

Abstract
Infection with human immunodeficiency virus (HIV) and treatment with HIV-protease inhibitor (PI)-based highly active antiretroviral therapies (HAART) is associated with dysregulated fatty acid and lipid metabolism. Enhanced lipolysis, increased circulating fatty acid levels, and hepatic and intramuscular lipid accumulation appear to contribute to insulin resistance in HIV-infected people treated with PI-based HAART. However, it is unclear whether currently prescribed HIV-PIs directly alter skeletal muscle fatty acid transport, oxidation, and storage. We find that ritonavir (r, 5 μmol/l) plus 20 μmol/l of atazanavir (ATV), lopinavir (LPV), or darunavir (DRV) reduce palmitate oxidation(16-21%) in differentiated C2C12 myotubes. Palmitate oxidation was increased following exposure to high fatty acid media but this effect was blunted when myotubes were pre-exposed to the HIV-PIs. However, LPV/r and DRV/r, but not ATV/r suppressed palmitate uptake into myotubes. We found no effect of the HIV-PIs on FATP1, FATP4, or FABPpm but both CD36/FAT and carnitine palmitoyltransferase I (CPTI) were reduced by all three regimens though ATV/r caused only a small decrease in CPT1, relative to LPV/r or DRV/r. In contrast, sterol regulatory element binding protein-1 was increased by all 3 HIV-PIs. These findings suggest that HIV-PIs suppress fatty acid oxidation in murine skeletal muscle cells and that this may be related to decreases in cytosolic- and mitochondrial-associated fatty acid transporters. HIV-PIs may also directly impair fatty acid handling and partitioning in skeletal muscle, and this may contribute to the cluster of metabolic complications that occur in people living with HIV.
Keywords: HIV-PIs; fatty acid uptake; fatty acid oxidation; CD36, CPT1; metabolic dysregulation
1. Introduction
Human immunodeficiency virus (HIV) -protease inhibitor (PI)-based highly active antiretroviral therapy (HAART) has reduced morbidity and mortality. However, PI-based HAART has been associated with a cluster of metabolic complications analogous to “the cardiometabolic syndrome” [1] including: peripheral lipoatrophy, visceral adiposity, hyperlipidemia, insulin resistance, hyperglycemia, and overt type 2 diabetes mellitus [2, 3]. The mechanisms responsible for these complications have only partially been elucidated [4-10]. HIV-PI induced disruptions in fatty acid (FA) and lipid partitioning and sensing might contribute to these complications, so we examined whether FA transport and/or oxidation are altered in murine skeletal muscle cells exposed to clinically relevant HIV-PI combinations.
Evidence supports the notion that, in addition to host, viral, and behavior factors, HIV-PI based HAART contributes to dysregulated FA and lipid metabolism. HIV-PI-based HAART increases whole body lipolysis and lipid oxidation rates [11, 12], increases intramuscular lipid accumulation [12], impairs adipocyte metabolism [13], and alters glucose homeostasis [5]. In adipocytes and rodent models, the HIV-PI ritonavir (r) inhibited human adipocyte differentiation [13], decreased LPL-mediated clearance of VLDL-TG, and impaired FA uptake into adipose tissue [14]. In rodent primary hepatocytes exposed to different HIV-PIs, lipid metabolism gene expression was reduced, and FA synthesis gene expression was increased [15]. In HIV-infected people treated with PI-based HAART (5), exercise-stimulated FA oxidation was blunted. This was attributed to impaired free FA (FFA) mobilization from adipose tissue, and not to defects in skeletal muscle FA transport and/or mitochondrial oxidation [16]. However, skeletal muscle FA transport and oxidation were not directly measured in this study. In addition, HIV-PI-based HAART-associated insulin resistance enhanced intramuscular lipid accumulation, which would reduce whole body lipid and muscle FA oxidation (17). In these human studies (5,17), it was not possible to determine the separate effects of HIV-PI, other anti-HIV medications, or other HIV-associated factors on muscle FA transport and oxidation. Therefore, we hypothesized that HIV-PIs impair skeletal muscle FA transport and oxidation. To test the effects of HIV-PIs on FA partitioning and sensing, we exposed C2C12 murine skeletal muscle cells to currently used HIV-PIs (ritonavir (r), lopinavir (LPV), atazanavir (ATV), darunavir (DRV) in combination (LPV/r, ATV/r, DRV/r)), and quantified FA transport and oxidation. We found that combined HIV-PI exposure (rather than individual PIs) reduces skeletal muscle FA oxidation by inhibiting FA transport protein expression.
2. Materials and Methods
2.1. Materials
HIV-protease inhibitors were supplied by The AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, and NIH (http://www.aidsreagent.org/). Rabbit polyclonal anti-FABPpm antiserum was generously provided by Dr. Jorge Calles-Escandon, Wake Forest University Health Sciences, Winston-Salem, North Carolina.). Other materials were (obtained from): C2C12 cells (American Type Culture Collection, CRL-1772; Manassas, VA); Biocoat tissue culture plates (Becton Dickinson, Bedford, MA); SDS gel supplies (Bio-Rad Laboratories, Hercules, CA); [14C]-palmitate or [14C]-acetyl-CoA (GE Healthcare, Piscataway, NJ); fetal bovine serum (FBS) and heat-inactivated horse serum (Invitrogen, Carlsbad, CA); chemiluminescent HRP substrate and Immoblolin-P polyvinylidene difluoride (PVDF) membranes (Millipore Corporation, Bedford, MA); Uniscint BD scintillation fluid (National Diagnostics, Atlanta, GA); TUNEL kit (Roche Diagnostic Corp., Indianapolis, IN) primary and secondary antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA); essentially fatty acid free-BSA, carnitine, and palmitic acid, and Dulbecco’s modified Eagles medium (DMEM), Oil Red O, and common laboratory reagents (Sigma Co., St. Louis, MO); and Hank’s balanced salt solution (Washington University School of Medicine Tissue Culture Support Center).
2.2. C2C12 skeletal muscle cell cultures
Mouse C2C12 cells were cultured, as described [17]. Briefly, monolayers of C2C12 myoblasts were grown in Dulbecco’s modified low-glucose Eagle’s medium (DMEM-LG; 1.5 g/l)/10% (v/v) FBS/4.0 mmol/l glutamine/100 U/ml penicillin/100 μg/ml streptomycin, in a humidified incubator (37 °C, 95% air/5% CO2). Cells were propagated in 75 mm flasks, subcultured at 60-80% confluence, and split using trypsin [0.25% (w/v) in MEM with 1.0 mmol/l EDTA]. Cells were subcultured into 12-well dishes coated with 0.01% (w/v) collagen. When cells were 70-80% confluent, myoblasts were induced to differentiate into myotubes by changing the medium to a low-serum differentiating medium (DFM) [DMEM-LG/2% (v/v) horse serum/4.0 mmol/l glutamine/100 U/ml penicillin/100 μg/ml streptomycin]. Differentiation medium was changed every 48 h. By day 6, confluent cells had differentiated into multinucleated contracting myotubes.
