

Abstract
A key unresolved question in the pathogenesis of phenotype development in nephropathic cystinosis is whether intralysosomal cystine, the hallmark of this lethal inborn error of metabolism, alters cytoplasmic redox potential. Variable findings on this issue have been reported. This study of fetal and non-fetal skin and lung-derived cystinotic fibroblasts compared to origin and age-matched normal control fibroblasts reveals that cystinotic cells do not exhibit redox perturbations. We find that the steady-state redox status as assessed by the [GSH]/[GSSG] ratio, an indicator of the intracellular redox poise, is unchanged in cystinotic cells. Furthermore, the dependence of the intracellular GSH and cysteine pool sizes and the [GSH]/[GSSG] ratio are similarly dependent on the two major sources of cysteine, i.e. the transsulfuration pathway and the plasma membrane cystine transporter, xc-, in both cystinotic and control cells, and the presence of lysosomal cystine has no measurable effect on the redox status of these cells. Hence, mechanisms other than cytosolic redox perturbations are involved in the etiology of nephropathic cystinosis.
Introduction
Understanding development of the cystinotic phenotype is complicated by the lysosomal location of the aberrant molecule in this disorder. Unlike other lysosomal storage diseases, cystinosis results in storage of a single amino acid, i.e. cystine, instead of a macromolecular polymer (1). The disorder results from failure of the lysosomal cystine transporter, cystinosin (2), to export cystine into the cytosol where it is reduced to cysteine and used for glutathione (GSH) (3) and protein synthesis (4). As a result, cystine accumulates to very high levels in the lysosomes from the degradation of cystine-containing proteins and by other, as yet not understood processes. The mechanism by which the lysosomal cystine pool can influence cytosolic functions leading to the disease phenotype remains to be elucidated.
A number of attempts have been made to explain development of the nephropathic cystinotic phenotype, which includes short stature, failure to thrive, the renal Fanconi syndrome, and renal failure by age 10 years, along with other cardinal finding such as crystalline keratopathy, retinopathy, photophobia, and later pancreatic and pulmonary insufficiency, proximal muscle weakness, and premature aging (1).
Beginning with the initial hypothesis of A. D. Patrick, who proposed poisoning of cytosolic enzymes as a mechanism for development of the cystinotic phenotype (5), attempts have been made to explain how cystine, isolated within lysosomes, could interact with other cell components in different compartments and lead to the phenotype. Absent a functioning cystine transporter, the only apparent route for cystine to exit lysosomes is via exocytosis, which results in cystine being deposited in the extracellular fluid where it may be taken up by the plasma membrane cystine/glutamate transporters and used for synthesis of GSH and proteins. Several groups have focused on potential changes in the cellular redox state in cystinotic cells and particularly on the intracellular pools of oxidized and reduced GSH. Baum et al (6) found that loading proximal tubular epithelial cells with cystine dimethylester (CDME), which leads to lysosomal cystine accumulation, resulted in a proximal tubular transport defect characteristic of the Fanconi syndrome. They determined that the transport defect was due to a decrease in active transport and that although Na-K-ATPase activity was unimpaired under maximal velocity conditions, the production of ATP was severely compromised. Levtchenko et al (7) reported elevated GSH disulfide (GSSG) in cystinotic fibroblasts, with a corresponding increase in the ratio of [GSSG]/total [GSH] (0.091 in cystinotic versus 0.047 in normal fibroblasts, p<0.05). Study of these metabolites in cultured proximal tubular epithelial cells obtained from the urine of cystinotic patients and controls, followed by immortalization with human papilloma virus, permitted measurement of intracellular cystine and reduced and oxidized GSH in transformed renal cells which were not pre-loaded with CDME. Two groups have reported studies on these cells. Wilmer et al found that oxidized GSH was increased to 1.16 ± 0.83 nmol mg−1 protein in cystinotic immortalized renal tubule cells versus 0.29± 0.18 nmol mg−1 protein in control cells (p=0.04). They found total GSH, free cysteine, and ATP contents were normal in these cells and concluded that the elevated GSSG to total GSH ratio might indicate increased oxidative stress (8). Laube et al. (9) found that cystinotic lines have a significant decrease in GSH content (6.8 nmol GSH mg−1 protein versus 11.8 nmol mg−1 in controls, p<0.001). The ATP content and mitochondrial activity were found to be normal. Chol, et al studied cystinotic fibroblasts derived from skin biopsies from three patients with nephropathc cystinosis. They found a moderate decrease (~30%) in GSH during the exponential growth phase (9). Mannucci et al found that cystinotic cells accumulate high levels of pyroglutamate after ATP depletion, and speculated that this decrease might be related to a relative cytosolic deficiency of cysteine normally derived from cystine, but which is unavailable due to lysosomal cystine storage (10).
