

Abstract
Ascorbate (vitamin C) recycling occurs when extracellular ascorbate is oxidized, transported as dehydroascorbic acid, and reduced intracellularly to ascorbate. We investigated microorganism induction of ascorbate recycling in human neutrophils and in microorganisms themselves. Ascorbate recycling was determined by measuring intracellular ascorbate accumulation. Ascorbate recycling in neutrophils was induced by both Gram-positive and Gram-negative pathogenic bacteria, and the fungal pathogen Candida albicans. Induction of recycling resulted in as high as a 30-fold increase in intracellular ascorbate compared with neutrophils not exposed to microorganisms. Recycling occurred at physiologic concentrations of extracellular ascorbate within 20 min, occurred over a 100-fold range of effector/target ratios, and depended on oxidation of extracellular ascorbate to dehydroascorbic acid. Ascorbate recycling did not occur in bacteria nor in C. albicans. Ascorbate did not enter microorganisms, and dehydroascorbic acid entry was less than could be accounted for by diffusion. Because microorganism lysates reduced dehydroascorbic acid to ascorbate, ascorbate recycling was absent because of negligible entry of the substrate dehydroascorbic acid. Because ascorbate recycling occurs in human neutrophils but not in microorganisms, it may represent a eukaryotic defense mechanism against oxidants with possible clinical implications.
Ascorbate is found in normal circulating human neutrophils in millimolar concentration and is accumulated by two mechanisms (1). In resting neutrophils, ascorbate is accumulated by a sodium- and energy-dependent transporter whose substrate is ascorbate (1–3). Ascorbate-specific transporter activity was expressed in Xenopus laevis oocytes injected with fractionated rabbit renal mRNA, but the transporter has not been isolated (4). Chemically activated neutrophils accumulate ascorbate by the mechanism of ascorbate recycling (1, 5). Chemically activated neutrophils produce diffusible oxidants that oxidize extracellular ascorbate to dehydroascorbic acid. Dehydroascorbic acid transport is at least 10- to 20-fold faster than ascorbate transport and is mediated by glucose transporters GLUT1 and GLUT3 (1, 5–7). Upon intracellular entry, dehydroascorbic acid is immediately reduced to ascorbic acid by the glutathione-dependent protein glutaredoxin (8). After extracellular ascorbate quenches diffusible oxidants, the oxidative metabolite dehydroascorbic acid is recycled intracellularly to increase intracellular ascorbate >10-fold within minutes (1, 5). Increased intracellular ascorbate is then available to quench diffusible oxidants.
In an era of emerging antibiotic resistance (9), ascorbate recycling has the potential to be a physiologic means of enhancing host defense. Because chemically activated neutrophils recycle ascorbate, we investigated whether clinically isolated microorganisms induced ascorbate recycling in normal human neutrophils. Ascorbate recycling by these pathogenic microorganisms themselves was also determined. The data indicate that Gram-positive bacteria, Gram-negative bacteria, and Candida albicans induced ascorbate recycling in human neutrophils but that these microorganisms themselves did not recycle ascorbate because they did not transport the substrate dehydroascorbic acid.
MATERIALS AND METHODS
Radiolabeled substrates, l-[1-14C]ascorbic acid (6.6 mCi/mmol), [14C(U)]glucose (265 mCi/mmol; 1 Ci = 37 GBq), or D[4,5-3H(N)]galactose (44 Ci/mmol) were obtained from DuPont/NEN. [14C]Dehydroascorbic acid was prepared from l-[1-14C]ascorbic acid immediately before use by bromine oxidation (5). Ascorbate was purchased from Sigma.
