

Abstract
We performed a whole-genome association study on HIV-1 viral load setpoint in an African American cohort (n=515), and an intronic SNP in the HLA-B gene showed one of the strongest associations. Using a subset of patients, we show that this SNP reflects the effect of the HLA-B*5703 allele, which shows a genome-wide significant association with HIV-1 VL setpoint (p=5.6×10−10). These analyses therefore confirm a member of the HLA-B*57 group of alleles as the most important common variant influencing viral load variation in African Americans, consistent with what is observed in individuals of European ancestry in which the most important common variant is HLA-B*5701.
Keywords: HIV, viral load setpoint, host genetics, association study, HLA
A recent-genome wide association studied carried out in individuals of European ancestry identified two polymorphisms associated with viral load at setpoint, and a third set of polymorphisms associated with a simple measure of disease progression [1]. One variant that was found to be associated with setpoint (rs2395029) encodes a nonsynonymous change in the HCP5 gene, and is also a tag for HLA-B*5701, which has been shown to associate with improved early outcomes after HIV-1 exposure [2,3]. The other variant that associated with setpoint (rs9264942) is 35kb upstream of the HLA-C locus and appears to be tagging a causative variant(s). A third variant (rs9261174) that associated with disease progression is located near the ZNRD-1 gene in the MHC region, although functional work on this gene has not yet identified a causal variant. Together, these variants are able to explain about 14% of the observed variation in outcome upon HIV-1 exposure.
A follow-up study investigated the impact of these same SNPs in an HIV-1 positive African-American cohort (n=121)[4]. Similar to the results in individuals of European ancestry, they found that the HLA-C associated variant (rs9264942) again associated with viral load, with the C “high expression” allele leading to lower viral load. They did not see an association with the G allele of rs2395029, however this allele is rare in African-Americans; the allele is in linkage disequilibrium (LD) with HLA-B*5701in people of European ancestry, which is also rare in people of African descent. However, an analysis of the HLA-B alleles present in the region showed an association between HLA-B*57 (comprised predominantly of HLA-B*5703) and favorable virologic outcome.
Although this and other studies [5,6] have assessed the impact of variants first identified in patients of European ancestry in African American individuals, there has not yet been any genome wide investigation of the most important common variants influencing viral load in patients of primarily African ancestry. Here we present the first genome-wide association study of determinants of HIV-1 control performed in a non-European population. Using a cohort of African American individuals (n=515), we sought to evaluate the associations previously reported and to discover novel or population-specific genetic variants that associate with HIV-1 control.
Methods
Samples
This study includesHIV-1 infected African American adult subjects enrolled in either the U.S. military (DoD) HIV Natural History Study (NHS)or in the Multicenter AIDS Cohort Study(MACS). This study was approved by local institutional review boards, and each subject provided written, informed consent.
The U.S. Military (DoD) HIV Natural History Study (NHS) (http://www.idcrp.org/hiv-natural-history-study.html) is an ongoing, prospective, continuous enrollment cohort study of consenting military personnel and beneficiaries with HIV infection and includes participants from the Army, Navy/Marines, Air Force, and their dependents. Since 1985, routine HIV testing (ELISA and confirmatory Western blot) has been used to exclude HIV-infected persons from enlisting for military service or from overseas deployment. Periodic testing among active duty members occurs every one to five years, resulting in a defined seroconversion window for incident HIV infection. Subjects with HIV infection are referred to military medical centers where they receive evaluation, and ongoing care, and are invited to enroll as participants in the DoD HIV NHS.
Those who consent to enroll in the DoD HIV NHS are seen every six months by an HIV specialist as part of the study, in addition to receiving routine clinical care. Data are collected on demographic characteristics, markers of HIV disease progression, medication use, and clinical events with medical record confirmation. Cells, plasma, and serum are collected at each visit and stored in a central repository.
Information was extracted from the database on HIV-infected African American individuals with 4 years between their last negative and first positive HIV tests, at least 5 million cells stored in the repository, and either one viral load result available between 3–12 months post seroconversion (n=140) or two viral loads within3 months -3 years post seroconversion (n=347). Ethnicity was self-identified. Seroconversion date was estimated as the midpoint between their last negative and first positive HIV tests.
