

Abstract
Dopamine released by amphetamine decreases the in vivo binding of PET radioligands to the dopamine D2 receptor. Although concentrations of extracellular dopamine largely return to baseline within one to two hours after amphetamine treatment, radioligand binding remains decreased for several hours. The purpose of this study was to determine whether the prolonged decrease of radioligand binding after amphetamine administration is caused by receptor internalization. To distinguish dopamine displacement from receptor internalization, we used wild-type and arrestin3 (arr3) knockout mice, which are incapable of internalizing D2 receptors. In addition, we used both the D2 selective agonist [11C]MNPA (which is thought to bind to the high affinity state of the receptor) and the D2 selective antagonist [18F]fallypride (which does not differentiate between high and low affinity state). After an initial baseline scan, animals were divided in three groups for a second scan: either 30 min or 4 hours after amphetamine administration (3 mg/kg, i.p.) or as retest. At 30 min, [11C]MNPA showed greater displacement than [18F]fallypride, but each radioligand gave similar displacement in knockout and wild-type mice. At 4 hours, the binding of both radioligands returned to baseline in arr3 knockout mice, but remained decreased in wild-type mice. Radioligand binding was unaltered on retest scanning. Our results suggest that the prolonged decrease of radioligand binding after amphetamine is mainly due to internalization of the D2 receptor rather than dopamine displacement. In addition, this study demonstrates the utility of small animal PET to study receptor trafficking in vivo in genetically modified mice.
Keywords: [11C]MNPA, [18F]fallypride, dopamine, D2 receptor, internalization, arrestin3, PET
INTRODUCTION
Agonist-induced internalization of G-protein coupled receptors, which include the D2 dopamine receptor, typically requires the proteins arrestin and dynamin (Pierce and Lefkowitz, 2001). Receptor internalization and desensitization is initiated after agonist binding through recruitment of an arrestin protein to the receptor. Arrestin targets the receptor for internalization by scaffolding the receptor to clathrin-coated pits. Dynamin pinches these pits from the membrane and thereby creates intracellular vesicles. Once internalized, D2 receptors can be sorted for recycling to the cell surface or degradation through lysosomes (Namkung et al., 2009). There are two known non-visual arrestins, referred to as arrestin2 and arrestin3, or β-arrestin1 and β-arrestin2 (Lefkowitz and Shenoy, 2005). Knockout of selective arrestin subtypes can block the internalization of subgroups of G-protein coupled receptors (Kohout et al., 2001). We recently demonstrated that knockout of the arrestin3 gene in mice blocks agonist-induced internalization of dopamine D2 receptors in striatal tissue slices (Skinbjerg et al., 2009). Thus, comparison of arrestin3 (arr3) knockout and wild-type mice can be used to examine the impact of internalization of D2 receptors.
Although internalization of dopamine D2 receptors has been extensively studied in vitro, its effect on in vivo receptor binding has largely been unexplored. Positron emission tomography (PET), an in vivo image technique, has been used for more than two decades to image and quantify dopamine D2 receptors in monkey and man. In addition, D2 receptor imaging has been combined with intravenous injection of psychostimulants (e.g., amphetamine or methylphenidate) to examine the interaction of endogenously released dopamine with the binding of PET radioligands to the D2 receptor. The synaptic release of dopamine decreases binding of the PET radioligand, which could be due one or more of the following three possibilities: direct displacement by dopamine, decreased affinity of the receptor, and/or internalization of the receptor. Decreased binding of the D2 radioligand persists for several hours after administration of the psychostimulant, although concentrations of extracellular dopamine largely return to baseline within one to two hours (Laruelle, 2000). Several investigators have speculated that the prolonged decrease of in vivo radioligand binding could be caused by internalization of the receptor, which would no longer be available to, or result in decreased affinity for, radioligand binding. This theory is supported by PET imaging studies in cats, which reported a decrease in Bmax of [11C]raclopride 2.5 hours after amphetamine and by ex vivo studies in rats, which reported decreased binding of [3H]raclopride in striatum up to 6 hours after amphetamine administration (Ginovart et al., 2004; Sun et al., 2003).