Human neutrophils were prepared from whole blood as described (2). Ascorbate and dehydroascorbic acid transport studies with and without bacteria were performed on plated neutrophils in Hepes-PO4 buffer (1, 5). Hepes-PO4 buffer contained 147 mM NaCl, 5 mM KCl, 1.9 mM KH2PO4, 1.1 mM Na2HPO4, 5 mM glucose, 0.3 mM MgSO4-7H2O, 1.0 mM MgCl2-6H2O, 1.5 mM CaCl2-2H2O, 10 mM Hepes (pH 7.4) adjusted with NaOH. Clinical isolates of Escherichia coli CP9 or CP922, Enterococcus faecalis, Moraxella catarrhlis, Klebsiella oxytoca, Acinetobacter baumanii, and C. albicans were grown overnight in Luria–Bertani broth shaking at 37°C, diluted 1/20 in Luria–Bertani broth, and incubated for 1 hr. Microorganisms were washed, diluted to 1 × 106/ml in Hepes-PO4 buffer, and added to plated neutrophils at the ratios shown at the same time as ascorbate or dehydroascorbic acid. After incubation at 37°C, neutrophils were washed three times with Hepes-PO4 buffer. To quantify radiolabel uptake, attached neutrophils were dissolved in 0.1 N NaOH, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. For HPLC analyses, ascorbate and dehydroascorbic acid were extracted as described (5). Bacterial titers were confirmed by plating diluted bacteria and counting colonies.
Bacterial transport of ascorbate, dehydroascorbic acid, and radiolabeled sugars was performed by using reported methods (10, 11). Before experiments, bacteria were diluted to A680 = 2, glycerol was added to a final concentration of 10 mM, and air was bubbled through the solution for 3 min. Radiolabeled ascorbic acid, dehydroascorbic acid, glucose, or galactose was added to the bacterial solution. At the times shown, 100 μl aliquots were removed and washed through a filter (Gelman; Supor-450, 0.45 μm) with 100–200 volumes of ice-cold KCl/Mes solution containing 150 mM KCl and 5 mM Mes, pH 6.5. Filters were collected and radioactivity was determined by scintillation spectrometry. To measure intracellular ascorbate or dehydroascorbic acid mass, bacteria were centrifuged for 30 sec (14,000 rpm) at 4°C and washed three times in ice-cold KCl/Mes buffer by resuspension and centrifugation. Ascorbate and dehydroascorbic acid were extracted by addition of 60% methanol, 1 mM EDTA to the bacterial pellet.
Ascorbate and dehydroascorbic acid were measured as described by HPLC with coulometric electrochemical detection or by scintillation spectrometry (1, 2, 12). Results of ascorbate accumulation were indistinguishable when measured by HPLC or by scintillation spectroscopy, except that HPLC accounted for endogenous neutrophil intracellular ascorbate of ≈1.5 mM. Intracellular neutrophil volume was determined as described, and measurement of intracellular ascorbate and [14C]ascorbate were corrected by protein concentration (2, 3). Protein was measured by using bicinchoninic acid (Pierce) (2, 3). Data points represent the mean of at least three samples ±SD; SD is not displayed when smaller than point size. Samples were from at least three different incubations of one subject’s neutrophils. All experiments were performed with neutrophil preparations from at least three different subjects, with similar results. Experiments from different subjects could not be combined because of the difficulty in predictably obtaining the same bacteria/neutrophil ratios in different experiments.
RESULTS AND DISCUSSION
Ascorbate recycling was determined by measuring intracellular ascorbate when neutrophils and microorganisms were incubated with extracellular ascorbate at 100 μM, a physiologic concentration for healthy humans (13). Neutrophils were incubated with Gram-negative or Gram-positive bacterial pathogens or C. albicans at target/effector ratios from 1:1 to 120:1. These ratios are within clinically observed ranges (14, 15). For E. coli, we used a wild-type pathogen (CP9) and an isogenic derivative (CP922) that lacked the group 3 capsule (K54) and the majority of O4 antigen of the lipopolysaccharide (16). Intracellular ascorbate in neutrophils was increased as much as 30-fold compared with control neutrophils incubated in the presence of ascorbate without microorganisms (Fig. 1). These data indicate that ascorbate recycling was induced by a wide range of microbial pathogens. Because recycling was induced by wild-type E. coli CP9 and the isogenic derivative CP922, induction of recycling did not require the group 3 capsule and the majority of 04 antigen.