2.3. Oil Red O staining of C2C12 myotubes
To determine the time- and concentration-dependence of fatty acid exposure, C2C12 myotubes were exposed to a high fatty acid media and Oil Red O stained, as reported [18], with modification. Briefly, C2C12 myoblasts were propagated and differentiated as described above. Upon differentiation, the myotubes were cultured overnight in a serum-free DFM (starvation media) followed by incubation with a serum-free high fatty acid media (50/25, 100/50, and 200/100 μmol/l oleic/palmitic acid; 0.5% BSA) for 5, 18, or 48 h. Lipid loaded myotubes were then washed twice (1X PBS), and fixed to the plates with 3.7% formaldehyde for 2 min. Following fixation, each well was washed one time with deionized water and Oil Red O stain applied at room temperature for 30 min. The cells were then washed three times with deionized water and stained lipid-loaded cell images were captured using a standard digital camera. These analyses revealed minimal Oil Red O staining above background following 5h fatty acid loading; dramatic increase in staining at 18h increased in myotubes exposed to 100/50 and 200/100 μmol/l oleic/palmitic acid; and modest increase or decrease after 48 h exposure. Based on these findings, myotube palmitate oxidation was performed following 18 h exposure to 200/100 μmol/l oleic/palmitic acid.
To get a quantitative measure of the effects of HIV-PIs on fatty acid uptake, the Oil Red O staining procedure [19, 20] was modified. Briefly, fully differentiated myotubes were exposed to combination HIV-PIs (20 μmol/l /5 μmol/l; ATV/r, LPVr, or DRV/r) dissolved in 0.1% DMSO or to 0.1% DMSO alone (Con) and incubated for 18 h in serum-free DFM-LG. The concentrations of HIV-PI’s were based on findings in our lab and reports from the Hruz group [21-23]. After washing the cells twice with Hank’s Balanced Salt Solution (HBSS), they were incubated in high fatty acid media (200/100 μmol/l oleic/palmitic acid; 0.5% BSA) for an additional 18h. The cells were washed again with HBSS prior to addition of 500 μl of 3% Oil Red O (3g/100 ml 70% Isopropanol) to each well. Following an 1 h incubation period at room temperature, the cells were rinsed twice with HBSS and 500 μl trypsin/EDTA was added to each well and the cells incubated for 10 min at 37 °C. The cells were harvested and trypsin was neutralized by the addition of media containing 10% serum. Then cells were centrifuged for 5 min at 3000 rpm (0 °C) and the supernatant was removed and the pellet rinsed with PBS. The cells were re-suspended in 70% Isopropanol and then briefly centrifuged (1 min at 3000 rpm); the supernatant was removed and OD measured at 492 nm in duplicate. The precipitated pellet was lysed and analyzed for total protein content using a BCA assay (Pierce) in duplicate.
2.4. Effects of protease inhibitor and high fat acid media exposure on fatty acid oxidation in C2C12 myotubes
To mimic the effects of intramyocellular lipid accumulation, and to distinguish the direct effects of HIV-PI exposure from those of high fatty acid media exposure, myotube fatty acid oxidation was measured following three different experimental manipulations: (A) Protease inhibitor exposure. Fully differentiated myotubes were exposed to combination HIV-PIs (20 μmol/l /5 μmol/l; ATV/r, LPVr, or DRV/r) dissolved in 0.1% DMSO or to 0.1% DMSO alone (Con) and incubated for 18 h in serum-free DFM-LG prior to quantifying [14C]-palmitate oxidation (described below). The concentrations of HIV-PI’s were based on our previous findings [4] that 20 μmol/l of HIV-PI alone and in combination with 5 μmol/l ritonavir induced suppressor of cytokine signaling-1 (SOCS-1) and impaired insulin signaling in cell cultures. (B) High fatty acid media exposure. Myotubes were incubated for 18 h with a high fatty acid media only, as described above (200/100 μmol/l oleic/palmitic acid; 0.5% BSA), prior to quantifying [14C]-palmitate oxidation. (C) Combined HIV-PI and high fatty acid media exposure. Myotubes were exposed to combination PIs (as above) for 18 h, followed by 18 h exposure to high fatty acid media (200/100 μmol/l oleic/palmitic acid; 0.5% BSA) prior to quantifying [14C]-palmitate oxidation.
2.5. Palmitate oxidation assays
Palmitate oxidation was determined as described [18]. Cells were incubated in the presence of [14C]-palmitate and [14C]-palmitate oxidation was measured as 14CO2 production (an indication of fatty acid oxidation) over a 3 h-period. 14CO2 released into the media was quantified using liquid scintillation counting in 3 ml of Uniscint BD. Following incubation with HIV-PIs, in the absence or presence of high fatty acid media, myotubes were washed twice with warmed (37 °C) PBS (21), and incubated in DFM plus 12.5 mmol/l HEPES, 0.2% BSA, 1.0 mmol/l carnitine, 100 μmol/l palmitic acid or acetyl-CoA, and 0.5 μCi/ml [14C]-palmitate or [14C]-acetyl-CoA. Oxidation of acetyl-CoA was used to assess oxidation downstream of the β-oxidation steps. After 3 h, 0.5 ml of incubation media was transferred to separate dishes, analyzed for labeled 14CO2 and quantitated by liquid scintillation. The myotubes were immediately placed on ice, washed 3 times with ice-cold PBS, and scraped into 2.0 ml centrifuge tubes in two additions of 0.075 ml 0.1% SDS lysis buffer and stored at −80 °C for subsequent protein and immunoblot analyses.