Agonist radioligands for the dopamine D2 receptor have recently been developed for in vivo PET imaging (Gao et al., 1990; Neumeyer et al., 1990; Steiger et al., 2009; Wilson et al., 2005). Based on in vitro binding studies, agonist PET radioligands are thought to bind in vivo preferentially to a high affinity state, whereas antagonists do not differentiate between high and low affinity states (Laruelle, 2000). The preferential binding of agonists, including endogenously released dopamine, to a high affinity state has been proposed as the reason that psychostimulants displace about two-fold greater percentage of the specific binding of agonist than of antagonist radioligands (Cumming et al., 2002; Laruelle, 2000; Narendran et al., 2004; Seneca et al., 2006). However, this proposal depends upon the existence of two affinity states in vivo, and recent PET and ex-vivo studies in monkeys and rats challenge the existence of two affinity states for the D2 receptor (Finnema et al., 2009; McCormick et al., 2009; McCormick et al., 2008).
The purpose of this study was to determine whether the prolonged decrease of in vivo radioligand binding induced by amphetamine is caused by receptor internalization. We assumed that the effects of amphetamine in wild-type mice would reflect both direct displacement and receptor internalization, whereas that in arr3 knockout mice would reflect only direct displacement. In addition, we used both an agonist, [11C]MNPA, and an antagonist, [18F]fallypride, radioligand. We predicted that PET radioligand binding in striatum would return to baseline more quickly in arr3 knockout mice compared to wild-type mice for both agonist and antagonist radioligands. However, because agonist radioligands are thought to bind to the high affinity (i.e. G-protein coupled) state of the D2 receptor and internalized receptors are uncoupled from G-proteins and thus in a low affinity state for agonist binding, we predicted that [11C]MNPA would show greater effects than [18F]fallypride for both direct displacement and internalization.
MATERIALS AND METHODS
Radioligand preparation
[18F]Fallypride was synthesized based on the literature method (Mukherjee et al., 1995), but using a microwave-accelerated Synthia radiosynthesis platform (Bjurling et al., 1995; Lazarova et al., 2007; Lu et al., 2009). No-carrier-added [18F]fluoride ion (~200 mCi) in [18O]water (250–400 μL) plus a solution of K2CO3 (5.5 mg/mL) and kryptofix 2.2.2 (30 mg/mL) in MeCN-H 2O (9: 1 v/v; 100 μL) was placed in a 1-mL V-vial. Water was removed by using four cycles of azeotropic evaporation with acetonitrile (600 μL each time) at 90 W for 2 min in a 521-type microwave cavity (Resonance Instrument Inc., IL). Tosyl-fallypride’ precursor (ABX, Germany, 1.0 mg) in acetonitrile (400 μL) was transferred into the V-vial containing anhydrous [18F]F−-K+-kryptofix 2.2.2. The mixture was irradiated with microwaves (60 W, 2 min × 2). The reaction mixture was diluted with water (700 μL) and injected onto a HPLC column (Luna C18, 5 μm, 250 × 10 mm; Phenomenex, CA) eluted with MeCN-aq. 25 mM HCOONH 4 (43: 57, v/v) at 3 mL/min. The HPLC fraction containing [18F]fallypride (tR = 16–18 min) was collected and diluted with water (25 mL). The aqueous solution was passed through a SPEC C-18 AR column (3 mL, Varian, CA) and further washed with water (8 mL). [18F]Fallypride for injection was formulated in physiological saline with 10% ethanol (10 mL) and filtered through a Millex-MP filter (0.22 um, Millipore, Carrigtwohill, Ireland). The decay-corrected radiochemical yield was 38.4 ± 5.8%, and the specific activity at time of injection was 151 ± 54 GBq/μmol (n = 22 syntheses). The chemical purity was >99%, and the radiochemical purity was >95%. The mean activity injected into mice was 5 ± 1 MBq, which was accompanied by 0.04 ± 0.02 nmol of carrier fallypride. [11C]MNPA was prepared by 11C-methylation of the precursor (R)-2-hydroxy-10,11-acetonide-NPA using a two-step labeling method (Gao et al., 1990; Steiger et al., 2009). Chemical purity was >98%, and radiochemical purity was >95%. The specific activity of [11C]MNPA at the time of injection into mice was 73 ± 23 GBq/μmol (n = 17 syntheses). The mean injected activity was 9 ± 1 MBq, which was accompanied by 0.14 ± 0.05 nmol of carrier.