Figure 1.
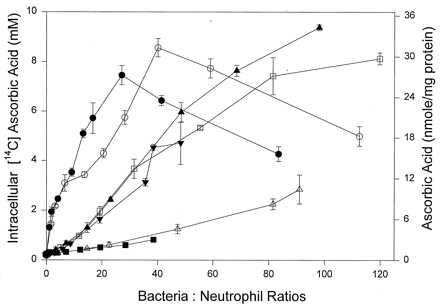
Induction of ascorbate recycling by different microorganisms. Neutrophils were incubated with 100 μM ascorbate for 45 min with the indicated microorganism/neutrophils (effector/target) ratios for the following microorganisms: E. coli CP9 (○) or CP922 (•), E. faecalis (▪), M. catarrhlis (□), K. oxytoca (▾), A. baumanii (▵), and C. albicans (▴). Neutrophils incubated with ascorbate and no microorganisms are indicated by ( ). Intracellular ascorbate was measured by scintillation spectrometry and is shown as mM (left axis) and nmol/mg protein (right axis).
By using E. coli (CP9), recycling was tested at five physiologic extracellular ascorbate concentrations (10–100 μM) and at one higher concentration (200 μM) that can be readily achieved by i.v. injection of vitamin C in humans (13). Control neutrophils were incubated with ascorbate and no bacteria. With bacteria, ascorbate accumulation increased at all time points as extracellular concentration increased (Fig. 2). Accumulation of 10–200 μM ascorbate is shown at one time point in the presence and absence of E. coli CP9 (Fig. 2 Inset). With E. coli, accumulation increased 3- to 6-fold at every ascorbate concentration. These data indicate that ascorbate recycling depends on extracellular ascorbate concentration across the physiologic range, and that recycling increases as concentration increases.
Figure 2.
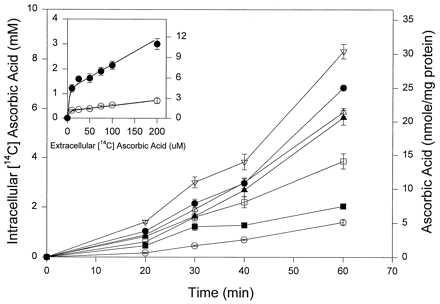
Dependence of ascorbate recycling on time and extracellular ascorbate concentrations. Intracellular ascorbate was measured by scintillation spectrometry and is shown as mM (left axis) and nmol/mg protein (right axis). Neutrophils were incubated with E. coli CP9 (effector/target ratio 28:1) for the indicated times with extracellular ascorbate concentrations of 10 μM (▪), 25 μM (□), 50 μM (▴), 75 μM (▵), 100 μM (•), and 200 μM (▿). Control neutrophils (○) were incubated without bacteria and 100 μM ascorbate for the times indicated. (Inset) Dependence of recycling on extracellular ascorbate concentrations at one fixed time. Neutrophils were incubated with (•) or without (○) E. coli CP9 (effector/target ratio, 28:1) and the indicated extracellular ascorbate concentrations for 30 min. The y axis is the same as in Fig. 2.
We predicted that recycling would depend on oxidation of extracellular ascorbate. As bacteria/neutrophil ratios increased, extracellular ascorbate concentrations progressively decreased, indicating ascorbate oxidation to dehydroascorbic acid (data not shown). When neutrophils, bacteria, and ascorbate were incubated with the oxidant quenchers superoxide dismutase (25 μg/ml) and catalase (600 units/ml), ascorbate recycling was completely inhibited (data not shown). These data provide evidence that ascorbate recycling depends on extracellular oxidants from neutrophils.