The Multicenter AIDS Cohort Study (MACS) (http://www.statepi.jhsph.edu/macs/macs.html) is an ongoing prospective study of the natural and treated histories of HIV-1 infection in homosexual and bisexual men conducted by sites located in Baltimore, Chicago, Pittsburgh and Los Angeles. A total of 6,973 men have been enrolled; 3, 427 subjects were HIV-seronegative at study entry and were tested for seroconversion semiannually by ELISA, with confirmation of positive tests by Western blotting. Of the seroincident subjects, African Americans with DNA and viral load data available before treatment initiation were selected for inclusion in the current study.
Other cohorts referenced in this analysis include HIV-1 infected adult subjects of European ancestry collected by Euro-CHAVI (http://www.chuv.ch/imul/imu_home/imu_collaborations/eurochavi.htm) and MACS who were included in a previous whole genome association study (n=2362) (Fellay et al, submitted). The Euro-CHAVI cohort represents a consortium of 8 European cohorts and 1 Australian cohort that agreed to participate in the Host Genetic Core initiative of the Center for HIV/AIDS Vaccine Immunology (CHAVI).
Genotyping
All samples were genotyped using Illumina Human Hap 1M (n=368), Human Hap 1M-Duo (n=135) or Illumina Human Hap 550K (n=12) bead chips. All samples are brought into a single Bead Studio file using the standard Illumina cluster file. For quality control purposes, any sample that has very low intensity or a very low call rate using the Illumina cluster (<99%) is deleted. All SNPs that have a call frequency below 99% are put into a filter and reclustered (excluding those on the X-chromosome). The reclustering step creates SNP calling errors, but the following procedures are used to prevent the errant calls from being released in the final report:1) the SNPs with a cluster separation value below 0.3 are deleted and 2) any SNPs with a Het Excess value between −1.0 to −0.1 and 0.1 to 1.0 are deleted. The filter is then released and any SNP with a call frequency below 99% is deleted. These procedures resulted in a success rate of genotyping calls ranging from 99.20%–99.999%, and 1,212,217 SNPs were included in the analysis. Ten samples were excluded because of insufficient call rate.
Specification of gender check, cryptic relatedness check, low minor allele frequency (MAF) check, the Hardy-Weinberg equilibrium check, and a recheck of the genotyping quality were all performed as described in [1], and no samples were omitted at these steps. 129,723 SNPs were dropped due to a low MAF. We also required that all SNPs included in the study were successfully genotyped in at least 50% of the samples, hence202, 676 SNPs were dropped at this point, many of which were those not genotyped on all of the chips.
We have used the Illumina 1M and 1M-Duo bead chip data as input into the Penn CNV program [7], which allows us to look at deletions (0, 1 copy) as compared to wild type (2 copies) and duplications (3, 4 copies) as compared to wild type (2 copies). Due to the complications of hemizygosity in males and X-chromosome inactivation in females, this analysis was restricted to autosomes. Additionally, to ensure that we worked with high-confidence CNVs, we excluded any CNV for which the difference of the log likelihood of the most likely copy number state and the less likely copy number state was less than10 (generated using the -conf function in Penn CNV). We limited our analysis to CNVs that occurred in at least 3 people (MAF>0.003). Further quality control thresholds used in Penn CNV are detailed in Ge et al [8]. In this analysis, 497 subjects were included.
HLA-B allotypes were assigned by DNA sequencing, beginning with the amplification of genomic DNA using primers that flank exons 2 and 3. PCR products were cleaned using Ampure (Beckman Coulter). The cleaned products were cycle sequenced on the ABI9700. The cycle sequenced products were cleaned using Clean SEQ (Beckman Coulter) and then run on the ABI PRIZM 3730. Sequence analysis was carried out using Assign (Conexio Genomics).
The EIGENSTRAT method [9] was used to control for population stratification. Assessment of population structure in 616African Americans using EIGENSTRAT results in 73 significant axes of stratification after the removal of 35 population outliers. The first axis makes a larger contribution to the proportion of variation (0.6%) explained than the second axis (0.2%) and reflects the degree of African versus European ancestry in individuals. We therefore used only the first axis as a covariate in our association analyses to control for population stratification.
Phenotype
Setpoint viral load was defined as1) the mean viral load for samples with two or more viral load values that were collected at least 30 days apart, within 3 months –3 years post seroconversion, and were within a 1 log range, similar to what was done in the previous study of determinants of setpoint in subjects of European ancestry [1], or2) the first available viral load within 3 –12 months post seroconversion, as long as there was a corresponding CD4+ T cell count greater than 350/ul. All viral loads were prior to the initiation of antiretroviral therapy. Viral loads from the two definitions were highly correlated (r2=0.74) and the first definition was preferentially used when sufficient data was available.