2.6. Effects of HIV-protease inhibitor exposure on palmitate uptake into C2C12 myotubes
C2C12 myotubes were exposed to the combination HIV-PIs ATV/r, LPV/r, and DRV/r (20 μmol/l/ 5 μmol/l) for 18 h. Following an overnight incubation in high fat media (DFM plus 200 μmol/l palmitic acid, 0.5% BSA, and 0.5 μCi/ml [14C]-palmitate), intracellular [14C]-palmitate uptake was measured. Briefly, 14C-containing media was removed from each well and placed in a corresponding 1.5 ml centrifuge tube. Tubes were centrifuged at 1,500 rpm for 5 min to remove detached cells. 14C-containing media was discarded and the cell pellet was washed twice with ice-cold PBS. The remaining plated cells were washed twice with ice-cold PBS and harvested in 2 × 100 μl aliquots of 0.05% SDS, and placed in corresponding 1.5 ml centrifuge tubes. Aliquots of cells (100 μl) were placed in scintillation vials and the uptake of [14C]-palmitate was determined by liquid scintillation.
2.7. Immunoblot analyses
C2C12 myotubes designated for Western blot analyses were harvested in cell lysis buffer (in mmol/l; 50 HEPES, 15 NaCl, 1 MgCl2, 1 CaCl2, 2 EDTA; 10% glycerol, 1% Triton X-100, 5 μl/ml protease and phosphatase inhibitor cocktail, 3 mg/ml benzamidine hydrochloride). Proteins were separated by 7.5% SDS-PAGE and the resolved proteins were transferred onto PVDF membranes. The blots were then prepared for probing with primary antibodies directed against fatty acid transport proteins 1 (FATP1, 1:100) and 4 (FATP4, 1:100); plasma membrane-associated fatty acid-binding protein (FABPpm, 1:30,000), fatty acid translocase CD36/FAT (1:200), carnitine palmitoyltransferase 1 (CPT1; 1:250), sterol regulatory element binding protein-1 (SREBP-1; 1:400), and GAPDH (1:500) or tubulin (1:1000) loading controls. Following subsequent exposure to secondary antibodies (1:2000 for FATPs and FABPpm; 1:10,000 for CD36, CPT1, and SREBP-1), immunoreactive protein bands were visualized using chemiluminescent HRP substrate.
2.8. Statistical Analysis
Data are expressed as mean ± SEM. Statistical analyses were performed using Statistical Package for the Social Sciences (SPSS, Inc. v. 14.0, Chicago, IL). A two-way repeated measures analysis of variance (ANOVA) was used to identify interactions or main effects for the dependent variables. The level of significance was set at p ≤ 0.05.
3. Results
3.1. HIV-protease inhibitor exposure reduces myotube palmitate oxidation
To determine the optimal concentrations of the HIV-PIs for testing, preliminary measurements of CO2 production were made in C2C12 or L6 skeletal muscle cells that had been exposed to varying concentrations of different HIV-PI regimens. As seen in Figure 1, 20 μmol/l of the HIV-PI alone (Panel A) or in combination with 5 μmol/l RTV (Panels B and C) caused near peak decreases in CO2 production. These findings are consistent with the concentration of HIV-PIs used alone or in combination with RTV in earlier studies in various cell systems [21-28] and 20 μmol/l of the HIV-PI alone ± 5 μM RTV were chosen for subsequent studies.
Fig. 1. Concentration-dependent effects of HIV-PIs on 14C]-palmitate oxidation in skeletal muscle cells.

L6 and C2C12 cells were exposed to HIV-PIs in the absence and presence of RTV (5 μM) and [14CO2] production (nmol/mg/h) measured. A. IDV. B. LPV/RTV. C. ATV/RTV. Results are mean ± SEM (n = 3 in each group). *PI groups significantly different from Con (DMSO) group (p ≤ 0.05).
Following 18 h exposure to different HIV-PI combinations, palmitate oxidation was quantified in fully differentiated myotubes maintained under basal conditions (Fig. 2). 14CO2 production from palmitate oxidation was significantly reduced (p ≤ 0.05) in myotubes exposed to all combinations of HIV-PIs (−15%, −50%, and −32%; ATV/r, LPV/r, and DRV/r, respectively) relative to DMSO-exposed control myotubes. In contrast, myotube acetyl CoA oxidation, which reflects fatty acid oxidation directly in the TCA cycle without passing through β-oxidation, was not significantly changed by exposure to any of the HIV-PI combinations (data not shown).
Fig. 2. HIV-protease inhibitor treatment reduces [14C]-palmitate oxidation in C2C12 myotubes.
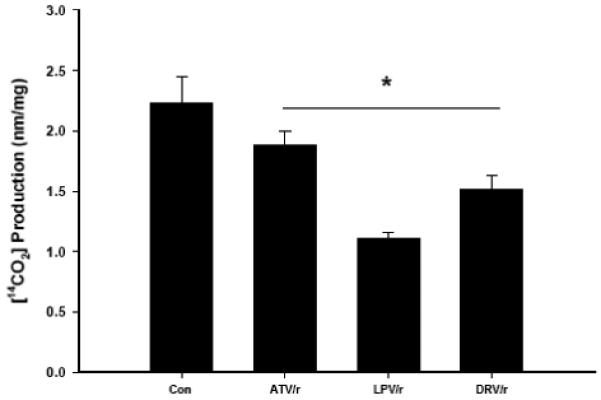
C2C12 myotubes were treated for 18 h with HIV-PIs in combination (ATV/r, LPV/r, and DRV/r; 20/5 μmol/l, respectively) and [14CO2] production measured. Results are mean ± SEM (n = 3 in each group). *PI groups significantly different from Con (DMSO) group (p ≤ 0.05).
3.2. High fatty acid media exposure increases myotube lipid storage and palmitate oxidation
Fully differentiated myotubes were exposed to fatty acid media (50/25 μmol/l, 100/50 μmol/l, and 200/100 μmol/l oleic/palmitic acid; 0.5% BSA) for 5, 18, or 48 h to confirm the temporal effects on neutral lipid storage, and to quantify palmitate oxidation (Fig. 3). Following 5 h of fatty acid loading, minimal Oil Red O staining was detected above background. Following 18 h, Oil Red O staining increased in myotubes exposed to 100/50 and 200/100 μmol/l oleic/palmitic acid. By 48 h, the staining was either unchanged or even modestly decreased. This confirmed that neutral lipids accumulate in C2C12 after 18h incubation in high fat media, so we examined myotube palmitate oxidation following 18 h exposure to 200/100 μmol/l oleic/palmitic acid (Fig. 4). Myotube [14C]-palmitate oxidation was significantly (p ≤ 0.05) higher following 18 h exposure to high fatty acid media than it was in control myotubes not exposed to high fatty acid media. Under these conditions, the increase in ß-oxidation was insufficient to handle the elevated fatty acid uptake, so some fatty acids were partitioned into neutral lipid synthetic pathways and stored. This provided an appropriate model system in which to challenge fatty acid loading (uptake and oxidation) in the presence of HIV-PIs.