If ascorbate oxidation were essential for recycling, bacteria should not induce ascorbate recycling in neutrophils unable to make oxidants. NADPH-oxidase is the primary neutrophil enzyme system responsible for generating reactive oxidants. Neutrophils were isolated from patients with chronic granulomatous disease, a genetic disorder characterized by defective NADPH-oxidase activity (17–19). Chronic granulomatous disease neutrophils were incubated with either E. coli CP9 or CP922 at target/effector ratios from 1:1 to 78:1 (Fig. 3). Neither CP9 nor CP922 induced any increase in ascorbate accumulation compared with neutrophils without bacteria. These data provide further evidence that ascorbate recycling depends on extracellular ascorbate oxidation.
Figure 3.
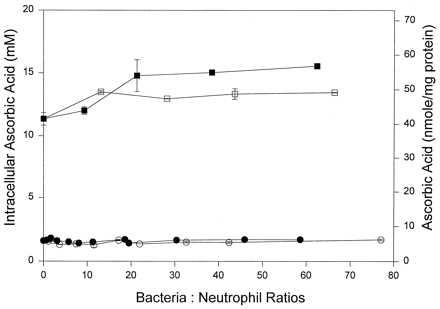
Ascorbate recycling in chronic granulomatous disease neutrophils. Chronic granulomatous disease neutrophils were incubated with 100 μM ascorbate (• and ○) for 45 min or 300 μM dehydroascorbic acid (▪ and □) for 5 min and the indicated target/effector ratios for E. coli CP9 (• and ▪) or E. coli CP922 (○ and □). Intracellular ascorbate was measured by HPLC and is shown as mM (left axis) and nmol/mg protein (right axis).
To test whether ascorbate recycling occurred if the substrate dehydroascorbic were available, chronic granulomatous disease neutrophils were incubated with dehydroascorbic acid and either E. coli CP9 or CP922 (Fig. 3). When the substrate was present, enhanced ascorbate accumulation occurred and was independent of bacteria. These findings show that chronic granulomatous disease neutrophils transport and reduce dehydroascorbic acid once it is provided. These neutrophils do not recycle ascorbate because extracellular ascorbate oxidation does not occur, and the substrate dehydroascorbic acid is not formed.
To determine whether ascorbate recycling occurred in bacteria, we tested bacterial transport of ascorbic acid and dehydroascorbic acid. Glucose and galactose transport served as controls because dehydroascorbic acid transport is mediated by glucose transporters in eukaryotes (7), and the bacterial glucose transporter GalP shares the most homology with mammalian glucose transporters (20, 21). Ascorbic acid was not transported by E. coli CP9 (Fig. 4A) or by other bacteria (data not shown). Dehydroascorbic acid entry in E. coli CP9 (Fig. 4A) or other microorganisms (Fig. 4B, see below) was less than could be accounted for by diffusion and was minimal compared with glucose and galactose transport (Fig. 4A; data not shown). These findings could not be explained by dehydroascorbic acid toxicity because galactose transport was similar in the presence and absence of dehydroascorbic acid (data not shown). Endogenous ascorbate and dehydroascorbic acid were absent from all microorganisms studied.
Figure 4.
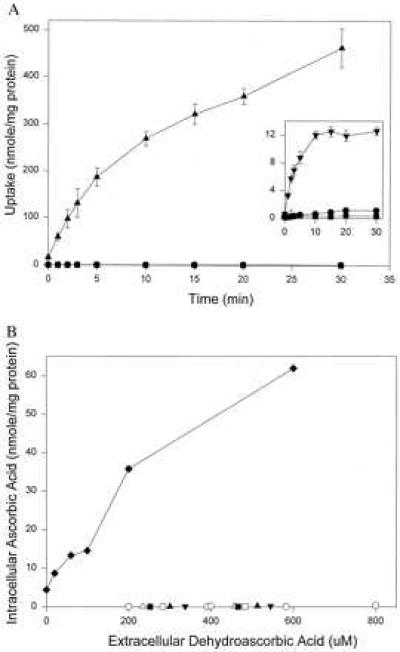
(A) Uptake of glucose, ascorbate, and dehydroascorbic acid by E. coli CP9. Bacteria were incubated with 400 μM [14C]ascorbate (▪), [14C]dehydroascorbic acid (•), [14C]glucose (▴), or [14C]galactose (▾) (Inset) for the indicated times. Uptake was measured by scintillation spectrometry. (B) Ascorbate recycling in either neutrophils or microorganisms. Different microorganisms or neutrophils were incubated with the indicated concentrations of extracellular dehydroascorbic acid for 5 min. Intracellular ascorbate was measured by HPLC. Symbols represent neutrophils (⧫), E. coli CP9 (○), E. faecalis (▪), M. catarrhlis (□), K. oxytoca (▾), A. baumanii (▿), and C. albicans (▾).
