

Abstract
Sulfur mustard (bis-(2-chloroethyl)sulfide) has been used in chemical warfare since World War I, and is well known as an acutely toxic vesicant. It has been implicated as a carcinogen after chronic low-level exposure, and is known to form inter-strand crosslinks in DNA. Sulfur and nitrogen mustards are currently of interest as potential chemical threat agents for terrorists due to ease of synthesis. Sulfur mustard and monofunctional analogs (half-mustards, 2-[chloroethyl] alkyl sulfides) react as electrophiles, damaging cellular macromolecules, and thus are potentially subject to scavenging by nucleophilic agents. We have determined rate constants for the reaction of four purine derivatives that contain nucleophilic thiol moieties with several sulfur-half-mustards. Three of these compounds, 2,6-dithiopurine, 2,6-dithiouric acid, and 9-methyl-6-mercaptopurine, exhibit facile reaction with the electrophilic mustard compounds. At near neutral pH, these thiopurines are much better nucleophilic scavengers of mustard electrophiles than other low molecular weight thiols such as N-acetyl cysteine and glutathione. Progress curves calculated by numerical integration techniques indicate that equimolar concentrations of thiopurine provide significant reductions in the overall exposure to the episulfonium ions, which are the major reactive, electrophiles produced when sulfur mustards are dissolved in aqueous solution.
Introduction
Sulfur mustard gas (SM1, bis-[2-chloroethyl]sulfide) and related chemical weapons have been used in warfare since World War I to maim and incapacitate enemy soldiers. Military exposures to the mustard agents have long been known to cause severe blistering of exposed skin, which is slow to heal, and damage to the cornea; the term vesicant is used to describe these actions. In addition, inhalation exposure of the aerosolized agent causes damage or destruction of the respiratory tract, including chronic bronchitis, bronchiectasis, and pulmonary fibrosis (1, 2). Despite extensive research efforts, little progress has been made on developing effective treatment regimens, and victims often require extended hospitalization.
There is increased concern in the Homeland Defense community (http://www.bt.cdc.gov/Agent/AgentlistChem.asp) that these agents or related chemicals could be used by terrorists to attack the civilian population of the United States, and thus there is need to develop strategies to ameliorate the effects of such an attack. The concerns extend not only to treating the exposed human population, but also to protecting emergency personnel called upon to treat the victims and decontaminate the affected area. SM, for example, is actually a liquid under ambient conditions that can contaminate clothing and surfaces after deployment and maintain its toxic activity for some time, thus posing a threat to rescue, medical and decontamination personnel (1). In addition, there is increasing evidence that a significant fraction of the SM dose remains biologically active within the victim's body for several days after exposure (3, 4).
The underlying cause of the various forms of toxicity is thought to be macromolecular damage. SM and the monofunctional analogs, 2-chloroethyl ethyl sulfide (CEES) and 2-chloroethyl methyl sulfide (CEMS) are direct acting, electrophilic agents that react with nucleophilic moieties in nucleic acids and proteins (5-9). The reaction of SM with DNA has been well-studied (5, 8). The initial reaction pathway in aqueous solution is spontaneous dechlorination of one of the chloroethyl substituents, resulting in the formation of a cyclic episulfonium ion (10, 11), which preferentially reacts at the N7 position of deoxyguanosine to give a mono adduct. Because of the reactivity of both chloroethyl groups, further reaction with an adjacent deoxyguanosine produces cross-links in the DNA. In particular, in both yeast (12) and mammalian cells (13) the cytotoxicity of SM is dramatically enhanced in cells that are deficient in the nucleotide excision repair pathway; in mammalian cells, cytotoxicity due to 2-(chloroethyl) ethyl sulfide, a monofunctional analog of SM, was also found to be enhanced by NER deficiency. An elegant host cell reactivation assay based in CHO cells (13), showed that this enhanced cytotoxicity correlated with an inability to functionally repair SM- or CEES-damaged DNA. Together, these data provide strong evidence that at the cellular level, DNA damage is the major determinant of mustard toxicity.
Numerous other chemical carcinogens also damage DNA through electrophilic intermediates that attack nucleophilic sites in DNA. Several strategies for scavenging these electrophilic carcinogens have been demonstrated previously. In particular, thiopurines have been shown to react facilely with a wide range of electrophilic toxicants (14-17), and have demonstrated effective suppression of cytotoxicity, mutagenesis and carcinogenesis (14, 18). Several of these compounds, notably 2,6-dithiopurine (DTP), are substrates for the cellular purine transport system (19), and can therefore be rapidly taken up by cells. These compounds, therefore, might be effective in scavenging reactive sulfur mustards.
The initial and rate-determining step in the nucleophilic substitution reactions of sulfur mustards in aqueous solution (Scheme 1)is dissociation of Cl- (10, 11, 20, 21). The episulfonium ion formed in this dissociation then reacts in a faster, bimolecular reaction with either water or an added nucleophile. An extensive series of anions was analyzed in early work for their ability to “compete” with water for reaction with the episulfonium ions derived from SM (20). Because the reaction with water is much faster than the initial dissociation, measurement of kW has not been reported, and the relative strengths of competing nucleophiles have been expressed as “competition factors”, which are actually the ratio k2/kW. Among the nucleophiles examined, one of the most reactive non-toxic compounds appears to be thiosulfate, with k2/kW ∼ 27,000 M-1 for SM and 19,000 M-1 for CEES; nucleic acids, cysteine and cysteine ethyl ester were reported to be at least an order of magnitude less reactive with SM (20).
Scheme 1.

The initial reaction of the half-mustards CEES and CEMS with nucleophiles in aqueous solution follows the same mechanism as the analogous reaction of SM. Thus, many studies have been done with the half-mustards, which are less toxic and can be utilized in a standard chemical fume hood, without the need for glove boxes and special facilities. We hypothesized that DTP and other thiopurines would likely show robust nucleophilic substitution reactions with the half-mustards CEES and CEMS, and therefore may be candidates for effective prophylactic and/or therapeutic agents against SM toxicity. In the present communication, we show that three non-toxic thiopurines, 2,6-dithiopurine (DTP), 2,6-dithiouric acid (DUA) and 9-methyl-6-mercaptopurine (MMP), exhibit facile substitution reactions with these sulfur half mustards. The “competition factors” for these thiopurines, a measure of the relative reactivity of a potential scavenging agent, are found to be comparable to thiosulphate, a well-known scavenger, whereas other commonly used thiol-containing reagents, N-acetyl cysteine (NAC) and reduced glutathione (GSH) are found to be much less active at near neutral pH. More importantly, we show that at equimolar concentrations these thiopurines significantly reduce overall exposure to the reactive episulfonium ions that cause DNA damage and genotoxicity.
