpr-mediated G(2) cell cycle arrest: Vpr interferes with cell cycle signaling cascades by interacting with the B subunit of serine/threonine protein phosphatase 2A. EMBO J. 2000;19:3956–3967. doi: 10.1093/emboj/19.15.3956. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [102].Elder RT, Yu M, Chen M, Zhu X, Yanagida M, Zhao Y. HIV-1 Vpr induces cell cycle G2 arrest in fission yeast (Schizosaccharomyces pombe) through a pathway involving regulatory and catalytic subunits of PP2A and acting on both Wee1 and Cdc25. Virology. 2001;287:359–370. doi: 10.1006/viro.2001.1007. [DOI] [PubMed] [Google Scholar]
- [103].Duong FH, Christen V, Berke JM, Penna SH, Moradpour D, Heim MH. Upregulation of protein phosphatase 2Ac by hepatitis C virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J Virol. 2005;79:15342–15350. doi: 10.1128/JVI.79.24.15342-15350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–1026. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005;19:827–839. doi: 10.1101/gad.1286005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- [108].Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- [109].Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lai CK, Jeng KS, Machida K, Cheng YS, Lai MM. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology. 2008;370:295–309. doi: 10.1016/j.virol.2007.08.037. [DOI] [PubMed] [Google Scholar]
- [112].Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- [113].Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- [114].Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Srivastava N, Gochhait S, de Boer P, Bamezai RN. Role of H2AX in DNA damage response and human cancers. Mutat Res. 2009;681:180–188. doi: 10.1016/j.mrrev.2008.08.003. [DOI] [PubMed] [Google Scholar]
- [116].Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK. Epigenetic reprogramming by adenovirus e1a. Science. 2008;321:1086–1088. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. Adenovirus small e1a alters global patterns of histone modification. Science. 2008;321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Poulin DL, Kung AL, DeCaprio JA. p53 targets simian virus 40 large T antigen for acetylation by CBP. J Virol. 2004;78:8245–8253. doi: 10.1128/JVI.78.15.8245-8253.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- [120].Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- [121].Lieberman PM. Chromatin regulation of virus infection. Trends Microbiol. 2006;14:132–140. doi: 10.1016/j.tim.2006.01.001. [DOI] [PubMed] [Google Scholar]
- [122].Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol. 2007;81:13248–13253. doi: 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol. 2003;77:11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ye J, Shedd D, Miller G. An Sp1 response element in the Kaposi’s sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J Virol. 2005;79:1397–1408. doi: 10.1128/JVI.79.3.1397-1408.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Speck SH, Chatila T, Flemington E. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 1997;5:399–405. doi: 10.1016/S0966-842X(97)01129-3. [DOI] [PubMed] [Google Scholar]
- [126].Everett RD. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000;22:761–770. doi: 10.1002/1521-1878(200008)22:8<761::AID-BIES10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- [127].Vissers JH, Nicassio F, van Lohuizen M, Di Fiore PP, Citterio E. The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div. 2008;3:8. doi: 10.1186/1747-1028-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Nathan D, Ingvarsdottir K, Sterner DE, Bylebyl GR, Dokmanovic M, Dorsey JA, Whelan KA, Krsmanovic M, Lane WS, Meluh PB, Johnson ES, Berger SL. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006;20:966–976. doi: 10.1101/gad.1404206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Varier RA, Kundu TK. Chromatin modifications (acetylation/ deacetylation/ methylation) as new targets for HIV therapy. Curr Pharm Des. 2006;12:1975–1993. doi: 10.2174/138161206777442092. [DOI] [PubMed] [Google Scholar]
- [130].He G, Ylisastigui L, Margolis DM. The regulation of HIV-1 gene expression: the emerging role of chromatin. DNA Cell Biol. 2002;21:697–705. doi: 10.1089/104454902760599672. [DOI] [PubMed] [Google Scholar]