We directly compared ascorbate recycling in eukaryotes and prokaryotes. Either neutrophils or different microorganisms were incubated with varying concentrations of dehydroascorbic acid, and internal ascorbate was measured. During 5 min incubations, neutrophils rapidly transported dehydroascorbic acid and reduced it to ascorbate (Fig. 4B). No internal dehydroascorbic acid was present (1, 5). In contrast, dehydroascorbic acid transport and reduction to ascorbic acid was negligible in microorganisms (<0.4 nmol/mg protein), ≈100-fold less than in neutrophils and less than could be accounted for by diffusion (Fig. 4B). Although absent in microorganisms, endogenous ascorbate in neutrophils was 4.35 nmol/mg protein (1.2 mM). Lysates of microorganisms reduced dehydroascorbic acid to ascorbate, with or without exogenous glutathione and/or NADPH (data not shown). Taken together, the data indicate that ascorbate recycling is negligible in microorganisms because entry of the substrate dehydroascorbic acid is less than can be accounted for by diffusion.
Neutrophils and bacteria have several distinct means of oxidant defense (22, 23). Neutrophils quench oxidants through the proteins catalase and superoxide dismutase. Glutathione reductase maintains glutathione as a chemical defense against oxidants. Bacteria have similar means of oxidant defense, some of which depend on transcriptional regulation and new protein synthesis.
The specific role of ascorbate recycling in neutrophil function is not certain, but there are several intriguing possibilities. Compared with bacterial defense mechanisms, ascorbate recycling by human neutrophils but not by microorganisms may be a unique system for oxidant protection. Because bacteria did not transport dehydroascorbic acid, ascorbate recycling may be a eukaryotic-specific system for oxidant defense. In neutrophils, dehydroascorbic acid reduction is immediate, an induction step is not required, and the dehydroascorbic acid-reducing protein glutaredoxin is present in large amounts in unactivated cells (1, 5, 8). Unlike bacterial mechanisms, ascorbate recycling is modulated by ascorbate itself, a vitamin that is obtained exogenously from our diets. Ascorbate recycling increases across the range of ascorbate concentrations that can occur in humans physiologically and pharmacologically (13). Higher concentrations of ascorbate within neutrophils might provide better protection against intracellular reactive oxygen intermediates. In neutrophils, these intermediates are an integral component of microbicidal pathways and mediate apoptosis (24–26). Increased intracellular ascorbate concentrations could be effective in quenching cytosolic and extracellular oxidants and possibly delay neutrophil apoptosis (27, 28). Prolongation of neutrophil survival could also translate into enhanced microbicidal activity.