Materials and Methods
Chemicals
2-chlorethyl methyl sulfide (CEMS) and 2-chlorethyl ethyl sulfide (CEES) were obtained from Aldrich Chemicals (St. Louis, MO), reduced glutathione (GSH), N-acetyl-cysteine (NAC), 2-mercaptoethanesulfonic acid (MESNA) and DTT were obtained from Sigma Chemical Co. (St. Louis, MO); all were used without further purification. Working stocks of the electrophiles were prepared in ethanol at 200 or 600 mM and stored at -20° C.
6MP, DTP, and 6TX were initially obtained from Sigma, and were used without further purification. Recent batches of DTP, obtained from Aldrich and nominally 97% pure, were found by HPLC to contain a contaminant amounting to about 40% by weight. Thus, DTP was routinely repurified by extraction with hot water. The lyophilized product gave a single HPLC peak with about 97% purity. DUA was prepared by published methods (Levin, 1960). MMP was synthesized by Chemsyn Laboratories (Lenexa, KS) based on published methods. Working stocks of the thiopurines were prepared at 10 mM in 0.05 N NaOH and stored at -20° C.
All other reagents were analytical reagent grade or better, and were obtained from standard suppliers. CEMS and CEES are toxic compounds with the potential to damage DNA, and must be handled with caution. All solutions containing these chemicals were treated with bleach prior to disposal, and solid waste was treated as biohazardous. In addition, CEMS and CEES are liquids at room temperature, and their vapors present an inhalation hazard. Absorbance measurements of solutions containing CEMS or CEES were made either in closely stoppered cuvets, or by locating the spectrophotometer inside a chemical fume hood.
Hydrolysis assay
Reaction of the sulfur half mustards (500 μM) with an equimolar amount of the nucleophile 6MP occurred rapidly at 23° C, as indicated by changes in the absorbance spectrum of the 6MP (vide infra). In a supporting buffer of 12.5 mM sodium phosphate buffer, pH 7.5 (PB), 50 mM NaCl, this reaction was complete within 10 minutes at 23° C. By varying the concentration of electrophile from 25-500 μM, a standard curve relating the absorbance ratio A320/A293 at completion of reaction to initial electrophile concentration could be constructed. The rate of hydrolysis of the electrophile could be measured by adding the electrophile, previously dissolved in ethanol, to an aqueous solution in the absence of nucleophile, and at timed intervals withdrawing aliquots of the hydrolysis reaction, mixing with 6MP under standard assay conditions and determining the concentration of electrophile remaining unhydrolyzed from the absorbance measurements and the standard curve. Absorbance measurements were made with either a Cary 50 UV-VIS spectrophotometer, or an HP8450 diode array spectrophotometer equipped with Peltier temperature-controlled cuvet holders. These instruments gave identical results.
Kinetics assays
Reactions were carried out in 3.1 mM PB, 12.5 mM NaCl. Standard reactions contained 500 μM electrophile and 50 μM thiopurine unless otherwise noted. The course of reaction was monitored by acquiring absorbance spectra with a diode-array spectrophotometer. Preliminary measurements were conducted to identify optimal wavelengths at which to measure changes in the absorbance of the thiopurines. Based on absorbance measurements in the absence of electrophile, and in the presence of electrophile but at the conclusion of reaction, absorbance measurements taken at multiple time points during the reaction could be converted to concentration of unreacted thiopurine remaining. The absorbance at the conclusion of the reaction was estimated by measuring absorbance at times corresponding to 5-10 half-lives, then extrapolating to infinite time from a linearized, double reciprocal plot.
As described in Supplementary Materials, the non-linear differential equations for this system cannot be solved in closed form. We have implemented the Runge-Kutta fourth order numerical integration algorithm on a Macintosh computer using the programming language in the Stata10 software system (StataCorp, College Station, TX). The error associated with this approach decreases as the fourth power of the step size, and can be made arbitrarily small by selecting a small step size. We have noted insignificant changes in the results of these progress-curve calculations with changes in step size below 0.01 s, and have used 0.01 s as the step size in all integrations reported. Progress curves for R(t) were calculated from 0 to 80 s of reaction given the measured value for k1, test values for k2 and kW, and the initial concentrations of R and M. The experimental values of R(t), determined at 10 s intervals between 5 and 80 s in 3-5 independent experiments, were compared with the integrated values by calculating the root mean squared error. For any given value of kW, the “best-fit” value for k2 (to three significant figures) was determined by minimizing this error function. To construct progress curves for M+, the episulfonium ion, best fit values for the rate constants were used and integration time was extended to 600 s.
HPLC
Separations were carried out with a Shimadzu dual pump HPLC equipped with autosampler/autoinjector (50 μL sample loop) and a variable wavelength absorbance detector. Aqueous samples were adjusted to 5 mM trifluoroacetic acid (TFA) and applied to a reverse phase (C8) column (Aquapore RP300, ABI) operated at room temperature and at a flow rate of 1.5 mL/min 1.7 mM TFA. Adducts were eluted using linear gradients of TFA/acetonitrile; the exact conditions were varied empirically depending on the separation needed. Alternatively, a mobile phase consisting of 5 mM Tris, pH 8.0, and acetonitrile was used.
Mass spectroscopic analysis
An electrospray, ion trap mass spectrometer (LCQ, Finnigan MAT, San Jose, CA) coupled on-line with a microbore HPLC (Magic 2002, Michrom BioResource, Auburn, CA) was used to acquire spectra of adducts formed between thiopurines and CEMS. Samples were loaded into the LCQ without a column through a small molecule desalt trap (Michrom BioResource) and desalted with 4× 50 μL mobile phase A (acetonitrile:water:formic acid:trifluoroacetic acid, 90:10:0.09:0.02) at a flow rate of 50 μL/min. Automated acquisition of full-scan spectra (either 100-1000 daltons or 150-1000 daltons) was controlled by Finnigan Excalibur™ software; the target number of ions was 1 × 108. The settings for the ESI were: spray voltage, 4.5kV; nitrogen sheath gas and auxiliary gas flow rates, 60 and 5 psi, respectively capillary temperature, 200° C, capillary voltage, 22 V; tube lens offset, 40 V.