Animals
Arrestin3 knockout and wild-type mice (2–6 month of age, 24 ± 6 g) were a kind gift from Robert J. Lefkowitz, (Duke University, Durham, NC, USA) and bred as described (Bjork et al., 2008). Arrestin3 knock out mice were healthy and viable and had no obvious phenotypic abnormalities. The mice were group housed (2–5 per cage) in a temperature and humidity controlled room with a 12 hour light-dark cycle and allowed free access to food and water. All animal procedures were in accordance with Guide for Care and Use of Laboratory Animals and were approved by National Institute of Mental Health Animal Care and Use Committee (Bethesda, MD, USA).
PET studies
A total of 60 mice were imaged twice with one week of rest between scans: 30 wild-type and 30 knockout mice (15 female and 15 male of each genotype). Mice were divided into three groups matched for sex and genotype and imaged with either [11C]MNPA or [18F]fallypride. After an initial baseline scan, animals had a second scan, either with drug treatment or as retest baseline. One group received amphetamine (3 mg/kg, i.p.) 30 min before the scan; another group received the same dose of amphetamine 4 hours before the scan; and the last group had a baseline retest and did not receive any treatment. Anesthesia was induced with 5% isoflurane and maintained with 1.5% isoflurane administered through a nose cone. Body temperature was maintained between 36.5 and 37°C with a heating pad or a heating lamp. Radioligands were administered through a tail vein catheter (PE-10, Becton Dickinson, NJ, USA) in a volume of 0.1 mL over 20 sec. Four to six mice were scanned simultaneously. To decrease variability of resolution in the field of view, the mice were positioned to be approximately equidistant from the central z-axis.
PET studies were performed on two different cameras: an Advanced Technology Laboratory Animal Scanner (ATLAS), which has a reconstructed resolution of 1.6 mm full-width at half maximum (Seidel et al., 2003), and a microPET Focus 120 scanner (Siemens Medical Solutions, Inc. Knoxville, TN, USA), which has a similar high resolution (Kim et al., 2007). These scanners can accommodate up to 4 and 6 mice, respectively, which allows pair-wise imaging of wild-type and knockout mice. Dynamic emission data were collected continuously for 90 min, with increasing duration of time frames from 20 s to 20 min. Data were constructed with an OSEM algorithm without attenuation or scatter correction.
Two cameras were used because the ATLAS broke after half of all scans were completed. We thought the results from the two cameras could be combined for three reasons. 1) The resolution of both devices was similar. 2) We scanned equal percentages of wild-type and knockout mice on each camera and both scans (baseline and treatment/retest) was performed on the same camera. 3) The scanner was included as covariate in the statistical model and showed no effect.
Image analysis
Images were analyzed with pixel-wise modeling software (PMOD Technologies, Zurich, Switzerland) using the two-parameter multilinear reference tissue model (MRTM2) (Ichise et al., 2003). The outcome measure was binding potential (BPND), which is the ratio at equilibrium of specific to nondisplaceable uptake (Innis et al., 2007). The cerebellum was used as the reference region to measure nondisplaceable uptake (free plus nonspecifically bound). Regions of interest were visually identified with guidance from a mouse brain stereotactic atlas (Paxinos and Franklin, 2001), and the regions were similar in size for the left and right striatum (total striatum 14.6 mm3). To reduce noise from bone uptake of [18F]fluoride ion, the cerebellar region for [18F]fallypride was more centrally located and smaller than that for [11C]MNPA (12.8 vs. 25.6 mm3).
Data analysis
Data were analyzed with repeated measures analysis of variance (rmANOVA) with BPND as the dependent variable, treatment as the within-subject variable, and time and genotype (knockout/wild-type) as between-subject variables. The cameras were included as a covariate. The values of BPND were tested and found to be normally distributed before the rmANOVA were performed. Significance was defined as P < 0.05.