A concern is that supplementation with massive amounts of ascorbate could protect pathogens from oxidants as well as host tissues. However, extracellular ascorbate concentrations are ≈80% saturated with ingestion of 200 mg ascorbate, the equivalent amount found in four to five servings of fruits and vegetables (13). Even with massive oral supplementation, plasma concentrations will not substantially increase above 100 μM. Higher ascorbate concentrations are only achieved with i.v. ascorbate, and only transiently. As shown here, extracellular ascorbate concentrations of 100 μM are as low as 1–2% of those within neutrophils when recycling occurs. Therefore, ascorbate recycling is expected to have predominantly intracellular effects. Although oxidants are generated within neutrophils in the phagosome for bacterial killing, neutrophils also generate extracellular oxidants. In theory, extracellular ascorbate could passively quench oxidants that otherwise mediate bacterial killing. Extracellular oxidant products could enhance bacterial killing but also mediate host tissue destruction (29). Physiologic concentrations of extracellular ascorbate protect collagen from oxidant degradation (30). Surprisingly, extracellular ascorbate may also specifically enhance extracellular oxidant production from neutrophils (31, 32). It is unknown whether extracellular ascorbate in vivo enhances oxidant production or whether there is a balance between oxidant protection and bacterial killing. Answers await better testing methods, including clinical experiments.
It is not known whether ascorbate recycling could have clinical application. One key issue is whether there is a relationship between ascorbate concentrations in humans and the clinical outcome in bacterial and fungal infection. However, only sparse data are available (33, 34), but based on new normative data, the problem can now be addressed experimentally (13). Because ascorbate has little toxicity (35) and can be increased by either enteral or parenteral administration (13), and because antibiotic resistance is increasing (9), it may be worthwhile to explore the clinical potential of ascorbate in bacterial and fungal infections.
Acknowledgments
We thank Dr. Harry L. Malech and his patients for providing blood for isolation of chronic granulomatous disease neutrophils, and the volunteers who provided blood for isolation of normal neutrophils. This work is supported in part by Takeda Chemical Industries (Y.W.) and Research for Health in Erie County (T.A.R.).
References
- 1.Welch R W, Wang Y, Crossman A, Jr, Park J B, Kirk K L, Levine M. J Biol Chem. 1995;270:12584–12592. doi: 10.1074/jbc.270.21.12584. [DOI] [PubMed] [Google Scholar]
- 2.Washko P, Rotrosen D, Levine M. J Biol Chem. 1989;264:18996–19002. [PubMed] [Google Scholar]
- 3.Washko P, Levine M. J Biol Chem. 1992;267:23568–23574. [PubMed] [Google Scholar]
- 4.Dyer D L, Kanai Y, Hediger M A, Rubin S A, Said H M. Am J Physiol. 1994;267:C301–C306. doi: 10.1152/ajpcell.1994.267.1.C301. [DOI] [PubMed] [Google Scholar]
- 5.Washko P W, Wang Y, Levine M. J Biol Chem. 1993;268:15531–15535. [PubMed] [Google Scholar]
- 6.Vera J C, Rivas C I, Fischbarg J, Golde D W. Nature (London) 1993;364:79–82. doi: 10.1038/364079a0. [DOI] [PubMed] [Google Scholar]
- 7.Rumsey S C, Kwon O, Xu G, Burant C F, Simpson I, Levine M. J Biol Chem. 1997;272:18982–18989. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- 8.Park J B, Levine M. Biochem J. 1996;315:931–938. doi: 10.1042/bj3150931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gold H S, Moellering R C J. N Engl J Med. 1996;335:1445–1453. doi: 10.1056/NEJM199611073351907. [DOI] [PubMed] [Google Scholar]