Effects of non-purine, thiol-containing compounds
To compare the reaction of other potential nucleophiles, a competition strategy was adopted. Initially, standard reaction mixtures contained 0.5 mM CEMS, 50 μM DTP and from 0-1000 μM of a competing nucleophile. The extent of reaction of CEMS with DTP was estimated by changes in the absorbance spectrum at completion of reaction. To obtain quantitative data, the standard reaction mixture contained 1000 μM DTP and 50 μM CEMS in the supporting buffer described for the kinetic experiments. This reaction was allowed to go to completion, and the major DTP adduct was quantitated by HPLC with absorbance detection. The competition reactions contained the same components, but an additional nucleophile was added at 1000 μM, equimolar to the DTP, prior to addition of CEMS. Three sulfhydryl-containing nucleophiles, sodium thiosulfate, NAC and GSH, were tested in this way. Additionally, purified salmon sperm DNA was tested. In order to analyze the DNA competition reactions, the high molecular weight DNA was removed by ethanol precipitation prior to HPLC analysis. The reaction mixtures were adjusted to 0.1 M NaCl, 2.5 vol ethanol were added, and the DNA was allowed to precipitate overnight at -20° C. Precipitated DNA was removed by centrifugation at 10,000 rpm for 30 min, and ethanol was removed from the supernate by evaporation under a stream of nitrogen at room temperature. Samples were reconstituted with 5 mM Tris, pH 8.0 and analyzed by HPLC. Control experiments demonstrated quantitative recovery of both DTP and the major DTP-CEMS adduct in the supernate.
Results
Reaction of sulfur half-mustards with thiopurines
In our previous studies, several mono- and di-thiopurines were identified that exhibited facile reaction with BPDE and other electrophiles (14-17); these compounds included 6MP, MMP, 6TX, DTP and DUA, the major in vivo metabolite of DTP (22). As shown in Figure 1A, addition of 500 μM CEMS to a solution containing 50 μM 6MP caused a time-dependent shift in the absorbance spectrum of 6MP (λmax ∼ 322 nm) towards lower wavelengths (shoulder at ∼ 295 nm) due to chemical adduct formation.
Figure 1.
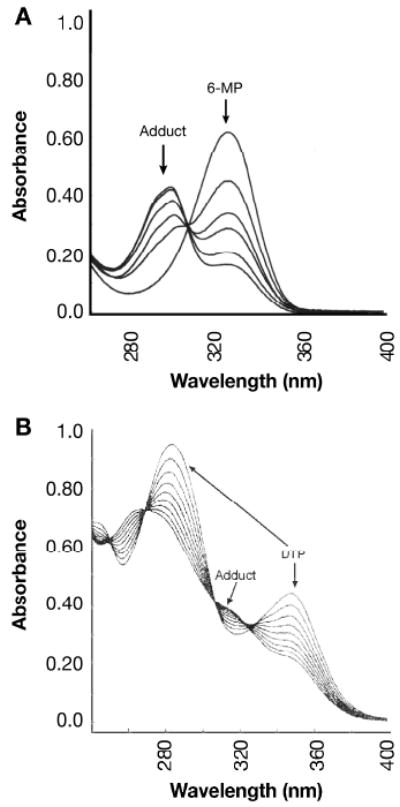
Reaction of thiopurines with CEMS: spectral shifts
A. Reactions were set up containing 500 μM CEMS and 50 μM 6MP in a screw-capped cuvet and absorbance spectra were recorded at intervals. The absorbance maximum for 6MP at 322 nm shifted to about 293 nm. Spectra shown were recorded at 0, 30, 60, 90, 180 and 600 s of reaction. B. Similar reactions were set up with 50 μM DTP. Spectra were recorded once every 8 s beginning at about 8 s of reaction.
Changes in the absorbance spectrum were complete within 15 min at 23° C. HPLC analysis of the spent reaction mixture at slightly alkaline pH gave a single predominant product with a retention time of 15.0 min (Figure 2A, trace 1). Control HPLC analyses without CEMS (trace 2) or without 6MP (trace 3) are shown for comparison. Absorbance spectroscopy indicated that this product exhibited a blue shift, with λmax ∼ 293 nm, and very low absorbance at 322 nm. Mass spectrometry of the isolated product indicated the addition of a single methyl ethyl sulfide to 6MP (Figure 2B), presumably at the 6-mercapto-moiety.
Figure 2.
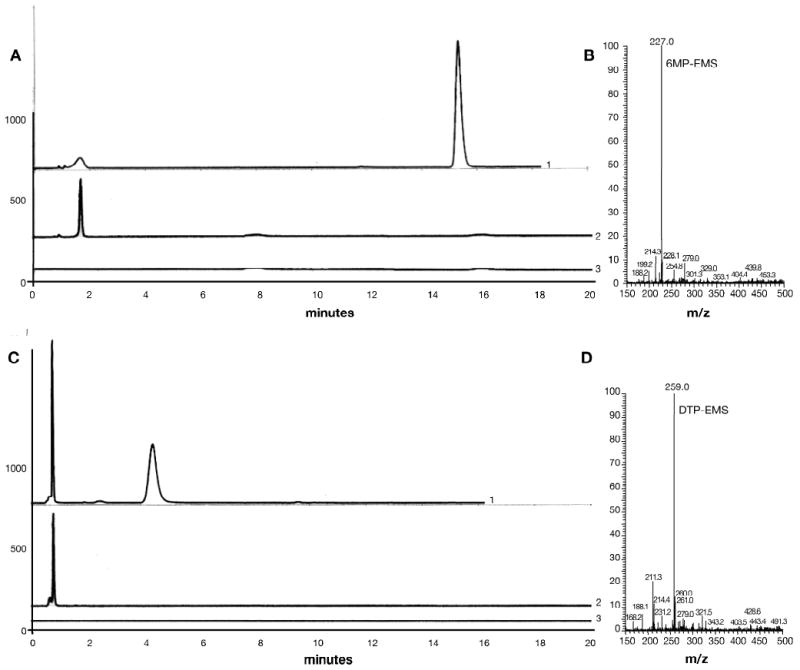
HPLC analysis of reaction products
Reaction mixtures contained 2 mM CEMS and 2 mM purinethiol. At completion of reaction, samples were diluted five-fold with 5.0 mM Tris, pH 8.0, and analyzed by HPLC. A. The reaction of CEMS with 6MP was analyzed with the absorbance detector set at 293 nm, selective for the expected adduct. Trace 1 is a representative chromatogram from the complete reaction mixture; trace 2, reaction mixture lacking CEMS; trace 3, reaction mixture lacking purinethiol; traces are offset by arbitrary amounts along the y-axis for clarity. B. The adduct eluting at 15.0 min was collected from multiple runs and analyzed by mass spectrometry as described in Materials and Methods. C. The reaction of CEMS with DTP was analyzed with the absorbance detector set at 320 nm, selective for the expected adduct. Traces labeled as in panel A. D. The adduct eluting at 4.4 min was collected form multiple runs and analyzed by mass spectrometry.