RESULTS
Highest radioactivity concentration was found in striatum after injection of both [11C]MNPA and [18F]fallypride (Fig 1), but the time course and striatum to cerebellum ratio were different. [18F]Fallypride washed out more slowly than [11C]MNPA, as expected from its higher affinity (Fig 2). Similar to studies in monkeys and humans, the ratio of target to background (striatum to cerebellum) was higher for [18F]fallypride than for [11C]MNPA (Fig 2). The ratio at equilibrium of specific to nondisplaceable uptake (BPND) was about 3 fold higher for [18F]fallypride (3.17 ± 0.93) than for [11C]MNPA (0.92 ± 0.16).
Fig. 1.
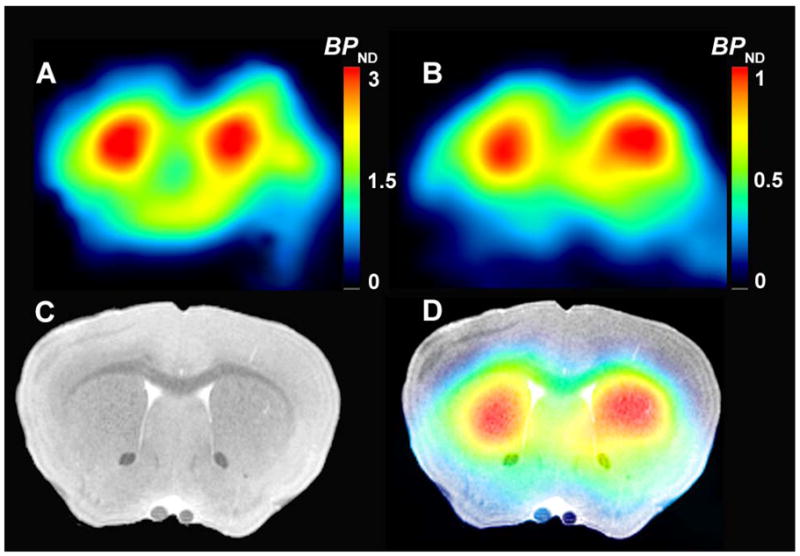
Representative parametric images of binding potential (BPND) from wild-type mice for [18F]fallypride (A) and for [11C]MNPA (B). The corresponding coronal MRI image (C) is fused with the image of BPND for [11C]MNPA (D) and shows high uptake in striatum.
Fig. 2.

Baseline BPND values and time course of radioactivity in striatum and cerebellum of arr3 wild-type (n = 15) and knockout mice (n = 15). Panels A & B are for [11C]MNPA. Panels C & D are for [18F]fallypride. The average BPND values (mean ± SD) for [11C]MNPA were 0.91 ± 0.18 in wild-type and 0.92 ± 0.14 in knockout mice (A). The average BPND values for [18F]fallypride were 3.08 ± 0.77 in wild-type and 3.31 ± 0.99 in knockout mice. As expected from its lower affinity, [11C]MNPA (B) washed out of striatum more quickly than did [18F]fallypride (D). Time activity curves show mean ± SD. Symbols: wild-type striatum (△), wild-type cerebellum (○), knockout striatum (■), knockout cerebellum (▼;).
BPND values of both [18F]fallypride and [11C]MNPA were similar between wild-type and knockout mice, even though the washout of [11C]MNPA was faster in knockout than in wild-type mice (Fig. 2). The faster washout of [11C]MNPA could have been caused by faster peripheral clearance. Nevertheless, BPND corrects for any differences in clearance and reflects the density of receptors.
Amphetamine decreased the binding of both radioligands. As expected from studies in monkeys (Seneca et al., 2006), amphetamine decreased the binding of the agonist [11C]MNPA (~63%) more than the antagonist [18F]fallypride (~35%). Of critical importance to the question of the study, D2 receptor binding returned more quickly to baseline levels in knockout than in wild-type mice (Fig. 3 and 4).
Fig. 3.