Fig. 3. Lipid accumulation in C2C12 myotubes following a fatty acid load.
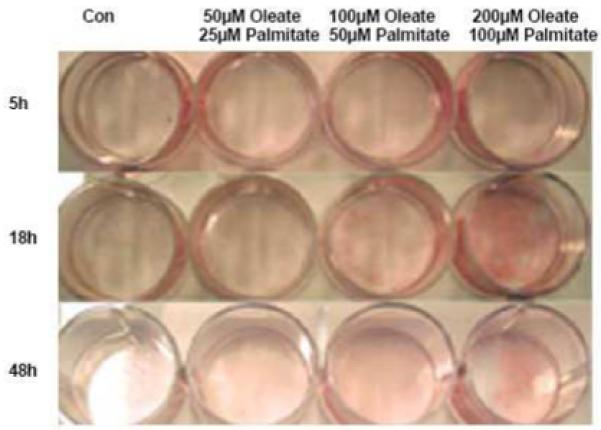
Oil red-O staining of neutral lipids in C2C12 myotubes treated with a fatty acid load (50/25 μmol/l, 100/50 μmol/l, and 200/100 μmol/l; oleate/palmitate, respectively) in a serum-free DFM for 5, 18, or 48 h.
Fig. 4. Fatty acid load increases [14C]-palmitate oxidation in C2C12 myotubes.
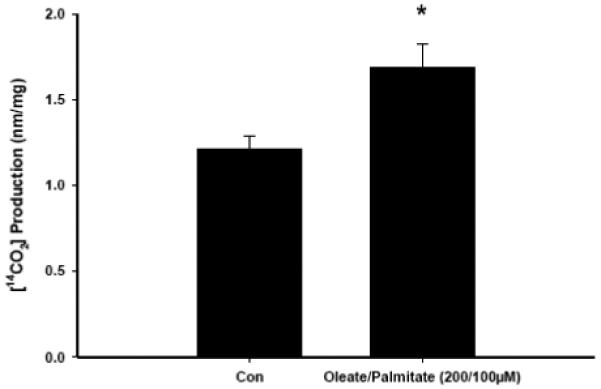
C2C12 myotubes were treated for 18 h with 200/100 μmol/l oleate /palmitate and [14CO2] production measured. Results are mean ± SEM (n = 3 in each group). *Oleate/palmitate group significantly different from Con group (p ≤ 0.05).
3.3. Combined HIV-protease inhibitor and high fatty acid media exposure reduces myotube palmitate oxidation
Myotubes were exposed to ATV/r, LPV/r, DRV/r (20 μmol/l/5 μmol/l) for 18 h followed by incubation with high fatty acid media (200/100 μmol/l oleic/palmitic acid) for 18 h (Fig. 5). These HIV-PI combinations significantly reduced [14C]-palmitate oxidation relative to control myotubes not exposed to HIV-PIs.
Fig. 5. HIV-protease inhibitor treatment followed by fatty acid load decreases [14C]-palmitate oxidation in C2C12 myotubes.
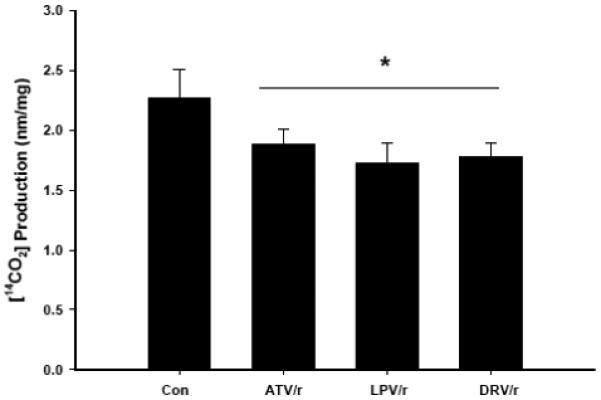
C2C12 myotubes were treated for 18h with PI’s in combination (ATV/r, LPV/r, and DRV/r; 20/5 μmol/l, respectively) followed by a fatty acid load (200/100 μmol/l oleate/palmitate) for 18 h and [14CO2] production measured. Results are mean ± SEM (n = 3 in each group). *PI groups significantly different from Con (DMSO) group (p ≤ 0.05).
3.4. Select HIV-protease inhibitor reduces lipid accumulation in myotubes
To address the issue of reduced palmitate oxidation following exposure of the myotubes to HIV-PIs, Oil Red O staining was quantitated in cells exposed to the high fatty acid media alone or after exposure to the HIV-PIs. As illustrated in Figure 6, exposure to fatty acids increased staining at 18 h with a modest further increase evident at 36 h, relative to cells treated with vehicle alone (Con) for 36 h. When the cells were first treated with LPV/r or DRV/r regimens before exposure to oleic/palmitic acid, the staining was similar to Con cells. However, the ATV/r combination had minimal effect on accumulation of the fatty acids in the cell.
Fig. 6. Lipid accumulation in C2C12 myotubes treated with HIV-PIs.
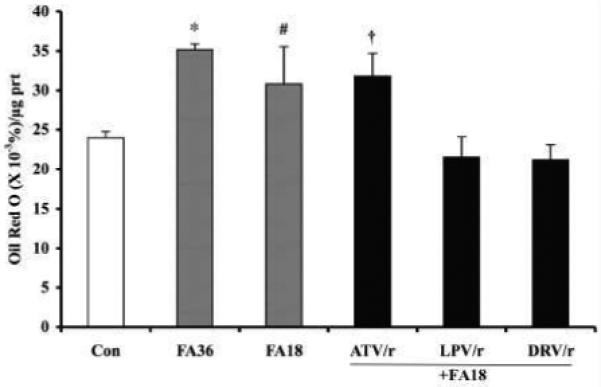
C2C12 myotubes were treated for 18 h in the presence of vehicle (DMSO) alone (Con) or with HIV-PIs in combination (ATV/r, LPV/r, and DRV/r; 20/5 μmol/l, respectively). The cells were then washed with HBSS and the Con cells were incubated with vehicle alone and the HIV-PI pretreated cells were incubated with 200/100 μmol/l oleate/palmitate for an additional 18 h. In some experiments, the myotubes were treated with only the fatty acids (FA) for 18 h (FA18) or 36 h (FA36). At the end of the incubation period, lipid in the cells was stained with 3% Oil Red O and quantitated as described in Methods section. The presented data are mean ± SEM of %Oil Red O stain/μg protein (n =3). (*.#FA groups significantly different from Con group, p < 0.0001 and p < 0.05, respectively).