Under the same conditions, reaction of DTP with CEMS also gave time-dependent changes in spectral characteristics (Figure 1B). The absorbance of DTP at 286 or 347 nm progressively decreased, shifting towards lower wavelengths and exhibiting apparent isosbestic points at ∼ 271 and 308 nm. HPLC analysis again indicated a single, predominant reaction product with tR=4.4 min at alkaline pH (Figure 2C, trace 1) and a blue shifted absorbance spectrum (local λmax at 321 and 262 nm). Mass spectrometry of this product (Figure 2D) again indicated the addition of a single methyl ethyl sulfide to DTP. The same methodology indicated facile formation of mono-adducts between CEMS and each of the other thiopurines tested, MMP (Supplementary Figure S1, panels A and B), 6TX (Supplementary Figure S1, panels C and D), and DUA (Supplementary Figure S1, panels E and F). In each case, the mass spectrum of the indicated HPLC-purified peak gave a major ion consistent with addition of a single methyl ethyl sulfide to the purinethiol.
Spectrophotometric measurement of half-mustard dissociation rates
To determine the scavenging potential of the thiopurines, it was first necessary to measure the rate constants for dissociation of CEMS and CEES when added to aqueous solution in the absence of any added nucleophile since this is the rate-determining step (Scheme 1; 10, 11, 20, 21). This rate is known to vary with the concentration of chloride ion, temperature and concentration of alcohol (10, 11, 20, 23). We decided to take advantage of the facile reactions of 6-MP with CEMS and CEES, characterized by an approximate 30 nm absorbance shift, to measure this rate constant under the conditions to be used for analyzing competition factors. As described in section 1 of the Supplementary Materials, measurement of the decay of the total reactive mustard concentration ([MCl] + [M+]) can provide a direct measurement of k1. By allowing the sulfur mustard to react in aqueous solution for varying times periods, and then diluting into a scavenging solution containing an excess of 6-MP, the time dependence of the total concentration of unreacted mustard ([MCl] + [M+]) can be determined. Standard curves for ([MCl] + [M+]) were developed by allowing different concentrations of CEMS or CEES (6.25– 50 μM) to react to completion with 50 μM 6-MP, and measuring the absorbance. Because of the spectral shift upon reaction (shown in Figure 1A), a standard curve relating [CEMS] or [CEES] to the ratio A320/A293 could be generated (Figure 3A).
Figure 3.
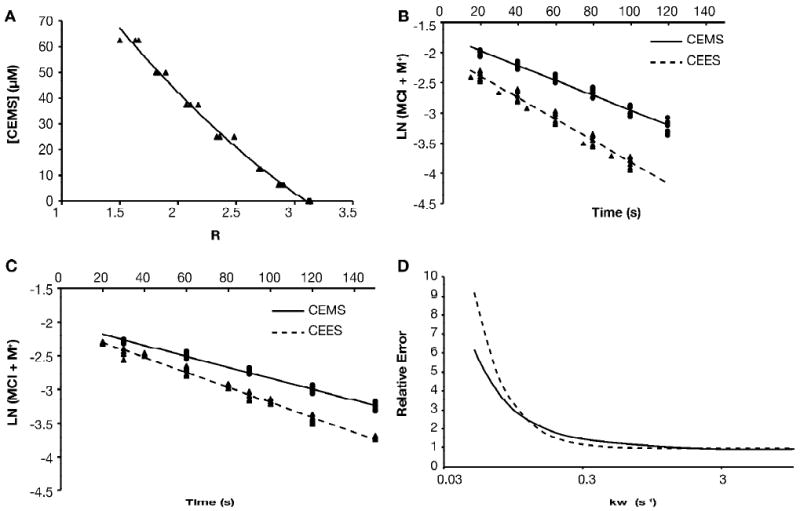
Measurement of hydrolysis rate constants
A. To prepare standard curves, reactions were set up containing 50 μM 6MP and from 6.25 to 62.5 μM CEMS and allowed to go to completion. Absorbance spectra were obtained and the ratio of A320/A293 =R was calculated. The smooth line represents the best fit of the data to a quadratic equation; the best-fit equation was then used to convert experimental R values to [CEMS]. Standard curves were prepared daily; all data points from a representative standard curve are shown. Similar curves were obtained for CEES. B. Ethanolic solutions of CEMS or CEES were diluted to 0.125 mM in aqueous solution with no added NaCl, and at the indicated times aliquots were mixed with 6MP and allowed to react to completion. R values were calculated from absorbance spectra and converted to give the concentration of CEMS remaining unhydrolyzed at the given time point; the natural logarithm of the unhydrolyzed mustard concentration is plotted. All data points from 8 independent determinations for CEMS (circles, solid line) and 8 independent determinations for CEES (triangles, dashed line) are shown, along with the linear least squares regression lines, calculated for all data. The regression analysis provides estimates of k1 (the slope of the line) and 95% confidence limits for that estimate. C. Data was obtained and analyzed as in panel B, but with 12.5 mM NaCl present in the initial hydrolysis reaction. The resulting linear relations between k-1 and kw were as follows: CEMS k-1 = (0.497 × kw - .00404)/[Cl-]; CEES k-1 = (0.583 × kw - .00648)/[Cl-] × kw. D. Numerical integration was used to calculate reaction progress curves for the hydrolysis of CEMS or CEES in the presence of 12.5 mM NaCl for a range of assumed values of kw from 0.05 to 10 s-1. The experimental data points (panel C) were compared to the calculated data by summing the squared deviations, and normalizing to that obtained with kw=5 s-1. The error functions calculated for CEMS (solid line) or CEES (dashed lines) are shown.
If CEMS hydrolysis is carried out in a buffer lacking added chloride, the concentration of chloride ion increases during the reaction to a level equal to the initial [CEMS]. By keeping this concentration less than about 1 mM, the effect of the back reaction (M+ + Cl-→MCl) is expected to be negligible based on previous work (10), and therefore the reaction can be described by first order kinetics. To measure the dissociation rate constant, k1, an ethanolic working stock of CEMS was diluted into 3.1 mM PB with no added NaCl at 23°C to initiate the hydrolysis reaction; the final concentration of CEMS was 0.125 mM. At intervals, aliquots of the hydrolysis reaction were removed and mixed with 6MP under “scavenging” conditions. After 30 min, the absorbance ratio was determined and the concentration of CEMS remaining unreacted at the time the aliquot was taken was calculated from the standard curve. The results of 8 independent determinations are plotted in Figure 3B (closed circles, solid line) as the natural logarithm of the unhydrolysed CEMS concentration versus time. Linear regression analysis (r2 = 0.972) gave an estimate of k1 for CEMS of 0.0122 ± 0.0006 s-1; similar analyses for CEES (N=8, r2=0.972; Figure 3B, closed triangles, dashed line) gave k1 = 0.0176 ± 0.0009 s-1. Allowing for differences in rate due to solvent composition, these data are in good agreement with previous measurements of the rate constants for these compounds (11, 23).