Striatal binding potential (BPND) of [11C]MNPA in arr3 wild-type (open bars) and knockout (solid bars) mice on retest or at 30 min and 4 hours after amphetamine. BPND of knockout mice returned to baseline at 4 hours, whereas that of wild-type mice was still decreased. The dashed line represents the average BPND of test scan expressed as 100%; the bars show the percentage of this baseline for retest, amphetamine 30 min, and amphetamine 4 hours. Bars represent mean ± SD, n = 5 of each genotype in each group.
Fig. 4.

Striatal binding potential (BPND) of [18F]fallypride in arr3 wild-type (open bars) and knockout (solid bars) mice on retest or at 30 min and 4 hours after amphetamine. BPND of knockout mice was slightly increased at 4 hours, whereas that of wild-type mice was slightly decreased. The dashed line represents the average BPND of test scan expressed as 100%; the bars show the percentage of this baseline for retest, amphetamine 30 min, and amphetamine 4 hours. Bars represent mean ± SD, n = 5 of each genotype in each group.
We found a significant 3-way interaction of treatment × genotype × time for both [11C]MNPA (F = 4.32, P = 0.026) and [18F]fallypride (F = 4.79, P = 0.018). That is, amphetamine decreased radioligand binding, and its effect was dependent upon genotype and time. At 30 min, amphetamine had a significant main effect for both [11C]MNPA and[18F]fallypride (F = 56.78, P = 0.001 for [11C]MNPA and F = 7.16, P = 0.004 for [18F]fallypride). For [11C]MNPA, amphetamine decreased BPND by 65 ± 2% in wild-type and by 60 ± 16% in knockout mice. For [18F]fallypride, amphetamine decreased BPND by 29 ± 13% in wild-type and by 41 ± 10% in knockout mice (Fig. 3 and 4).
To determine whether genotype affected the time course of recovery of radioligand binding, we separately analyzed the three groups (baseline, 30 min, and 4 hours, n = 5 of each genotype in each group). For both radioligands, BPND was insignificantly different between wild-type and knockout mice at baseline and at 30 min. Only the values at 4 hours were affected by genotype. Specifically, in comparison to wild-type mice, the arr3 knockout mice had slower recovery to baseline values (F = 12.91, P = 0.009 for [11C]MNPA, F = 6.88, P = 0.034 for [18F]fallypride). At 4 hours, [11C]MNPA BPND was reduced only in the wild-type group (−29 ± 8%), while it remained unchanged in the knockout mice (0 ± 13%). For [18F]fallypride, BPND was slightly reduced in the wild-type mice (−20 ± 16%), while it was slightly increased in the knockout group (+12 ± 15%). Thus, knockout of arr3 allowed faster recovery of agonist and antagonist radioligand binding to striatal D2 receptors (Fig. 3 and 4).
DISCUSSION
We used both an agonist and an antagonist radioligand for the D2 receptor in mice that are incapable of internalization to separate the effects of dopamine displacement from receptor internalization after amphetamine challenge. Our results are consistent with the idea that receptor internalization prolongs the reduction in radioligand binding previously reported in humans and animals (Cardenas et al., 2004; Ginovart et al., 2004; Narendran et al., 2007). That is, radioligand binding returned to baseline levels more quickly in arr3 knockout than in wild-type mice once baseline dopamine levels are restored.
Since internalization occurs quickly after agonist exposure, the results at 30 min may reflect both displacement and internalization. However, BPND at 30 min was equally reduced in knockout and wild-type mice, suggesting that the reduction of radioligand binding at this time point was primarily caused by displacement rather than by internalization. Since the extracellular concentrations of dopamine return to baseline within a few hours, the results at four hours almost exclusively reflect receptor internalization. Consistent with our hypothesis, striatal BPND was reduced at four hours for both radioligands only in wild-type mice. Although the prolonged decrease of radioligand binding reflects the receptor internalization in wild-type mice, it is not possible to determine whether the receptor internalization prevents radioligand binding or decreases the affinity of the radioligand for the relocated receptor. However, previous studies suggest that the latter is the most likely possibility (Laruelle et al., 2008).