3.5. Select HIV-protease inhibitor exposure reduces palmitate uptake into myotubes
One explanation for the observed HIV-PI mediated reductions in [14C]-palmitate oxidation may be that palmitate uptake by differentiated myotubes is reduced. To test this possibility, myotubes were exposed to ATV/r, LPV/r, DRV/r (20 μmol/l/5 μmol/l) for 18 h, followed by high fatty acid media exposure (200 μmol/l palmitate; 1.0 μCi/ml [14C]-palmitate) for 18 h (Fig. 7). Consistent with the Oil Red O staining, LPV/r and DRV/r significantly reduced (p ≤ 0.05) [14C]-palmitate uptake, whereas ATV/r appeared to have no affect on palmitate uptake.
Fig. 7. HIV-protease inhibitor treatment decreases [14C]-palmitate uptake in C2C12 myotubes.
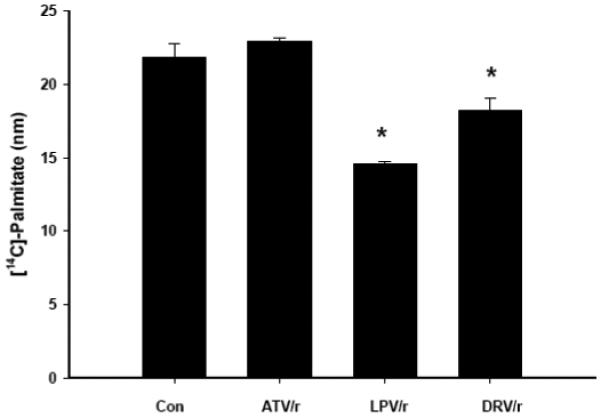
C2C12 myotubes were treated for 18 h with HIV-PIs in combination (ATV/r, LPV/r, and DRV/r; 20/5 μmol/l, respectively) followed by a fatty acid load (200/100 μmol/l oleate/palmitate) for 3 h. Myotubes were harvested and assayed for [14C]-palmitate incorporation. Results are mean ± SEM (n = 3 in each group). *PI groups significantly different from Con (DMSO) group (p ≤ 0.05).
3.6. Select HIV-protease inhibitors reduce expression levels for proteins involved in fatty acid uptake and oxidation
To determine which components in the myotube fatty acid uptake and oxidation pathways are affected by HIV-PI exposure, we examined myotube protein expression for fatty acid transporters in the plasma membrane (FATP1, FATP4, and FABPpm), in the plasma membrane and mitochondria (CD36) and the outer mitochondria (CPT1), and the lipogenic protein SREBP-1 following 18h exposure to combination HIV-PIs under basal conditions. As shown by the ratio of target transporter protein to tubulin in Figure 8A, FABPpm was relatively unchanged and the FATPs were modestly increased following exposure of the myotubes to the HIV-PIs for 18h. However, relative to control conditions, CD36 (Fig. 8B) was reduced by ATV/r (−16%), LPV/r (−17%), and DRV/r (−21%); CPT1 (Fig. 8C) was reduced by ATV/r (−4%), LPV/r (−26%), and DRV/r (−28%); but SREBP-1 (Fig. 8D) was increased ATV/r (71%), LPV/r (75%), and DRV/r (47%). These changes are summarized as fold-change, relative to control, in Figure 8E.
Fig. 8. Effects of HIV-protease inhibitor treatment on proteins involved in fatty acid transport and metabolism in C2C12 myotubes.
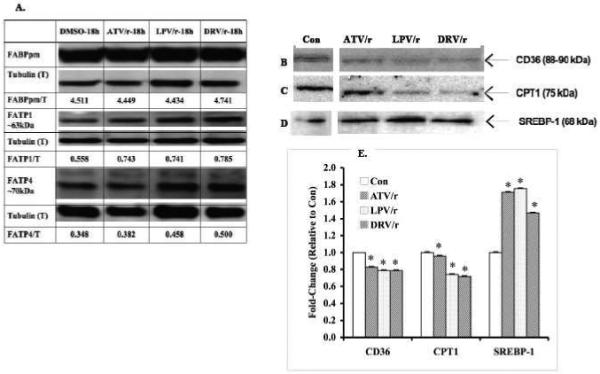
Representative immunoblots for FABPpm, FATP1, and FATP4 (A), CD36 (B), CPT1 (C), and SREBP-1 (D) in whole cell lysates of C2C12 myotubes following treatment for 18 h with vehicle (DMSO) alone (Con) or with PI’s in combination (ATV/r, LPV/r, and DRV/r; 20/5 μmol/l, respectively). Tubulin (A) or GAPDH (B-D) were used as loading controls. E. Quantitation of immunoreactive CD36, CPT1, and SREBP-1 bands. The target immunoreactive bands were normalized to GAPDH loading control and the data are presented as fold-change relative to untreated Con sample. (*Significantly different from corresponding Con group, p < 0.0001, n =3).
4. Discussion
The purpose of this study was to examine the effects of clinically relevant HIV-PI combinations on palmitate oxidation and transport into skeletal muscle cells using basal and lipid loaded C2C12 murine myotubes as a model system. The major findings were: (1) HIV-PI exposure reduces palmitate oxidation in differentiated myotubes incubated under basal conditions and after increased palmitate availability (lipid loading); (2) HIV-PI exposure reduces palmitate uptake/transport into myotubes; (3) HIV-PI mediated reductions in myotube palmitate transport and oxidation appear not to be due to decreases in plasma membrane-associated fatty acid transporters FATP1, FATP4, or FABPpm; and (4) HIV-PI mediated reductions in myotube palmitate transport and oxidation are associated with reductions in the fatty acid transport proteins CD36 and CPT1, and increases in the lipogenic protein SREBP-1.
These findings suggest that, along with other factors that dysregulate fatty acid and lipid metabolism in HIV-infected people [29], several of the currently prescribed ritonavir-boosted HIV-PIs directly impair the ability of skeletal muscle cells to transport, partition, and oxidize palmitate (i.e., fatty acids), especially if neutral lipids have accumulated in the muscle cells (intramyocellular lipid accumulation), as has been reported to occur in HIV-infected people with insulin resistance [12]. Cade et al. [16] and others have postulated that HIV-PIs impair skeletal muscle fatty acid oxidation, but to our knowledge, this is the first direct in vitro evidence that HIV-PIs impair skeletal muscle palmitate uptake and oxidation.