At higher salt concentrations, the effect of the back reaction of M+ with Cl- cannot be neglected (10). The altered kinetics in the presence of NaCl can be used to obtain estimates for the rate constants for the back reaction, k-1, and for the hydrolysis step, kw, in a two-step analysis. In the first step, we used matrix algebra to obtain an approximation for the kinetics in closed form (Supplementary Materials, Section 2) that allows us to express k-1 as a linear function of kw, based on the apparent first-order rate constant for the reaction, determined as above but in the presence of a non-negligible concentration of NaCl. As shown in Figure 3C, kinetic determinations for CEMS at [Cl-] = 12.5 mM (N = 6, closed circles, solid line) closely approximated a first order decay (r2=0.987) with kapp = 0.0081 ± 0.0004 s-1. Similar analysis for CEES (N=8, Figure 3C closed triangles, dashed line, r2=0.987) gave kapp = 0.0111 ± 0.0004 s-1. The linear relationships between k-1 and kw as determined by these analyses are given explicitly in the Figure Legend.
In the second step of this analysis, we used numerical integration to obtain solutions of the kinetics equations starting with estimates of all rate constants, and compared the computed solutions to the data obtained at [NaCl] = 12.5 mM. In this analysis, we started with the experimental estimate of k1 and the relation between k-1 and kw as determined above, and allowed kw to vary. We searched for the best fit to the data by minimizing the sum of squares error function. As shown in Figure 3D for both CEMS and CEES, the error function was relatively constant for values of kw ≥ 1, but increased sharply at values of kw less than about 0.2. Conservatively, these data suggest that kw ≥ 0.2. This is consistent with the concept that dissociation is the rate-determining step (10, 11, 20, 21), which implies that kw ≫ k1.
Determination of thiopurine competition factors
The efficacy of nucleophiles as reactants for sulfur mustards has classically been described by the “competition factor” (20); in the nomenclature of Scheme 1 this is k2/kW. As described in Supplementary Materials and noted above, the differential equations describing these reactions cannot be solved in closed form, but can of course be solved by numerical integration. Using the Runge-Kutta fourth order method, reaction progress curves can be calculated for any given set of values of the relevant rate constants and initial conditions. In practice, we used the experimentally determined values for k1 and assumed a trial value for kW; as described above, this allowed us to calculate k-1. We then assumed a trial value for k2 and compared the calculated progress curve to experimental data for the reaction of CEES or CEMS with the chosen thiopurine. By allowing k2 to vary systematically and minimizing the root mean square deviation between the experimental data and the calculated progress curve, we could obtain best-fit values for k2/kW.
As shown in Figure 4A, the numerical integration produced reaction progress curves which closely fit the experimental data. In this experiment, 50 μM DTP was allowed to react with an excess of CEMS, and the concentration of DTP remaining unreacted (solid triangles) was determined spectrophotometrically as described in Materials and Methods. With kW set at 1.0 s-1, the best fit of the calculated points (solid line) to the experimental data was obtained with k2 = 8340 M-1 −s-1. When kW was allowed to vary over the range 0.1 to 10 s-1, the best fit value of k2 for this reaction increased linearly with kW (Supplementary Figure S2; r2 = 1.00). Thus, values of k2/kW are essentially invariant over this range, and in Table 1, we have chosen to present least-squares best-fits of k2/kW with kW set at 1.0 s-1.
Figure 4.
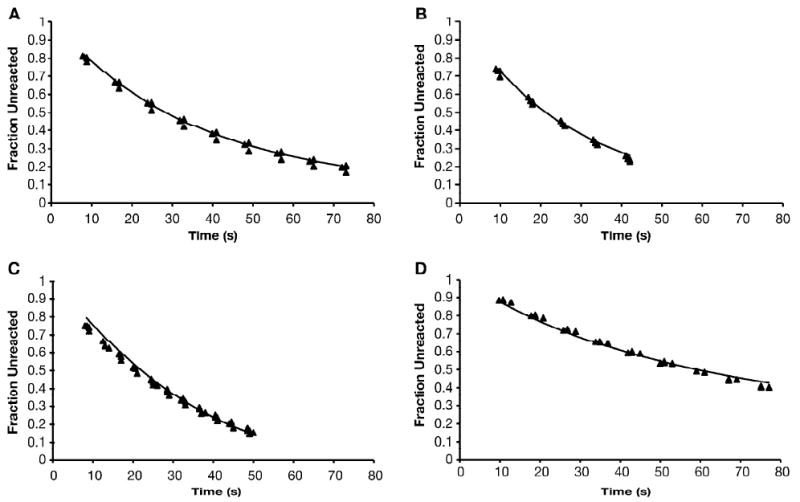
Reaction of purinethiols with CEMS
A small aliquot of CEMS (2.5 μL) diluted in EtOH was placed in the bottom of a plastic tube, and diluted and rapidly mixed with an aqueous purinethiol solution under conditions described in Materials and Methods. The reaction mixture was transferred to a stoppered quart cuvet and absorbance spectra were recorded at 10 s intervals beginning about 8 s after mixing. The changes in absorbance were used to calculate the concentration of purinethiol remaining unreacted at each time point. The fraction of the original purinethiol concentration remaining at each time point is shown as closed triangles, with all data points from 3-5 independent determinations plotted. The solid line represents the progress curve obtained by numerical integration of the differential equations, utilizing the known initial values, the measured value of k1, and the best-fit value of k2; kW was conservatively assumed to be 1.0 s-1. A. DTP; B. DUA; C. MMP; D. TX.
Table 1.
Thiopurine competition factors
| CEMS | ||
|---|---|---|
| kW = 1 s-1 | ||
|
k2/kW (95% CI) (L/mol) |
Fractional Protection | |
| DTP | 8340 (8200,8700) | 0.707 |
| MMP | 33600 (32000,35000) | 0.877 |
| DUA | 11000 (10900,11600) | 0.751 |
| TX | 4000 (3870,4120 | 0.626 |
| CEES | ||
| kW = 1 s-1 | ||
|
k2/kW (95% CI) (L/mol) |
Fractional Protection | |
| DTP | 6240 (6100,6500) | 0.663 |
| MMP | 9900 (9600,10600) | 0.756 |
| DUA | 7780 (7600,8300) | 0.682 |
| TX | 1780 (1740,1850) | 0.408 |
Similar analyses were performed for DUA (Figure 4B), MMP (Figure 4C), and TX (Figure 4D) reacting with CEMS, and for all four purinethiols reacting with CEES (Supplementary Figure S3). In all cases the fit between the theoretical and experimental reaction progress curves was excellent, similar to that shown in Figure 4A. For both CEMS and CEES, competition factors in the range 6240 to 33,600 M-1 were obtained with kW = 1.0 s-1 for DTP, DUA and MMP (Table 1). The competition factors for TX were somewhat lower, but still in excess of 1700 M-1. Thus, the non-toxic thiopurines DTP, DUA and MMP are comparable in strength to the well-known nucleophilic scavenger thiosulphate (k2/kW ∼ 19,000 M-1). The competition factors determined in this way for DTP reacting with CEES and CEMS exhibited little pH dependence between about pH 7 and 9, and decreased at lower pH values (Supplementary Figure S4).