Agonist vs. Antagonist Radioligand
Consistent with prior PET studies in monkeys and cats, the agonist radioligand showed greater sensitivity to amphetamine-induced displacement than the antagonist radioligand (Ginovart et al., 2006; Narendran et al., 2004; Seneca et al., 2006). That is, the acute effects of amphetamine (at 30 min), which reflect direct displacement, were greater for the agonist [11C]MNPA than for the antagonist [18F]fallypride radioligand. This differential effect is consistent with the hypothesis of agonist radioligand binding to a high affinity state of the receptor. Although not immediately obvious, effects of internalization would also be expected to be more marked for agonist than antagonist radioligand because the internalized receptors are in the low affinity state (i.e. uncoupled from G-protein). Furthermore, the antagonist [18F]fallypride is likely to have access to the internalized receptor, since it readily crosses membranes, like those at the blood-brain barrier. Using cultured cells, Laruelle and colleagues (2008) recently confirmed that [3H]fallypride can access internalized D2 receptors, but the affinity of the radioligand is decreased 2 fold. Thus, binding of [18F]fallypride should be affected by internalization, because of its lower affinity for this relocated target.
Limitations of Imaging in Small Animals
Two important limitations of PET imaging in small animals, such as mice, are the spatial resolution of the camera and the effect of the co-injected nonradioactive ligand. Because of the limited resolution of PET (about 1.6 mm for our device), radioactivity can both “spill in” and “spill out” of the region of interest. This so-called partial volume effect blunts the true ratio of radioactivity in striatum to that in cerebellum, because radioactivity in striatum spills out, or is blurred into, the surrounding brain. Despite this blurring effect, the primary conclusion of this paper remains unchanged, since the effect of blurring was similar for wild-type and for knockout mice.
Another important limitation for PET imaging in mice is the percentage occupancy of the receptor by the co-injected nonradioactive ligand. Tracer kinetic modeling is based on the assumption that the concentration of receptor available to bind radioligand is constant during the entire scan. In other words, receptor occupancy by the injected ligand is negligible (often defined as <10%) throughout the scan, and virtually all receptors (>90%) remain available to bind radioligand. To achieve low receptor occupancy in small animals such as mice, the radioligand must have high specific radioactivity. In our study, we injected 0.14 nmol [11C]MNPA and 0.04 nmol [18F]fallypride, which corresponds to 5.8 and 1.7 nmol/kg, respectively, in a 24 g mouse. These injected doses likely caused significant occupancy of striatal D2 receptors for [11C]MNPA but not for [18F]fallypride. The maximal specific binding in striatum for [11C]MNPA was ~2.5 nM, calculated as striatum-cerebellum. Based on a Bmax value ~25 nM in rat striatum (Malmberg et al., 1996), this amount of specific binding corresponds to ~ 10% receptor occupancy (2.5/25), which is the upper limit for tracer kinetic modeling. In addition, agonist are thought to bind to the subset of D2 receptors in the high affinity state, which cause receptor occupancy of [11C]MNPA to be even more than 10%. For [18F]fallypride the maximum specific binding in striatum was 1.6 nM, resulting in 6.4% occupancy (1.6/25). Thus estimated receptor occupancy was high (>10%) for [11C]MNPA but acceptable (<10%) for [18F]fallypride. Nevertheless, the receptor occupancies were the same for the important comparison between wild-type and knockout mice. That is, the injected mass doses of each radioligand were insignificantly different for these two genotypes: 0.13 ± 0.04 and 0.14 ± 0.04 nmol for [11C]MNPA and 0.04 ± 0.01 and 0.04 ± 0.01 nmol for [18F]fallypride in wild-type and arr3 knockout mice, respectively.
In conclusion, our results suggest that prolonged decrease of radioligand binding to dopamine D2 receptors induced by amphetamine is mainly due to receptor internalization rather than direct displacement by dopamine. In addition, this study demonstrates the utility of PET to image genetically modified mice and explore the in vivo effects of receptor trafficking.