Statistical Analysis
Viral load at setpoint was used as a quantitative trait in a linear regression using additive allelic effects. Because previous studies have found that gender and age may be associated with viral load, these factors were used as covariates the model [10,11]. The first Eigenstrat axis was used to control for population stratification and the cohort was also included in the model given baseline differences between the two groups. Individual regressions for each SNP were carried out using PLINK v1.06 [12,13]. Bonferroni correction was used to control for multiple comparisons; a p-value < 5×10−8 was considered genome wide significant.
All HLA-B allotypes were tested for association with viral load set point in a linear regression model, and were also evaluated to determine if they were responsible for associations observed for SNPs in the genome-wide SNP association analyses.
Results
From the DoD HIV NHS, 487subjects met inclusion criteria and 471 were successfully genotyped. From the MACS cohort, 158 subjects met the inclusion criteria and 145 were successfully genotyped. Thirty-five subjects were dropped due to the EIGENSTRAT correction for ancestry, and 66 subjects were not included because their viral load results did not meet the definition of setpoint described in the Methods section, leaving 515 subjects in the final dataset. Table 1 shows the baseline characteristics of subjects in the DoD and MACS cohorts as well as the bead chips which were used for genotyping. There were more females in the DoD cohort (p=0.006) and subjects in this cohort were on average younger at seroconversion (p<0.001); however, there was no difference between the cohorts in mean set point viral load (p=0.13).
Table 1.
Baseline characteristics of subjects in the MACS and DoD cohorts (n=515)
| MACS | DoD | |
|---|---|---|
| Number of subjects | 118 | 397 |
| % Male | 100% | 94% |
| Mean Age at Seroconversion (range) | 32.0 (20–55) | 27.5(18–55) |
| Mean set point VL (log10, range) | 4.00 (1.91–6.01) | 4.12 (1.91–5.97) |
| Mean Year of Seroconversion (range) | 1991 (1984–2005)a | 1996 (1986–2003) |
| MAF for rs2523608 | 39.4%b | 35.4% |
| HLA-B*5703 carriers | 7.5%b | 8.3% |
| Illumina 1M | 53% | 77% |
| Illumina 1M-Duo | 37% | 23% |
| Illumina 550K | 10% | 0% |
Serocoversion date was only available for 52 of the 118 MACS subjects.
p>0.05 comparing MACS with DoD cohort
MAF = minor allele frequency
No single SNP had a genome-wide significant association (p<5×10−8) with viral load at setpoint. The tables list the top 20 genome-wide associations with setpoint (Table 2), the top 10 associations from the MHC region (Table 3A), and the top 10 functional SNPs that associate with set point (Table 3B). A functional SNP was defined as a SNP that would cause the gain or loss of a stop codon, a nonsynonymous coding change, or one that occurred in a splice site.
Table 2.
Top 20 setpoint associated outcomes in African Americans (n=515)
| SNP | Rank | CHR | P-Value | Closest gene | Type | Frequency |
|---|---|---|---|---|---|---|
| rs454422 | 1 | 20 | 1.49E-06 | MCM8 | Intronic | 0.258 |
| rs2523608 | 2 | 6 | 2.29E-06 | HLA-B | Intronic | 0.366 |
| rs6948404 | 3 | 7 | 3.41E-06 | AOAH | Intronic | 0.074 |
| rs558718 | 4 | 19 | 3.71E-06 | EVI5L | Intronic | 0.110 |
| rs1357339 | 5 | 11 | 4.58E-06 | N/A | Intergenic | 0.035 |
| rs1413191 | 6 | 13 | 4.61E-06 | GPC5 | Intronic | 0.181 |
| rs236104 | 7 | 20 | 6.72E-06 | MCM8 | Intronic | 0.252 |
| rs7998089 | 8 | 13 | 7.41E-06 | GPC5 | Intronic | 0.191 |
| rs2593321 | 9 | 3 | 7.70E-06 | AC023798.16 | Intergenic | 0.231 |
| rs6492611 | 10 | 13 | 7.97E-06 | GPC5 | Intronic | 0.208 |
| rs4872511 | 11 | 8 | 9.25E-06 | PPP3CC | Downstream | 0.011 |
| rs2280890 | 12 | 8 | 9.25E-06 | SORBS3 | Upstream | 0.011 |
| rs2789066 | 13 | 6 | 9.41E-06 | RP11-100A16.1 | Upstream | 0.127 |
| rs430374 | 14 | 18 | 1.01E-05 | ST8SIA5 | Intergenic | 0.196 |
| rs9910853 | 15 | 17 | 1.07E-05 | ZNF652 | Intronic | 0.085 |
| rs762372 | 16 | 21 | 1.10E-05 | U6 | Upstream | 0.470 |
| rs236106 | 17 | 20 | 1.23E-05 | MCM8 | Intronic | 0.252 |
| rs1348478 | 18 | 5 | 1.26E-05 | PRR16 | Intergenic | 0.337 |
| rs12103812 | 19 | 17 | 1.34E-05 | ZNF652 | 3′ UTR | 0.084 |
| rs8014482 | 20 | 14 | 1.44E-05 | AL355773.4-1 | Intergenic | 0.256 |
Age, gender, cohort, and one significant EIGENSTRAT axis were used as covariates.