Since M+, the episulfonium ion produced in the initial dissociation event, is the electrophile that ultimately damages DNA and other tissue nucleophiles, the utility of a scavenging agent in protecting biological systems lies in the ability of the scavenger to decrease the overall exposure to M+. This effectiveness can be expressed as the area-under-the-curve (AUC) for the reaction progress curve expressed as [M+](t). The Runge-Kutta numerical integration method used to estimate the competition factors can also be used to produce reaction progress curves for [M+], using the parameters obtained in the best-fit analysis given above. In Figure 5A, we illustrate this with [M+] reaction curves for an initial CEMS concentration of 500 μM, in the presence of 0, 250, 500 or 1000 μM DTP. Increasing concentrations of DTP significantly decrease [M+] at all reaction times. At equimolar levels of DTP and CEMS, the AUC for [M+] is decreased by about 70%; for MMP the fractional protection from CEMS is about 88% (see Figure 5B). Similar analyses indicate that each of the three most reactive thiopurines decrease the integrated exposure to M+ by 65 – 88% when present initially at a concentration equimolar to CEMS or CEES (Table 1). Thus, these thiopurines exhibit excellent potential to act as chemoprotective agents in aqueous solution.
Figure 5.
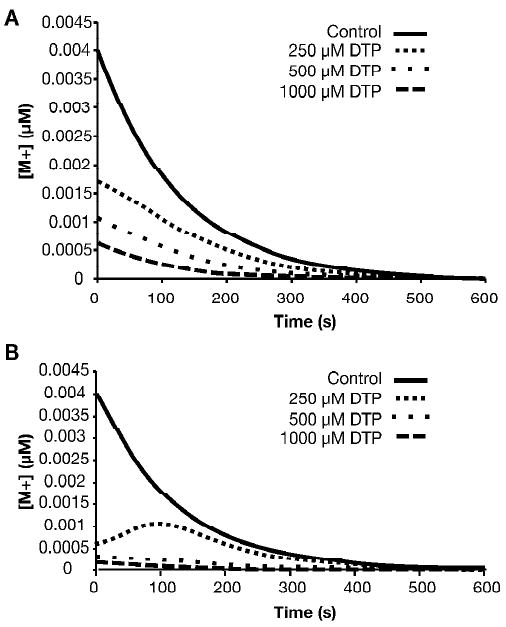
Reaction progress curves for [M+]
Numerical integration was used to calculate time-dependent changes in the concentration of M+, for CEMS in the presence of the indicated concentrations of (A) DTP or (B) MMP, utilizing the best fit values of k2 with kW set at 1.0 s-1.
Reaction of CEMS with other thiol-containing compounds
Initially, several low molecular weight thiol-containing compounds (MESNA, DTT, NAC and GSH) were assayed for possible scavenging potential by determining their ability to compete with DTP for reaction with CEMS. Standard reaction conditions included 500 μM electrophile, 50 μM DTP and from 0 to 1000 μM of the thiol competitor, buffered at pH 7.5 with 10 mM sodium phosphate. Reactions proceeded to completion, and the change in the absorbance spectrum of DTP due to reaction with the electrophile was recorded. In this assay, MESNA and DTT gave no observable inhibition of the reaction of DTP with CEMS, even at 20-fold molar excess over DTP. NAC and GSH gave only about 20% inhibition of the DTP reaction by the spectrophotometric assay at 20-fold molar excess over DTP. As shown in Supplementary Figure S5, when either GSH or NAC were added at 50 μM, equimolar to DTP, little or no inhibition of adduct formation was seen. However, raising the pH in the reaction mixture to 10.2 (buffered with sodium bicarbonate), well above the pK for the sulfhydryl of the cysteine moiety (pK∼8.75), increased the ability of both NAC and GSH to block the reaction of DTP with CEMS (Supplementary Figure S5).
To study the possible competition of GSH with DTP at physiological pH in more detail, HPLC methods were employed to determine the amount of DTP-CEMS adduct formed at completion of the reaction. To provide a quantitative comparison of scavenging rates, reactions must be carried out under conditions where the electrophile is limiting. Accordingly, we analyzed reaction mixtures containing an excess of DTP (1000 μM) over CEMS (50 μM) in the presence or absence of 1000 μM GSH. At the completion of reaction, the reaction mixtures were analyzed by HPLC under slightly alkaline conditions, and the yield of the predominant DTP-CEMS mono-adduct (tR=4.4 min; see Figure 2C) was determined. Theoretical analysis (Supplementary Materials section 4) indicated that if GSH acts as a competitive inhibitor of this reaction, the inhibitory rate constant could be estimated from the decrease in yield of the DTP reaction product. No inhibition of DTP-CEMS adduct formation was observed (Figure 6). Furthermore, HPLC analysis of reaction mixtures of GSH and CEMS without DTP failed to give any indication of a putative GSH-CEMS reaction product. Similarly, competition experiments with 1000 mM NAC or purified DNA did not give significant inhibition of DTP-CEMS adduct formation (Figure 6). In a similar experiment with sodium thiosulfate as competitor, about a six-fold inhibition of DTP-CEMS monoadduct formation was observed (Figure 6). This is in general agreement with previous reports of rapid reaction of thiosulfate with SM and CEES (20, 24). We conclude that DTP is at least 8-fold more active than GSH as a chemical scavenger of CEMS at near-neutral pH.
Figure 6.

Analysis of competition between DTP and other nucleophiles
Control reactions containing 1.0 mM DTP and 50 μM CEMS (light grey bars) were allowed to go to completion, and the amount of the major DTP-EMS adduct formed was analyzed by HPLC (4.4 min peak, see Figure 2C). Competition reactions contained a second nucleophile, also at 1.0 mM, in addition to DTP and CEMS (solid bars) and were analyzed identically. TS, sodium thiosulfate. *, competition reaction is significantly different from control (p < 0.05).