- 10.Henderson P J F, Giddens R A, Jones-Mortimer M C. Biochem J. 1977;162:309–320. doi: 10.1042/bj1620309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson P J F, McDonald T P, Steel A, Litherland G J, Cairns M T, Martin G E M. Biochem Soc Trans. 1994;22:643–646. doi: 10.1042/bst0220643. [DOI] [PubMed] [Google Scholar]
- 12.Dhariwal K R, Hartzell W O, Levine M. Am J Clin Nutr. 1991;54:712–716. doi: 10.1093/ajcn/54.4.712. [DOI] [PubMed] [Google Scholar]
- 13.Levine M, Conry-Cantilena C, Wang Y, Welch R W, Washko P W, Dhariwal K R, Park J B, Lazarev A, Graumlich J, King J, Cantilena L R. Proc Natl Acad Sci USA. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamm W E, Counts G W, Running K R, Fihn S, Turck M, Holmes K K. N Engl J Med. 1982;307:463–468. doi: 10.1056/NEJM198208193070802. [DOI] [PubMed] [Google Scholar]
- 15.Kunin C M, White L V, Hua T. Ann Int Med. 1993;119:454–460. doi: 10.7326/0003-4819-119-6-199309150-00002. [DOI] [PubMed] [Google Scholar]
- 16.Russo T A, Sharma G, Brown C R, Campagnari A A. Infect Immun. 1995;63:1263–1269. doi: 10.1128/iai.63.4.1263-1269.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curnutte J T. Clin Immunol Immunopathol. 1993;67:S2–S15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 18.Thrasher A, Keep N, Wientjes F, Segal A. Biochim Biophys Acta. 1994;1227:1–24. doi: 10.1016/0925-4439(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 19.Roos D, de Boer M, Kuribayashi F, Meischl C, Weening R S, Segal A W, Ahlin A, Nemet K, Hossle J P, Bernatowska-Matusziewicz E, Middleton-Price H. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 20.Walmsley A R, Lowe A G, Henderson P J F. Eur J Biochem. 1994;221:513–522. doi: 10.1111/j.1432-1033.1994.tb18763.x. [DOI] [PubMed] [Google Scholar]
- 21.Henderson P J F, Martin G E M, McDonald T P, Steel A, Walmsley A R. Antonie Van Leeuwenhoek. 1994;65:349–358. doi: 10.1007/BF00872218. [DOI] [PubMed] [Google Scholar]
- 22.Storz G, Tartaglia L A. J Nutr. 1992;122:627–630. doi: 10.1093/jn/122.suppl_3.627. [DOI] [PubMed] [Google Scholar]
- 23.Miller R A, Britigan B E. Clin Microbiol Rev. 1997;10:1–18. doi: 10.1128/cmr.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halliwell B, Wasil M, Grootveld M. FEBS Lett. 1987;213:15–17. doi: 10.1016/0014-5793(87)81456-4. [DOI] [PubMed] [Google Scholar]
- 25.Liles W C, Kiener P A, Ledbetter J A, Aruffo A, Klebanoff S J. J Exp Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segal A, Gelsow M, Garcia R, Harper A, Miller R. Nature (London) 1981;290:406–408. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 27.Whyte M K B, Meagher L C, MacDermot J, Haslet C. J Immunol. 1993;150:5124–5134. [PubMed] [Google Scholar]
- 28.Kasahara Y, Iwai K, Yachie A, Ohta K, Konno S, Seki H, Miyawaki T, Taniguchi N. Blood. 1997;89:1748–1753. [PubMed] [Google Scholar]
- 29.Weiss S J. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 30.Mukhopadhyay C K, Chatterjee I B. J Biol Chem. 1994;269:30200–30205. [PubMed] [Google Scholar]
- 31.Miller T E. J Bacteriol. 1969;98:949–955. doi: 10.1128/jb.98.3.949-955.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hemila H, Roberts P, Wikstrom M. FEBS Lett. 1984;178:25–30. doi: 10.1016/0014-5793(84)81232-6. [DOI] [PubMed] [Google Scholar]
- 33.Clemetson C A. In: Vitamin C. Clemetson C A, editor. I. Boca Raton, FL: CRC; 1989. pp. 65–76. [Google Scholar]
- 34.Pitt H A, Costrini A M. J Am Med Assoc. 1979;241:908–911. [PubMed] [Google Scholar]
- 35.Levine M, Rumsey S, Wang Y, Park J B, Kwon O, Xu W, Amano N. In: Present Knowledge in Nutrition. 7th Ed. Filer L J, Ziegler E E, editors. Washington, DC: ILSI; 1996. pp. 146–159. [Google Scholar]