Table 3.
Other setpoint associated outcomes in African Americans (n=515)
| A. Top 10 setpoint associated outcomes in the MHC region in African Americans | |||||
|---|---|---|---|---|---|
| SNP | Overall Rank | P-Value | Gene | Type | Frequency |
| rs2523608 | 2 | 2.29E-06 | HLA-B | Intronic | 0.366 |
| rs34548063 | 23 | 1.57E-05 | STK19 | Stop Gained | 0.028 |
| rs2523933 | 29 | 2.03E-05 | HLA-80 | Intergenic | 0.205 |
| rs2844538 | 53 | 5.07E-05 | ZDHHC20P2 | Downstream | 0.492 |
| rs2596503 | 81 | 8.15E-05 | HLA-B | Downstream | 0.162 |
| rs9266689 | 96 | 9.86E-05 | ZDHHC20P2 | In non-coding gene | 0.485 |
| rs4151650 | 99 | 0.0001 | CFB, C2 | Synonymous Coding | 0.030 |
| rs1736936 | 99 | 0.0001 | HLA-G | Upstream | 0.492 |
| rs9378200 | 150 | 0.0002 | UQCRHP | Intergenic | 0.013 |
| rs4713213 | 150 | 0.0002 | OR5V1;OR12D3 | Intronic | 0.331 |
| B. Top 10 functional SNPs that associate with setpoint | |||||
| rs34548063 | 23 | 1.57E-05 | STK19 | Stop Gained | 0.028 |
| rs1034405 | 91 | 9.65E-05 | C3orf18 | Nonsynonymous Coding | 0.332 |
| rs4838865 | 99 | 0.0001 | TUBGCP6 | Nonsynonymous Coding | 0.048 |
| rs6542522 | 99 | 0.0001 | C2orf76 | Nonsynonymous Coding, Splice site | 0.196 |
| rs2280801 | 332 | 0.0004 | BAT2 | Nonsynonymous Coding | 0.012 |
| rs13146272 | 332 | 0.0004 | CYP4V2 | Nonsynonymous Coding | 0.394 |
| rs2273549 | 332 | 0.0004 | TPC11L1 | Nonsynonymous Coding | 0.092 |
| rs2278329 | 332 | 0.0004 | OSMR | Nonsynonymous Coding | 0.042 |
| rs10423723 | 435 | 0.0005 | AC020907.3 | Nonsynonymous Coding | 0.625 |
| rs7258700 | 435 | 0.0005 | AC020907.3 | Nonsynonymous Coding | 0.625 |
Age, gender, cohort and one significant EIGENSTRAT axis were used as covariates.
The most significant SNP in the MHC region for association with viral load setpoint in this African American cohort was rs2523608, located in the HLA-B gene (p=2.3×10−06, Figure 1A, Table 3A). We found that the same SNP was also significantly associated with HIV-1 set point in a large sample of individuals of European ancestry (p = 1.1×10−06, corrected for age and gender, Figure 1B). This association remains nominally significant (p=0.0083) after accounting for variants in the MHC region previously shown to associate with HIV-1 outcomes (rs2395029, rs9264942, rs9261174), and 12 significant EIGENSTRAT axes to control for population stratification in this cohort.