Acknowledgments
This research was supported by the Intramural Program of NIMH (project #Z01-MH-002795-07 and Z01-MH-002793) and NINDS (project #NS002263-33). We thank Cheryl Morse and Yi Zhang for assistance on radiotracer production; Jussi Hirvonen for assistance on data analysis; and PMOD Technologies (Zurich, Switzerland) for providing its image analysis and modeling software.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bjork K, Rimondini R, Hansson AC, Terasmaa A, Hyytia P, Heilig M, Sommer WH. Modulation of voluntary ethanol consumption by beta-arrestin 2. Faseb J. 2008;22:2552–2560. doi: 10.1096/fj.07-102442. [DOI] [PubMed] [Google Scholar]
- Bjurling P, Reineck R, Westerburg G, Gee AD, Sutcliffe J, Långström B. In: Link JM, Ruth TJ, editors. Synthia, a compact radiochemistry system for automated production of radiopharmaceuticals; Proceedings - Sixth Workshop on Targetry and Target Chemistry; TRIUMF, Vancouver. 1995. pp. 282–284. [Google Scholar]
- Cardenas L, Houle S, Kapur S, Busto UE. Oral D-amphetamine causes prolonged displacement of [11C]raclopride as measured by PET. Synapse. 2004;51:27–31. doi: 10.1002/syn.10282. [DOI] [PubMed] [Google Scholar]
- Cumming P, Wong DF, Gillings N, Hilton J, Scheffel U, Gjedde A. Specific binding of [11C]raclopride and N-[3H]propyl-norapomorphine to dopamine receptors in living mouse striatum: occupancy by endogenous dopamine and guanosine triphosphate-free G protein. J Cereb Blood Flow Metab. 2002;22:596–604. doi: 10.1097/00004647-200205000-00011. [DOI] [PubMed] [Google Scholar]
- Finnema SJ, Halldin C, Bang-Andersen B, Gulyas B, Bundgaard C, Wikstrom HV, Farde L. Dopamine D(2/3) receptor occupancy of apomorphine in the nonhuman primate brain-A comparative PET study with [11C]raclopride and [11C]MNPA. Synapse. 2009;63:378–389. doi: 10.1002/syn.20615. [DOI] [PubMed] [Google Scholar]
- Gao YG, Baldessarini RJ, Kula NS, Neumeyer JL. Synthesis and dopamine receptor affinities of enantiomers of 2-substituted apomorphines and their N-n-propyl analogues. J Med Chem. 1990;33:1800–1805. doi: 10.1021/jm00168a040. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, Houle S, Kapur S, Wilson AA. Binding characteristics and sensitivity to endogenous dopamine of [11C]-(+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem. 2006;97:1089–1103. doi: 10.1111/j.1471-4159.2006.03840.x. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-Receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry. 2004;55:1188–1194. doi: 10.1016/j.biopsych.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Ichise M, Liow JS, Lu JQ, Takano A, Model K, Toyama H, Suhara T, Suzuki K, Innis RB, Carson RE. Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab. 2003;23:1096–1112. doi: 10.1097/01.WCB.0000085441.37552.CA. [DOI] [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee JS, Im KC, Kim SJ, Kim SY, Lee DS, Moon DH. Performance measurement of the microPET Focus 120 scanner. J Nucl Med. 2007;48:1527–1535. doi: 10.2967/jnumed.107.040550. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423–451. doi: 10.1097/00004647-200003000-00001. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Guo N, Guo W, Jiang M, Schieren I, Abi-Dargham A, Javitch JA, Rayport S. Impact of dopamine D2 receptor internalization on binding parameters of D2 PET radiotracers. NeuroImage. 2008;41:T36. doi: 10.1038/npp.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarova N, Siméon FG, Musachio JL, Lu SY, Pike VW. Integration of a microwave reactor with Synthia to provide a fully automated radiofluorination module. J Label Compd Radiopharm. 2007;50:463–465. [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lu SY, Giamis AM, Pike VW. Synthesis of [18F]fallypride in a micro-reactor: rapid optimization and multiple-production in small doses for micro-PET studies. Curr Radiopharm. 2009;2:49–55. doi: 10.2174/1874471010902010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg A, Jerning E, Mohell N. Critical reevaluation of spiperone and benzamide binding to dopamine D2 receptors: evidence for identical binding sites. Eur J Pharmacol. 1996;303:123–128. doi: 10.1016/0014-2999(96)00080-5. [DOI] [PubMed] [Google Scholar]