In order to further understand the cytosolic redox status in cystinotic cells, we have evaluated the GSSG and GSH concentrations and their ratio, in age and passage number-matched nephropathic cystinotic and control normal human diploid epithelial fibroblasts.
METHODS
Cystinotic skin fibroblasts (GM00008 and GM00046, donor ages 5 and 3 years, respectively), normal skin fibroblasts (GM00498, donor aged 3 years), cystinotic fetal lung fibroblasts (GM00090, donor aged 24 weeks) and normal fetal lung fibroblasts (GM01379, donor aged 12 weeks) were purchased from the Coriell Institute Biorepository (Camden, New Jersey). These cells were cultured in Ham's F-12 (Gibco 11765) media supplemented with 15% FBS, 1% penicillin (5000 U/mL)/streptomycin (5000 μg/mL) and amphotericin B (250 μg/mL) by volume, and 1 mM L-glutamine (3). Cells were grown to full confluency over a period of 7–14 days and media was changed as needed. Table 1 provides a description of the cells lines used.
Table 1.
Summary of cell lines used in this study.
| Line/passage # when used | Sex | Age | Phenotype | Genotype |
|---|---|---|---|---|
| GM01379 8 , 12,19 |
M | 12 FW | Apparently healthy fetal lung fibroblasts. | None given. |
| GM00090 11,14,11,12 |
M | 24 FW | Fetal lung fibroblasts, increased intracellular cystine. | None given, diagnosis made on basis of increased intracellular cystine and positive family history. |
| GM00498 14,17,17,16 |
M | 3 YR | Apparently healthy skin fibroblasts. | None given. |
| GM00008 13,14,13,13 |
F | 5 YR | Skin fibroblasts. Increased intracellular cystine. | Homozygous for 57kb deletion that removes first 10 exons of CTNS gene. |
| GM00046 9,12,16,11 |
M | 3 YR | Skin fibroblasts. Increased intracellular cystine. | Homozygous for a 5bp deletion in the CTNS gene (545delTCCTT) resulting in the substitution of arginine for isoleucine at codon 69 and causing termination at codon 73. |
The incubation medium was changed 24–28 h before sample collection. An inhibitor of g-cystathionase, propargylglycine (11) and an inhibitor of the cystine transporter xc-, sulfasalazine (12)were added 12 h after medium change and samples were collected 15−16 h later. Propargylglycine was added as a sterile 250 mM stock solution in PBS to a final concentration of 2.5 mM. Sulfasalazine was added as a sterile 10 mM stock solution in PBS to a final concentration of 0.5 mM. For sample collection, the cell culture media was aspirated and cells were washed twice with cold PBS, scraped and suspended in 100 μl PBS. For analysis of thiols, an aliquot of the cell suspension was fixed with metaphosphoric acid solution and after 2,4dinitrofluorobenzene derivatization, the concentrations of cystine, cysteine, reduced and oxidized glutathione were measured using an HPLC method as described previously (13,14). The protein concentration in cell suspension samples was measured using the Bradford reagent (Bio-Rad) as described (14) and thiol concentrations were normalized to protein levels.
RESULTS AND DISCUSSION
In our experiments, the cystinotic cells exhibited a greater than 10-fold increase in intracellular cystine concentration versus normal age-matched fibroblast cells (Table 2), as expected (1). In contrast, the concentrations of cysteine, GSH, and GSSG were not statistically significantly different between normal and cystinotic cells, (Table 2). In one skin cystinotic cell line (GM00008), an increase in the [GSH]/[GSSG] ratio was observed compared to corresponding normal fibroblast cell line, and this was due primarily to a lower GSSG concentration. In contrast, the [GSH]/[GSSG] ratio in the second skin cystinotic cell line, GM00046, was comparable to that in the normal fibroblast. No difference in the [GSH]/[GSSG] ratio was observed between normal and cystinotic fetal lung fibroblast lines.
Table 2.