Figure 1. Distribution of mean viral load (VL) setpoint according to genotype.
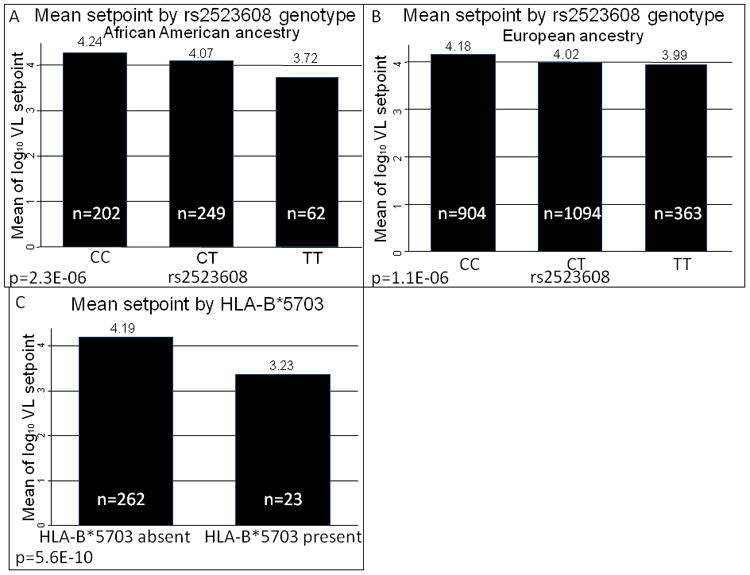
The direction of the effect of the rs2523608 genotype is consistent in A) African Americans and B) Europeans. C) Samples with HLA-B*5703 have a lower HIV-1 viral load setpoint.
The rs2523608 variant is located in intron 5 (according to Ensembl transcript ENST00000376228), over 100bp from the nearest exon. Analysis of the HLA-B allotypes from 285 genotyped study subjects showed that this association was due to the association between rs2523608 and HLA-B*5703(D′=1, r2=0.075. The degree of linkage disequilibrium can be quantified using the D′ statistic [14]. This statistic compares the ancestral recombination patterns between two variants by standardizing allele frequencies. A D′=1 indicates that one variant always appears on the background of the other. On the other hand, the r2 statistic is sensitive to allele frequency differences and assesses the degree to which the two variants appear together [15,16]). When considered alone, HLA-B*5703 was by far the strongest association with HIV-1 viral load setpoint for any HLA-B allotype, showing genomewide significance (p=5.6×10−10, with age, gender, cohort and one EIGENSTRAT axis as covariates)(Table 4, Figure 1C). Moreover, when HLA-B*5703 was included as a covariate, it was able to account for the effect of the rs2523608 genotype. This analysis shows that HLA-B*5703 is the most important common variant in influencing viral load in African Americans, explaining about 10% of the variation in viral load setpoint in this dataset, with an allele frequency of about 4.0%.
Table 4.
Top associations between HLA-B allotypes and setpoint(n=285)
| Allotype | Rank | P-value | Frequency |
|---|---|---|---|
| HLA-B*5703 | 1 | 5.6E-10 | 0.040 |
| HLA-B*3910 | 2 | 0.00032 | 0.006 |
| HLA-B*1517 | 3 | 0.00040 | 0.006 |
| HLA-B*4501 | 4 | 0.00084 | 0.062 |
| HLA-B*1302 | 5 | 0.020 | 0.005 |
| HLA-B*580101 | 6 | 0.021 | 0.016 |
| HLA-B*5802 | 7 | 0.022 | 0.040 |
| HLA-B*4201 | 8 | 0.022 | 0.040 |
| HLA-B*140201 | 9 | 0.024 | 0.020 |
| HLA-B*1801 | 10 | 0.025 | 0.016 |
Age, gender, cohort and one significant EIGENSTRAT axis were used as covariates.
CNV analysis
There were 8,724 SNPs that showed evidence of a duplication and 16,778 SNPs that showed evidence of a deletion. The CNV calls for each SNP were then run as genotypes in a regression using an additive genetic model, testing for association with HIV-1 setpoint. Gender, cohort and the first EIGENSTRAT axis were used as covariates. Using a Bonferroni correction (6×10−6 for duplications and 3×10−6 for deletions), no SNPs reached genome-wide significance for either deletions or duplications. Furthermore, when the association results from these two models were compared, no SNP associated with both deletions and duplications with a p-value of less than p=0.05.