Secondary reactions
Reaction of DTP or DUA (1.0 mM) at neutral pH and 23°C with an excess of CEMS or CEES (19-20 mM) resulted in precipitate formation. This may be due in part to acidification of the reaction due to release of HCl in the hydrolysis reaction (20). The precipitates could be dissolved in base, and analyzed by mass spectroscopy. For DTP and CEMS, products consistent with the addition of two and three methyl ethylsulfide moieties (Supplementary Figure S7A) were observed; for DUA and CEMS, products representing addition of two, three and four methyl ethylsulfide moieties (Supplementary Figure S7B) were observed. Similar addition products, with addition of several ethyl ethylsulfide moieties to the thiopurine were observed in the reaction of CEES with DTP and DUA (data not shown). Further analysis of the products of reaction of DTP and CEMS at high CEMS concentration was carried out by HPLC. When the precipitated reaction products were dissolved in .05N NaOH, then diluted into 5 mM Tris and analyzed under our standard conditions, a new adduct with tR = 27.9 min was found in good yield. Mass spectral analysis of the compound indicated the addition of two methyl ethylsulfide groups to DTP, presumably at the two thiol moieties (Supplementary Figure S7C). Similar methods yielded a di-adduct of DUA in good yield with tR=23.5 min (Supplementary Figure S7C).
Discussion
Previous work has established that thiopurines are excellent nucleophilic scavengers of several electrophilic carcinogens that react via SN2 pathways (14-16) and via SN1 pathways (17). In the current work, we have established facile reaction of DTP, DUA and MMP with two SM analogs. Facile reaction was observed over a range of temperatures from 4-50° C (data not shown), and under pH conditions from about 7 to 9. Based on our earlier work, we suggest that the primary site of reaction with DTP is the 2-mercapto moiety. However, for DTP and DUA secondary reactions also occur with the 6-mercapto group and one or more of the ring nitrogens, presumably N7 and/or N9. Thus in principle, one mole of DTP or DUA is capable of scavenging more than one mole of mustard. Nevertheless, the fastest rate constants were observed for MMP. Since optimal scavenging requires higher concentrations of nucleophile than electrophile, conditions that do not favor formation of di-adducts or higher order adducts, it is likely that MMP is intrinsically the better scavenger. However, it remains to be seen in the context of the mammalian cell, where transport into the cell may be a determining factor, whether MMP will retain this intrinsic advantage over DTP and DUA.
We wish to put these findings into perspective in two ways. First, the analogous reaction rate constant for DTP reacting with the carcinogen BPDE was reported previously as 22.2 L/M-1 S-1 (16), more than three orders of magnitude lower than the rate constants for CEMS and CEES. To compare the effects of DTP on the potential damage produced by the different carcinogens we have used numerical integration to model the time course of exposure in the case of BPDE under the same conditions used above for CEMS (i.e. 500 μM each reactant, see Table 1 and Figure 5). The fractional protection from BPDE exposure is calculated to be 0.78, only slightly higher than for CEMS (0.707, Table 1). Since we have demonstrated significant biological activity of DTP against the DNA damaging, cytotoxic and carcinogenic effects of BPDE (14, 18), this comparable protection from CEMS suggests that DTP may have potent protective effects against biological damage caused by mustard agents.
Secondly, DTP, DUA and MMP can be compared to other common nucleophiles that have been suggested as scavengers of electrophilic carcinogens. The data presented indicate that GSH and N-acetyl-cysteine are significantly less efficient at scavenging the mustard analogs than DTP at physiological pH. This is in general agreement with previous results of Ogston (20) who found relatively low competition factors for cysteine-compounds reacting with SM or CEES. The pKa for the sulfhydryl moiety of GSH is ∼8.75, and thus at near-neutral pH the concentration of reactive thiolate anion is low compared to the neutral thiol. In contrast, the more acidic of the two sulfhydryls in DTP has a pKa below 6, and thus is predominantly in the thiolate form at near-neutral pH. Consistent with this interpretation, k2/kw for DTP decreases as the pH is lowered from 7 to 6.2 (Supplementary Figure S4). Conversely, the ability of GSH and NAC to compete with DTP increases when the pH is raised to 10.2 (Supplementary Figure S5). Thus, for these compounds to be protective in the context of biological systems, either the mechanism will not be direct scavenging of the electrophile, or the scavenging reaction will require the enzymatic participation of glutathione S-transferases or other biological catalysts. On the other hand, the thiopurines exhibit reactivity with CEES and CEMS comparable to that of thiosulfate, long known to be an effective scavenger in vitro.
The studies of Ludlum and co-workers (13) clearly indicate that the formation of DNA adducts represents the major determinant of cytotoxicity of both SM and CEES in culture, and that nucleotide excision repair is important in the repair of DNA adducts derived from both toxicants. Interestingly, double stranded DNA was found to be a poor competitor for reaction with CEES, producing only a slight and statistically not-significant decrease in the formation of the DTP-CEES adduct at equimolar concentration (Figure 6). This suggests that DTP may have in vitro protective activity for mammalian cells. In the accompanying paper, we show that indeed DTP protects cells from the cytotoxic and mutagenic actions of CEMS and CEES. It remains to be determined whether in vivo protection can also be demonstrated.
Supplementary Material
Acknowledgments
We thank Dr. Weiguo Qing for the synthesis of DUA, K. Leslie Powell and Tammy Fields for technical assistance, Dr. Herng-Hsiang Lo for mass spectroscopic analyses, and Rebecca Deen, Joi Holcomb, and Chris Brown for manuscript preparation. This work was funded by the National Institutes of Health CounterACT Program through the National Institute of Neurological Disorders and Stroke (award #U01NS058191) and by an NIEHS Center grant (P30ES007784). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Footnotes
Non-standard abbreviations used are: SM, bis-(2-chloroethyl)sulfide; CEMS, 2-chloroethyl methyl sulfide; CEES, 2-chloroethyl ethyl sulfide; DTP, 2,6-dithiopurine; MMP, 9-methyl-6-mercaptopurine; DUA, 2,6-dithiouric acid; 6MP, 6-mercaptopurine; 6TX, 6-thioxanthine; GSH, glutathione; NAC, N-acetyl-cysteine; MESNA, 2 mercapto ethane sulfonic acid; PB, phosphate buffer; TFA, trifluoroacetic acid; AUC, area-under-the-curve; BPDE, 7r,8t-dihydroxy-9t,10t-oxy-7,8,9,10-tetrahydrobenzo[a]pyrene.