The mechanism by which these HIV-PIs impair skeletal muscle palmitate transport and oxidation has been partially elucidated. CD36 (also referred to as fatty acid translocase; FAT) is a transmembrane protein involved in the transport of long-chain fatty acids (LCFA) across cellular membranes. It has been shown that CD36 is expressed in skeletal muscle plasma membrane of C2C12 myocytes and that overexpression of this transport protein increased fatty acid uptake and utilization by myotubes [30]. It has also been demonstrated that CD36 exists in the mitochondrial outer membrane of human skeletal muscle and was shown to participate in transport and subsequent oxidation of LCFA [31, 32]. Using immunoblotting techniques, we found reduced expression of CD36 in myotubes exposed to combination HIV-PIs. It has been demonstrated that antiretroviral therapy, specifically ritonavir, nelfinavir, and the combination LPV/r, decreased expression of CD36 in HIV-infected and healthy human subject monocytes as well as in human THP-1 monocyte and human C32 melanoma cell lines [33]. Our findings indicate that ATV/r, LPV/r, and DRV/r reduce CD36 protein expression in C2C12 myotubes by 16%, 17%, and 21% , respectively, suggesting that this contributes to the observed decrease in palmitate oxidation (ATV/r, LPV/r, and DRV/r; 15%, 50%, and 32%, respectively).
It might be suggested that these drugs may induce apoptosis of the myotubes, similar to their effects in pancreatic β-cells [28], however, TUNEL analyses of cells treated with the HIV-PI regimens alone or with the HIV-PI regimens and then fatty acids revealed no incidence of TUNEL-positive cells (data not shown). These findings indicate that the concentrations and duration of treatments used in the present studies do not induce apoptosis of the murine skeletal muscle cells.
Carnitine palmitoyltransferase I (CPT1) is a mitochondrial transport protein that traverses the outer mitochondrial membrane. Malonyl-CoA is a known intracellular antagonist of CPT1. Increased concentrations of acetyl-CoA and intracellular CO2 lead to the formation of malonyl-CoA which directly inhibits CPT1 expression and in turn, decreases fatty acid uptake into the mitochondria. It has been suggested that mitochondrial CD36 and CPT1 interact to transport acylcarnitines across the outer mitochondrial membrane to carnitine-acylcarnitine translocase (CAT), then on to CPTII, and finally to the intermitochondrial space for oxidation [31]. Despite 16-21% reductions in CD36 protein levels among the HIV-PI combinations, only LPV/r and DRV/r significantly reduced CPTI protein levels in myotubes. This implies that ATV/r reduces palmitate oxidation through a mechanism that differs from that of LPV/r and DRV/r. We cannot discount the possibility that LPV/r and DRV/r decreased CD36 protein levels, which reduced fatty acid availability to CPTI, and this subsequently reduced mitochondrial fatty acid uptake and oxidation by the muscle cells.
It is not unexpected that the ATV/r regimen has different effects than the other regimens studied. It is well-recognized that different HIV-PIs have variable effects on cellular lipid/cholesterol metabolism and circulating lipid/lipoprotein levels [15, 34-37]. In clinical trials, ATV/r and LPV/r are well-tolerated, have equivalent virologic and immunologic potencies, but ATV/r is associated with a more favorable impact on serum lipid levels, and switching from a regimen that includes LPV/r, to one that includes ATV/r is associated with improvements in serum lipid/lipoprotein profiles. In lipid-loaded C2C12 cells (Fig 7), CPT-1 expression was not reduced as much by ATV/r (−4%) as it was by LPV/r (−26%) or DRV/r (−28%) exposures. We suggest that LPV/r and DRV/r exposure reduce ß-oxidation in C2C12 by inhibiting both palmitate uptake and CPT-1-associated ß-oxidation. However, as noted for cholesterol metabolism in rat hepatocytes and macrophages [37], ATV/r does not disrupt fatty acid uptake or ß-oxidation as much as LPV/r and DRV/r exposure. Instead, ATV/r must mediate its effects on C2C12 lipid metabolism at sites downstream from cellular uptake and CPT-1 expression. This may reflect a difference in ATV-protein binding, lipid membrane solubility, or affinity for lipid transporters among the different PIs.
Sterol regulatory element binding proteins (SREBPs) regulate transcription of genes involved in fatty acid synthesis [38] (fatty acid synthase, and acetyl-CoA carboxylase) as well as triglyceride synthesis [39]. SREBP-1 protein levels are reduced in 3T3-F442A adipocytes [40] and increased in primary rat hepatocytes [36] exposed to the HIV-PI indinavir. Interestingly, ritonavir administration to mice increased SREBP-1 protein expression in liver and adipose tissue [41]. Indinavir administration to ZDF fa/fa rats increased SREBP-1 protein levels in skeletal muscle, liver, and adipose tissue, and this was associated with induction of the suppressor of cytokine signaling-1 (SOCS-1) cascade [4]. Also, SREBP-1 protein levels were increased in L6 myotubes exposed to ritonavir, ATV, and LPV alone or ATV/r, and LPV/r in combination. Together, these findings indicate that indinavir, ritonavir, and ATV/r, LPV/r, and DRV/r increase SREBP-1 protein levels in skeletal muscle cells from several mammalian species.
There were some limitations to this in vitro study. Several factors, in addition to HIV-PI, contribute to dysregulated fatty acid and lipid metabolism in HIV-infected people. We cannot account for all these factors (e.g., genetics, physical activity, diet, HIV replication, host response to HIV-infection, other HAART components) in our model system. The primary goal was to isolate the effects of select HIV-PIs that are commonly used on palmitate metabolism. Also, myotubes were acutely exposed to HIV-PIs, and more chronic exposures should be examined to evaluate the durability/sustainability of these acute effects on myotube fatty acid metabolism.
The cellular mechanism(s) by which combination HIV-protease inhibitors dysregulate substrate oxidation in various tissues are unclear. People living with HIV and treated with HIV-PI-based HAART experience dyslipidemia and impairments in lipid metabolism, partitioning, and storage. The current observations extend our understanding by demonstrating that combination HIV-PI regimens that are currently in clinical use, decrease palmitate uptake and oxidation in murine skeletal muscle cells, and that this dysregulation is associated with reduced CD36 protein levels in muscle cells. Further studies are required to identify the precise molecular mechanisms by which combination HIV-PIs reduce CD36 expression and downstream regulators of fatty acid oxidation.