Association of previously implicated variants
We also analyzed genetic variants that have previously been shown to have an effect on HIV-1setpoint. First, we tested the association of rs2395029, a nonsynonymous SNP in the HCP5 gene that is a tag for the functional allele HLA-B*5701 and found this SNP to show a weak association with viral load setpoint (p=0.030, Table 5). This SNP has a very low minor allele frequency in African Americans (MAF=0.008) due its virtual absence in West African populations. Power to detect an association in this cohort is, therefore, only 81% at p<0.05, assuming that the effect size in our cohort is comparable to that seen for individuals of European descent [1].
Table 5.
Association outcomes for genetic variants that have been previously reported to associate with setpoint
| SNP | Gene | Location | Pa(AA) | MAF (AA) | Pb (European) | MAF (European) |
|---|---|---|---|---|---|---|
| rs2395029 | HCP5 | Nonsynonymous | 0.030(n=513) | 0.008 | 4.6×10−35 (n=2362) | 0.048 |
| rs9264942 | HLA-C | Upstream | 0.018(n=515) | 0.286 | 6.0×10−32 (n=2362) | 0.412 |
| rs9261174 | ZNRD1 | Intergenic | 0.352(n=511) | 0.245 | 1.1×10−04 (n=2362) | 0.141 |
| rs333 (Δ32) | CCR5 | Genic Deletion | 0.484(n=502) | 0.017 | 1.9×10−10 (n=2334) | 0.099 |
P-values are corrected for age, gender, cohort, and one significant EIGENSTRAT axis.
P-values are corrected for age, gender and 12 significant EIGENSTRAT axes.
We then tested the association between rs9264942, a C → T polymorphism 35kb upstream of the HLA-C gene. This SNP itself is not causal, but is a tagging SNP for unknown causal variant(s). We observed a weak association between rs9264942 and setpoint in African Americans (p = 0.018, Table 5).
We also tested the associations between viral load at setpoint and rs9261174, a SNP located near the ZNRD1 gene in the MHC region, and CCR5-Δ32, a 32bp deletion in the CCR5 gene (rs333)that is rare in non-European populations. In our African American cohort, neither rs9261174 (p=0.352) nor CCR5-Δ32 (p=0.484) showed an association with viral load at setpoint (Table 5).
Discussion
This was the first genome-wide association study on HIV-1 outcomes to be carried out in an African-American cohort, the majority of who were infected with HIV-1 subtype B. We have shown that the intronic SNP rs2523608, the top associated SNP in the MHC region, is tagging HLA-B*5703. The D′ =1 between rs2523608 and HLA-B*5703, and a regression model shows that the HLA-B*5703 genotype is able to account for the effect of this intronic SNP. HLA-B*5703 strongly associates with viral load at setpoint, and reached whole genome-significance in the subset of samples for which HLA-B allotype data is available (p=5.6×10−10 in a model that also included age, gender, cohort, and the first EIGENSTRAT axis, n=285).
HLA-B*5701 is an important mechanism of HIV-1 viral control in the European population. It has an allele frequency of about 6.1% in a European population, but was not observed in a Yoruban population [17]. Its close relative HLA-B*5703 is absent in a European population, but has an allele frequency of about 5.8% in a Yoruban population [17]. Here, we have shown that African-American individuals who have HLA-B*5703 also show improved viral control, of a similar magnitude to that afforded by HLA-B*5701 in people of European descent. Thus, our results indicate that the general mechanism of genetic control of the HIV-1 virus is similar between African-Americans and Europeans: HLA-B*5701 accounts for about 6% of the observed variation in viral load setpoint in Europeans (Fellay et al, submitted), and HLA-B*5703 accounts for about 10% of the observed variation in viral load setpoint in African-Americans. There was also a small contribution to HIV-1 control by HLA-B*5701 (frequency=0.3%) in our African-American dataset, due to admixture.
In addition to HLA-B*5703, we also found that HLA-B*3910 and HLA-B*1517 may be playing a lesser role in viral control, although additional studies would be needed to confirm these observations. This pattern was similar to that observed in people of European descent, where HLA-B*5701 is the largest determinant of HIV-1 control but other HLA-B alleles (HLA-B*27, HLA-B*35 and others) also play a role (Fellay et al, submitted). The alleles that we have identified are further supported by an analysis of the MHC region conducted in southern African populations infected with HIV-1 subtype C, where HLA-B*5703 was found to be the HLA allele most strongly associated with a decreased viral load setpoint [3,18], with a weaker contribution by HLA-B*39[18].