Supporting Information Available: Theoretical analysis of the reaction kinetics is given in Supplementary Materials. Six Supplementary Figures are also given. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dacre JC, Goldman M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol Rev. 1996;48:289–326. [PubMed] [Google Scholar]
- 2.Emad A, Rezaian GR. Immunoglobulins and cellular constituents of the BAL fluid of patients with sulfur mustard gas-induced pulmonary fibrosis. Chest. 1999;115:1346–1351. doi: 10.1378/chest.115.5.1346. [DOI] [PubMed] [Google Scholar]
- 3.Drasch G, Kretschmer E, Kauert G, von Meyer L. Concentrations of mustard gas [bis(2-chloroethyl)sulfide] in the tissues of a victim of a vesicant exposure. J Forensic Sci. 1987;32:1788–1793. [PubMed] [Google Scholar]
- 4.Hattersley IJ, Jenner J, Dalton C, Chilcott RP, Graham JS. The skin reservoir of sulphur mustard. Toxicol In Vitro. 2008;22:1539–1546. doi: 10.1016/j.tiv.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Fidder A, Moes GW, Scheffer AG, van der Schans GP, Baan RA, de Jong LP, Benschop HP. Synthesis, characterization, and quantitation of the major adducts formed between sulfur mustard and DNA of calf thymus and human blood. Chem Res Toxicol. 1994;7:199–204. doi: 10.1021/tx00038a013. [DOI] [PubMed] [Google Scholar]
- 6.Lawley PD, Brookes P. Interstrand cross-linking of DNA by difunctional alkylating agents. J Mol Biol. 1967;25:143–160. doi: 10.1016/0022-2836(67)90285-9. [DOI] [PubMed] [Google Scholar]
- 7.Lawley PD, Lethbridge JH, Edwards PA, Shooter KV. Inactivation of bacteriophage T7 by mono- and difunctional sulphur mustards in relation to cross-linking and depurination of bacteriophage DNA. J Mol Biol. 1969;39:181–198. doi: 10.1016/0022-2836(69)90341-6. [DOI] [PubMed] [Google Scholar]
- 8.Ludlum DB, Austin-Ritchie P, Hagopian M, Niu TQ, Yu D. Detection of sulfur mustard-induced DNA modifications. Chem Biol Interact. 1994;91:39–49. doi: 10.1016/0009-2797(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 9.Noort D, Benschop HP, Black RM. Biomonitoring of exposure to chemical warfare agents: a review. Toxicol Appl Pharmacol. 2002;184:116–126. [PubMed] [Google Scholar]
- 10.Bartlett PD, Swain CG. Kinetics of hydrolysis and displacement reactions of beta, beta1-dichlorodiethyl sulfide and of beta-chloro-beta1-hydroxydiethyl sulfide. J Am Chem Soc. 1949;71:1406–1415. doi: 10.1021/ja01172a076. [DOI] [PubMed] [Google Scholar]
- 11.Yang YC, Ward JR, Luteran T. Hydrolysis of mustard derivatives in aqueous acetone-water and ethanol-water mixtures. J Org Chem. 1986;51:2756–2759. [Google Scholar]
- 12.Kircher M, Fleer R, Ruhland A, Brendel M. Biological and chemical effects of mustard gas in yeast. Mutat Res. 1979;63:273–289. doi: 10.1016/0027-5107(79)90059-9. [DOI] [PubMed] [Google Scholar]
- 13.Matijasevic Z, Precopio ML, Snyder JE, Ludlum DB. Repair of sulfur mustard-induced DNA damage in mammalian cells measured by a host cell reactivation assay. Carcinog. 2001;22:661–664. doi: 10.1093/carcin/22.4.661. [DOI] [PubMed] [Google Scholar]
- 14.MacLeod MC, Humphrey RM, Bickerstaff T, Daylong A. Inhibition by 6-mercaptopurine of the binding of a benzo(a)pyrene diol- epoxide to DNA in Chinese hamster ovary cells. Cancer Res. 1990;50:4355–4359. [PubMed] [Google Scholar]
- 15.MacLeod MC, Stewart E, Daylong A, Lew LK, Evans FE. Reaction of a chemotherapeutic agent, 6-mercaptopurine, with a direct- acting, electrophilic carcinogen, benzo[a]pyrene-7,8-diol 9,10-epoxide. Chem Res Toxicol. 1991;4:453–462. doi: 10.1021/tx00022a009. [DOI] [PubMed] [Google Scholar]
- 16.MacLeod MC, Qing WG, Powell KL, Daylong A, Evans FE. Reaction of nontoxic, potentially chemopreventive purinethiols with a direct-acting, electrophilic carcinogen, benzo[a]pyrene-7,8-diol 9,10- epoxide. Chem Res Toxicol. 1993;6:159–167. doi: 10.1021/tx00032a004. [DOI] [PubMed] [Google Scholar]
- 17.Qing WG, Powell KL, MacLeod MC. Kinetics of the reaction of a potential chemopreventive agent, 2,6-dithiopurine, and its major metabolite, 2,6-dithiouric acid, with multiple classes of electrophilic toxicants. Chem Res Toxicol. 1996;9:1298–1304. doi: 10.1021/tx960088n. [DOI] [PubMed] [Google Scholar]
- 18.MacLeod MC, Mann KL, Thai G, Conti CJ, Reiners JJ., Jr Inhibition by 2,6-dithiopurine and thiopurinol of binding of a benzo(a)pyrene diol epoxide to DNA in mouse epidermis and of the initiation phase of two-stage tumorigenesis. Cancer Res. 1991;51:4859–4864. [PubMed] [Google Scholar]
- 19.Plagemann PG, Marz R, Wohlhueter RM, Graff JC, Zylka JM. Facilitated transport of 6-mercaptopurine and 6-thioguanine and non- mediated permeation of 8-azaguanine in Novikoff rat hepatoma cells and relationship to intracellular phosphoribosylation. Biochim Biophys Acta. 1981;647:49–62. doi: 10.1016/0005-2736(81)90294-7. [DOI] [PubMed] [Google Scholar]
- 20.Ogston AG, Holiday ER, Philpot JSL, Stocken LA. The replacement reactions of β-β′-dichlorodiethyl sulphide and of some analogues in aqueous solution: The isolation of β-chloro-β′-hydroxydiethyl sulphide. Trans Faraday Soc. 1946;44:45–52. [Google Scholar]
- 21.Tilley RI. The hydrolysis of Bis(2-chloroethyl) sulfide (sulfur mustard) in aqueous mixtures of ethanol, acetone and dimethyl sulfoxide. Aust J Chem. 1993;46:293–300. [Google Scholar]
- 22.Qing WG, Powell KL, Stoica G, Szumlanski CL, Weinshilboum RM, Macleod MC. Toxicity and metabolism in mice of 2,6-dithiopurine, a potential chemopreventive agent. Drug Metab Dispos. 1995;23:854–860. [PubMed] [Google Scholar]
- 23.Yang YC, Szafraniec LL, Beaudry WT, Ward JR. Kinetics and mechanism of the hydrolysis of 2-chloroethyl sulfides. J Org Chem. 1988;53:3293–3297. [Google Scholar]
- 24.Stein WH, Fruton JS. Chemical reactions of mustard gas and related compounds. IV. Chemical reactions of β-chloroethyl-β′-hydroxyethylsulfide. J Org Chem. 1946;11:686–691. doi: 10.1021/jo01176a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.