Acknowledgements
The work was supported by grants from The National Institutes of Health R01-DK69455, P30-DK056431, P60-DK020579, T32-DK007296-27 (to S.R.R. and M.J.C.), DK074345, DK049393, DK059531, Bristol-Myers Squibb (to KEY), and the Campbell Foundation (to S.R.). The NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH provided the following protease inhibitors: (ritonavir, reagent #4622); (lopinavir, reagent #9481); (atazanavir, reagent #10003); and (darunavir, reagent #11447).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cade WT, Yarasheski K. Cardiometabolic disease in the human immunodeficiency virus: the tip of the iceberg ? J. Cardio. Metab. Syndr. 2008;3(2):77–78. doi: 10.1111/j.1559-4572.2008.07204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12:F51–58. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- [3].Yarasheski KE, Tebas P, Sigmund C, Dagogo-Jack S, Bohrer A, Turk J, Halban PA, Cryer PE, Powderly WG. Insulin resistance in HIV protease inhibitor-associated diabetes. J. Acquir. Immune Defic. Syndr. 1999;21:209–216. doi: 10.1097/00126334-199907010-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carper MJ, Cade WT, Cam M, Zhang S, Shalev A, Yarasheski K, Ramanadham S. HIV-Protease inhibitors induce expression of suppressor of cytokine signaling-1 in insulin-sensitive tissues and promote insulin resistance and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2008 doi: 10.1152/ajpendo.00167.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hruz PW. Molecular Mechanisms for Altered Glucose Homeostasis in HIV Infection. Am. J. Infect. Dis. 2006;2:187–192. doi: 10.3844/ajidsp.2006.187.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hruz PW, Murata H, Mueckler M. Adverse metabolic consequences of HIV protease inhibitor therapy: the search for a central mechanism. Am. J. Physiol. Endocrinol. Metab. 2001;280:E549–553. doi: 10.1152/ajpendo.2001.280.4.E549. [DOI] [PubMed] [Google Scholar]
- [7].Hruz PW, Murata H, Qiu H, Mueckler M. Indinavir induces acute and reversible peripheral insulin resistance in rats. Diabetes. 2002;51:937–942. doi: 10.2337/diabetes.51.4.937. [DOI] [PubMed] [Google Scholar]
- [8].Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 2000;275:20251–20254. doi: 10.1074/jbc.C000228200. [DOI] [PubMed] [Google Scholar]
- [9].Murata H, Hruz PW, Mueckler M. Indinavir inhibits the glucose transporter isoform GLUT4 at physiologic concentrations. AIDS. 2002;16:859–863. doi: 10.1097/00002030-200204120-00005. [DOI] [PubMed] [Google Scholar]
- [10].Reeds DN, Yarasheski KE, Fontana L, Cade WT, Laciny E, DeMoss A, Patterson BW, Powderly WG, Klein S. Alterations in liver, muscle, and adipose tissue insulin sensitivity in men with HIV infection and dyslipidemia. Am. J. Physiol. Endocrinol. Metab. 2006;290:E47–E53. doi: 10.1152/ajpendo.00236.2005. [DOI] [PubMed] [Google Scholar]
- [11].Cade WT, Reeds DN, Lassa-Claxton S, Davila-Roman VG, Waggoner AD, Powderly WG, Yarasheski KE. Post-exercise heart rate recovery in HIV-positive individuals on highly active antiretroviral therapy. Early indicator of cardiovascular disease ? HIV Med. 2008;9:96–100. doi: 10.1111/j.1468-1293.2007.00524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luzi L, Perseghin G, Tambussi G, Meneghini E, Scifo P, Pagliato E, Del Maschio A, Testolin G, Lazzarin A. Intramyocellular lipid accumulation and reduced whole body lipid oxidation in HIV lipodystrophy. Am. J. Physiol. Endocrinol. Metab. 2003;284:E274–280. doi: 10.1152/ajpendo.00391.2001. [DOI] [PubMed] [Google Scholar]
- [13].Kim RJ, Wilson CG, Wabitsch M, Lazar MA, Steppan CM. HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity (Silver Spring, Md. 2006;14:994–1002. doi: 10.1038/oby.2006.114. [DOI] [PubMed] [Google Scholar]
- [14].den Boer MA, Berbee JF, Reiss P, van der Valk M, Voshol PJ, Kuipers F, Havekes LM, Rensen PC, Romijn JA. Ritonavir impairs lipoprotein lipase-mediated lipolysis and decreases uptake of fatty acids in adipose tissue. Arterioscler. Thromb. Vasc. Biol. 2006;26:124–129. doi: 10.1161/01.ATV.0000194073.87647.10. [DOI] [PubMed] [Google Scholar]
- [15].Zhou H, Gurley EC, Jarujaron S, Ding H, Fang Y, Xu Z, Pandak WM, Jr., Hylemon PB. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Amer. J. Physiol. 2006;291:G1071–1080. doi: 10.1152/ajpgi.00182.2006. [DOI] [PubMed] [Google Scholar]
- [16].Cade WT, Reeds DN, Mittendorfer B, Patterson BW, Powderly WG, Klein S, Yarasheski KE. Blunted lipolysis and fatty acid oxidation during moderate exercise in HIV-infected subjects taking HAART. Am. J. Physiol. Endocrinol. Metab. 2007;292:E812–819. doi: 10.1152/ajpendo.00300.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Muoio DM, Seefeld K, Witters LA, Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 1999;338(Pt 3):783–791. [PMC free article] [PubMed] [Google Scholar]
- [18].Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, Kraus WE, Dohm GL. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes. 2002;51:901–909. doi: 10.2337/diabetes.51.4.901. [DOI] [PubMed] [Google Scholar]
- [19].Beattie JH, Duthie SJ, Kwun IS, Ha TY, Gordon MJ. Rapid Quantification of Aortic Lesions in ApoE --/-- Mice. J. Vasc. Res. 2009;46:347–352. doi: 10.1159/000189795. [DOI] [PubMed] [Google Scholar]
- [20].Zhang C, Teng L, Shi Y, Jin J, Xue Y, Shang K, Gu J. Effect of emodin on proliferation and differentiation of 3T3-L1 preadipocyte and FAS activity. Chin Med J. 2002;115:1035–1038. [PubMed] [Google Scholar]