- McCormick PN, Kapur S, Reckless G, Wilson AA. Ex vivo [11C]-(+)-PHNO binding is unchanged in animal models displaying increased high-affinity states of the D2 receptor in vitro. Synapse. 2009;63:998–1009. doi: 10.1002/syn.20671. [DOI] [PubMed] [Google Scholar]
- McCormick PN, Kapur S, Seeman P, Wilson AA. Dopamine D2 receptor radiotracers [11C](+)-PHNO and [3H]raclopride are indistinguishably inhibited by D2 agonists and antagonists ex vivo. Nucl Med Biol. 2008;35:11–17. doi: 10.1016/j.nucmedbio.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Mukherjee J, Yang ZY, Das MK, Brown T. Fluorinated benzamide neuroleptics--III. Development of (S)-N-[(1-allyl-2-pyrrolidinyl)methyl]-5-(3-[18F]fluoropropyl)-2, 3-dimethoxybenzamide as an improved dopamine D2 receptor tracer. Nucl Med Biol. 1995;22:283–296. doi: 10.1016/0969-8051(94)00117-3. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Dipace C, Javitch JA, Sibley DR. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J Biol Chem. 2009;284:15038–15051. doi: 10.1074/jbc.M900388200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A, Laruelle M. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]-raclopride. Synapse. 2004;52:188–208. doi: 10.1002/syn.20013. [DOI] [PubMed] [Google Scholar]
- Narendran R, Slifstein M, Hwang DR, Hwang Y, Scher E, Reeder S, Martinez D, Laruelle M. Amphetamine-induced dopamine release: duration of action as assessed with the D2/3 receptor agonist radiotracer (−)-N-[(11)C]propyl-norapomorphine ([11C]NPA) in an anesthetized nonhuman primate. Synapse. 2007;61:106–109. doi: 10.1002/syn.20346. [DOI] [PubMed] [Google Scholar]
- Neumeyer JL, Gao YG, Kula NS, Baldessarini RJ. Synthesis and dopamine receptor affinity of (R)-(−)-2-fluoro-N-n-propylnorapomorphine: a highly potent and selective dopamine D2 agonist. J Med Chem. 1990;33:3122–3124. doi: 10.1021/jm00174a002. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 4. Academic Press; San Diego: 2001. [Google Scholar]
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- Seidel J, Vaquero JJ, Green MV. Resolution uniformity and sensitivity of the NIH ATLAS small animal PET scanner: comparison to simulated LSO scanners without depth-of-interaction capability. IEEE Trans Nucl Sci. 2003;50:1347–1350. [Google Scholar]
- Seneca N, Finnema SJ, Farde L, Gulyas B, Wikstrom HV, Halldin C, Innis RB. Effect of amphetamine on dopamine D2 receptor binding in nonhuman primate brain: a comparison of the agonist radioligand [11C]MNPA and antagonist [11C]raclopride. Synapse. 2006;59:260–269. doi: 10.1002/syn.20238. [DOI] [PubMed] [Google Scholar]
- Skinbjerg M, Ariano MA, Thorsell A, Heilig M, Halldin C, Innis RB, Sibley DR. Arrestin3 mediates D2 dopamine receptor internalization. Synapse. 2009;63:621–624. doi: 10.1002/syn.20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger C, Finnema SJ, Raus L, Schou M, Nakao R, Suzuki K, Pike VW, Wikstrom HV, Halldin C. A two-step one-pot radiosynthesis of the potent dopamine D2/D3 agonist PET radioligand [11C]MNPA. J Label Compd Radiopharm. 2009;52:158–165. [Google Scholar]
- Sun W, Ginovart N, Ko F, Seeman P, Kapur S. In vivo evidence for dopamine-mediated internalization of D2-receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol. 2003;63:456–462. doi: 10.1124/mol.63.2.456. [DOI] [PubMed] [Google Scholar]
- Wilson AA, McCormick P, Kapur S, Willeit M, Garcia A, Hussey D, Houle S, Seeman P, Ginovart N. Radiosynthesis and evaluation of [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9 -ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J Med Chem. 2005;48:4153–4160. doi: 10.1021/jm050155n. [DOI] [PubMed] [Google Scholar]