We found a reduced or absent association with viral load setpoint when we explicitly checked variants that had been shown to associate in a cohort of European descent. Similar outcomes were seen in Shrestha et al. [4], who saw a reduced association between rs9264942 and setpoint, and no association between rs2395029 and setpoint. Our sample size was over four times larger than Shrestha et al, and revealed only weak associations between both variants and setpoint. So while both rs9264942 and rs2395029 definitively associate with viral load setpoint in European populations, neither study was able to replicate these associations in an African American cohort.
It is worth noting that it is only the HLA-B*5701 association in the previous study [1]where the causal site is thought to have been identified. In the cases of rs9264942 and rs9261174, it is not likely that the associated variants are themselves causal, but are rather markers of an as yet unidentified causal site or sites. It may therefore not be a coincidence that the HLA-B*57 association is the only one to show a similar effect in African Americans as in individuals of European ancestry. For the other two, the different and generally lower linkage disequilibrium in this region in African Americans (Figure 2) could mean that the causal sites are no longer being tagged by these variants.
Figure 2. Average linkage disequilibrium (LD) in the MHC region in people of European descent versus African Americans.
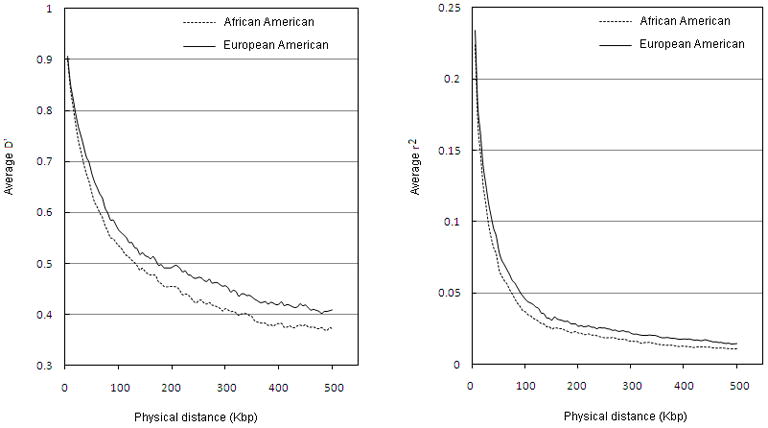
The average D′ and the average r2 are both shorter in the African American genome as compared to the European genome.
A potential limitation of this study is that HIV-infected persons with rapid disease progression may have been excluded. Rapid progressors may not have had many research visits and therefore may not have had enough cells in the repository to be included in this study. Also, because of their rapid progression, they may not have had viral loads available that satisfied the definition of set point. Approximately ten percent of the subjects in this study progressed to a CD4 count < 200 cells/mm3 within 2years of seroconversion; therefore, there were at least some rapid progressors included.
Earlier studies have likewise suggested that HLA-B*5703 may also be involved in HIV-1 control in African-Americans, but our study shows that it is indeed the most significant common genetic factor affecting early viral control in this population. By using a genome-wide scan to implicate another allele of HLA-B*57 in HIV-1 control, in an entirely different ancestral background from where this association had previously been observed, we provide further support as to the important role of HLA-B*57 in HIV-1 control and the decreased fitness level of the viral mutants that are selected for by HLA-B*57. Given the increased burden of disease in African and African-American populations, and the paucity of common variants that clearly influence HIV-1 control, it is important to continue to investigate rare variants that may function specifically in these populations.
Acknowledgments
We would like to thank all of the patients in the DoD HIV NHS, MACS, and Euro-CHAVI cohorts. The members of the IDCRP HIV Working Group (by site) are: National Institute of Allergy and Infectious Diseases, Bethesda, MD: M. Polis, J. Powers, E. Tramont.
Naval Medical Center, Portsmouth, VA: J. Maguire.
Naval Medical Center, San Diego, CA: M. Bavaro, N. Crum-Cianflone, H. Chun.
National Naval Medical Center, Bethesda, MD: C. Decker, A. Ganesan, T. Whitman.
San Antonio Military Medical Center, San Antonio, TX: W. Bradley, V. Marconi, S. Merritt, J. Okulicz.