- [21].Koster JC, Remedi MS, Qiu H, Nichols CG, Hruz PW. HIV protease inhibitors acutely impair glucose-stimulated insulin release. Diabetes. 2003;52:1695–1700. doi: 10.2337/diabetes.52.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yan Q, Hruz PW. Direct comparison of the acute in vivo effects of HIV protease inhibitors on peripheral glucose disposal. J. Acquir. Immune Defic. Syndr. 2005;40:398–403. doi: 10.1097/01.qai.0000176654.97392.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carper MJ, Cade WT, Cam M, Zhang S, Shalev A, Yarasheski KE, Ramanadham S. HIV-protease inhibitors induce expression of suppressor of cytokine signaling-1 in insulin-sensitive tissues and promote insulin resistance and type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2008;294:E558–567. doi: 10.1152/ajpendo.00167.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bode H, Lenzner L, Kraemer OH, Kroesen AJ, Bendfeldt K, Schulzke JD, Fromm M, Stoltenburg-Didinger G, Zeitz Mullrich R. The HIV protease inhibitors saquinavir, ritonavir, and nelfinavir induce apoptosis and decrease barrier function in human intestinal epithelial cells. Antivir. Ther. 2005;10:645–655. [PubMed] [Google Scholar]
- [25].Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008;4:107–109. doi: 10.4161/auto.5224. [DOI] [PubMed] [Google Scholar]
- [26].Hampson L, Kitchener HC, Hampson IN. Specific HIV protease inhibitors inhibit the ability of HPV16 E6 to degrade p53 and selectively kill E6-dependent cervical carcinoma cells in vitro. Abtivir. Ther. 2006;11:813–825. [PubMed] [Google Scholar]
- [27].Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, Wang X, Hylemon PB, Pandak WM, Jr, Sanyal AJ, Zhang L, Wang G, Chen J, Wang J-Y, Zhou H. HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology. 2009;138:197–209. doi: 10.1053/j.gastro.2009.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang S, Carper MJ, Lei X, Cade WT, Yarasheski KE, Ramanadham S. Protease inhibitors used in the treatment of HIV+ induce β-cell apoptosis via the mitochondrial pathway and compromise insulin secretion. Am. J. Physiol. Endocrinol .Metab. 2009;296:E925–935. doi: 10.1152/ajpendo.90445.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med. 2005;352:48–62. doi: 10.1056/NEJMra041811. [DOI] [PubMed] [Google Scholar]
- [30].Bastie CC, Hajri T, Drover VA, Grimaldi PA, Abumrad NA. CD36 in myocytes channels fatty acids to a lipase-accessible triglyceride pool that is related to cell lipid and insulin responsiveness. Diabetes. 2004;53:2209–2216. doi: 10.2337/diabetes.53.9.2209. [DOI] [PubMed] [Google Scholar]
- [31].Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: essential role in fatty acid oxidation. Am. J. Physiol. Endocrinol. Metab. 2006;290:E509–515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- [32].Coburn CT, Hajri T, Ibrahimi A, Abumrad NA. Role of CD36 in membrane transport and utilization of long-chain fatty acids by different tissues. J. Mol. Neurosci. 2001;16:117–121. doi: 10.1385/JMN:16:2-3:117. discussion 151-117. [DOI] [PubMed] [Google Scholar]
- [33].Serghides L, Nathoo S, Walmsley S, Kain KC. CD36 deficiency induced by antiretroviral therapy. AIDS. 2002;16:353–358. doi: 10.1097/00002030-200202150-00006. [DOI] [PubMed] [Google Scholar]
- [34].Mallolas JMDP, Podzamczer DMD, Milinkovic AMD, Domingo PMD, Clotet BMD, Ribera EMD, Gutierrez FMD, Knobel HMD, Cosin JMD, Ferrer EMD, Arranz JAMD, Roca VMD, Vidal FMD, Murillas JMD, Pich JP, Pedrol EMD, Llibre JMMD, Dalmau DMD, Garcia IMD, Aranda MMD, Cruceta AMD, Martinez EMD, Blanco JLMD, Lazzari E.d.M.S., Gatell JMMD, the Atazip Study Group Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/r-containing HAART: The ATAZIP Study. J. Acquir. Immune Defic. Syndr. 2009;51:29–36. doi: 10.1097/QAI.0b013e31819a226f. A.S.G. [DOI] [PubMed] [Google Scholar]
- [35].Nguyen ST, Eaton SA, Bain AM, Rahman AP, Payne KD, Bedimo R, Herrington JD, Maclayton DO, Rodriguez-Barradas MC, Busti A. Lipid-lowering efficacy and safety after switching to atazanavir-ritonavir-based highly active antiretroviral therapy in patients with human immunodeficiency virus. Pharmacotherapy. 2008;28:323–330. doi: 10.1592/phco.28.3.323. [DOI] [PubMed] [Google Scholar]
- [36].Williams K, Rao YP, Natarajan R, Pandak WM, Hylemon PB. Indinavir alters sterol and fatty acid homeostatic mechanisms in primary rat hepatocytes by increasing levels of activated sterol regulatory element-binding proteins and decreasing cholesterol 7alpha-hydroxylase mRNA levels. Biochem. Pharmacol. 2004;67:255–267. doi: 10.1016/j.bcp.2003.08.044. [DOI] [PubMed] [Google Scholar]
- [37].Zhou H, Jarujaron S, Gurley EC, Chen L, Ding H, Studer E, Pandak WM, Jr, Hu W, Zou T, Wang J-Y, Hylemon PB. HIV protease inhibitors increase TNF-[alpha] and IL-6 expression in macrophages: Involvement of the RNA-binding protein HuR. Atherosclerosis. 2007;195:e134–e143. doi: 10.1016/j.atherosclerosis.2007.04.008. [DOI] [PubMed] [Google Scholar]
- [38].Magana MM, Osborne TF. Two tandem binding sites for sterol regulatory element binding proteins are required for sterol regulation of fatty-acid synthase promoter. J. Biol. Chem. 1996;271:32689–32694. doi: 10.1074/jbc.271.51.32689. [DOI] [PubMed] [Google Scholar]
- [39].Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J. Biol. Chem. 1997;272:7298–7305. doi: 10.1074/jbc.272.11.7298. [DOI] [PubMed] [Google Scholar]
- [40].Caron M, Auclair M, Vigouroux C, Glorian M, Forest C, Capeau J. The HIV protease inhibitor indinavir impairs sterol regulatory element-binding protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistance. Diabetes. 2001;50:1378–1388. doi: 10.2337/diabetes.50.6.1378. [DOI] [PubMed] [Google Scholar]
- [41].Riddle TM, Kuhel DG, Woollett LA, Fichtenbaum CJ, Hui DY. HIV protease inhibitor induces fatty acid and sterol biosynthesis in liver and adipose tissues due to the accumulation of activated sterol regulatory element-binding proteins in the nucleus. J. Biol. Chem. 2001;276:37514–37519. doi: 10.1074/jbc.M104557200. [DOI] [PubMed] [Google Scholar]