Tripler Army Medical Center, Honolulu, HI: A. Johnson.
Uniformed Services University of the Health Sciences, Bethesda, MD: B. Agan.
Walter Reed Army Institute of Research, Rockville, MD: C. Eggleston, L. Jagodzinski, R. O’Connell, S. Peel.
Walter Reed Army Medical Center, Washington, DC: C. Hawkes, G. Wortmann, M. Zapor.
University of Minnesota: L Eberly, A. Lifson.
Funding:
Support for this work was provided by the Infectious Disease Clinical Research Program (IDCRP), a Department of Defense (DoD) program executed through the Uniformed Services University of the Health Sciences. This project has been funded in whole, or in part, with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), under Inter-Agency Agreement Y1-AI-5072. The content of this publication is the sole responsibility of the authors and does not necessarily reflect the views or policies of the NIH or the Department of Health and Human Services, the DoD or the Department of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government. The authors have no commercial or other associations that might pose a conflict of interest.
Funding was provided by the NIAID Center for HIV/AIDS Vaccine Immunology grant AI067854. K.P. was funded by NIH Genetics Training Grant 5T32 GM007754-29.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The MACS is funded by the National Institute of Allergy and Infectious Diseases, with additional supplemental funding from the National Cancer Institute; and the National Heart, Lung, and Blood Institute: UO1-AI-35042, 5-M01-RR-00052 (GCRC), UO1-AI-35043, UO1-AI-37984, UO1-AI-35039, UO1-AI-35040, UO1-AI-37613, and UO1-AI-35041.
Footnotes
Conflict of Interest: The authors do not have any commercial or other association that might pose a conflict of interest.
This work has not previously been presented at any scientific meeting.
No additional changes to the author affiliations that are listed on the first page.
MACS centers are located at: The Johns Hopkins Bloomberg School of Public Health, Baltimore, MD: J. Margolick.
Howard Brown Health Center and Northwestern University Medical School, Chicago, IL: J. Phair.
University of California, Los Angeles, Los Angeles, CA: R. Detels.
University of Pittsburgh, Pittsburgh, PA: C. Rinaldo.
Data Analysis Center, Baltimore, MD: L. Jacobson.
References
- 1.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Migueles SA, Sabbaghian MS, Shupert WL, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA. 2000;97:2709–14. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiepiela P, Leslie AJ, Honeyborne I, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–75. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 4.Shrestha S, Aissani B, Song W, Wilson CM, Kaslow RA, Tang J. Host genetics and HIV-1 viral load set-point in African-Americans. AIDS. 2009;23:673–7. doi: 10.1097/QAD.0b013e328325d414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shrestha S, Strathdee SA, Galai N, et al. Behavioral Risk Exposure and Host Genetics of Susceptibility to HIV-1 Infection. J Infect Dis. 2006;193:16–26. doi: 10.1086/498532. [DOI] [PubMed] [Google Scholar]
- 6.Lama J, Planelles V. Host factors influencing susceptibility to HIV infection and AIDS progression. Retrovirology. 2007;4:52. doi: 10.1186/1742-4690-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K, Li M, Hadley D, et al. Penn CNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 9.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 10.Napravnik S, Poole C, Thomas JC, Eron JJ. Gender difference in HIV RNA levels: a meta-analysis of published studies. J Acquir Immune Defic Syndr. 2002;31:11–9. doi: 10.1097/00126334-200209010-00002. [DOI] [PubMed] [Google Scholar]
- 11.Quinn TC, Wawer MJ, Sewankwambo N, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N Engl J Med. 2000;342:921–9. doi: 10.1056/NEJM200003303421303. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plink… [Accessed 15 June 2009];Whole genome association analysis toolset. Available at: http://pngu.mgh.harvard.edu/purcell/plink/
- 14.Lewontin RC. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nature Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- 16.Pritchard JK, Przeworski M. Linkage Disequilibrium in Humans: Models and Data. Am J Hum Genet. 2001;69:1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Bakker PI, Mc Vean G, Sabeti PC, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38:1166–72. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang J, Tang S, Lobashevsky E, et al. Favorable and Unfavorable HLA CLass I Alleles and Haplotypes in Zambians Predominantly Infected with Clade C Human Immunodeficiency Virus Type 1. J Virol. 2002;76:8276–8284. doi: 10.1128/JVI.76.16.8276-8284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]