Thiol metabolite concentrations in normal and cystinotic fibroblastsa.
| Cell line | GM00498 Normal Skin | GM00008 Cystinotic Skin | GM00046 Cystinotic Skin | GM01379 Normal Fetal lung | GM00090 Cystinotic Fetal lung |
|---|---|---|---|---|---|
| [Cystine] | 0.22±0.16 (n=10) | 2.0±0.6 (n=10, p<10−7) | 2.4±1.0 (n=10, p<10−5) | 0.08±0.01 (n=2) | 1.18±0.05 (n=2, p<0.002) |
| [Cysteine] | 2.1±1.2 (n=10) | 1.8±0.7 (n=10, p>0.5) | 1.5±0.7 (n=10, p>0.2) | 1.05±0.01 (n=2) | 1.29±0.14 (n=2, p>0.1) |
| [GSH] | 23±10 (n=11) | 27±7 (n=11, p>0.2) | 21±14 (n=11, p>0.7) | 8.9±1.4 (n=2) | 10.8±1.0 (n=2, p>0.2) |
| [GSSG] | 0.35±0.09 (n=11) | 0.33±0.12 (n=11, p>0.6) | 0.32±0.17 (n=11, p>0.6) | 0.07±0.01 (n=2) | 0.08±0.00 (n=2, p>0.09) |
| [GSH]/[GSSG] | 64±19 (n=11) | 92±34 (n=11, p<0.04) | 63±19 (n=11, p>0.8) | 141±13 (n=2) | 138±8 (n=2, p>0.8) |
Values are expressed in μmoles gm−1 of protein. Data are presented as mean±standard deviation. The number of measurements (n) and the statistical significance (p value) of the difference between normal and corresponding cystinotic cell lines are shown in parentheses.
Cysteine is the limiting reagent in the synthesis of GSH, and is obtained either via import of extracellular cystine, that is rapidly reduced to cysteine, or from methionine via the transsulfuration pathway (Fig. 1). To further assess potential dysregulation of cytosolic cysteine homeostasis in native, non-transformed cystinotic cells, we have examined the effects of pharmacological inhibition of both the cysteine synthesis and transport processes in skin (Fig. 2) and fetal lung (Fig. 3) fibroblasts. Inhibition of γ-cystathionase with the irreversible inactivator propargylglycine (PPG) inhibits metabolic flux through the transsulfuration pathway whereas sulfasalazine (SAS) is a reversible inhibitor of the transmembrane cystine transporter, xC- (Fig.1).
Fig. 1.
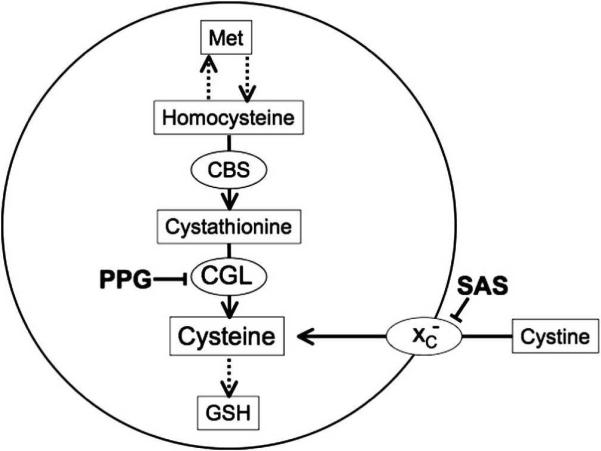
Outline of the GSH biosynthetic pathway and the sites of action of the inhibitors used in this study. Propargylglycine (PPG) inhibits cystathionine γ-lyase CGL) preventing cysteine synthesis from methionine (Met) via the transsulfuration pathway. Sulfasalazine (SAS) inhibits the transmembrane cystine transporter xc-. Both inhibitors can decrease the intracellular cysteine concentration and thus cause a decrease in intracellular GSH concentration. CBS denotes cystathionine
CGL) preventing cysteine synthesis from methionine (Met) via the transsulfuration pathway. Sulfasalazine (SAS) inhibits the transmembrane cystine transporter xc-. Both inhibitors can decrease the intracellular cysteine concentration and thus cause a decrease in intracellular GSH concentration. CBS denotes cystathionine  -synthase, the first enzyme or transsulfuration pathway.
-synthase, the first enzyme or transsulfuration pathway.
Fig. 2.

The influence of propargylglycine and sulfasalazine on thiols in normal and cystinotic non-fetal human skin fibroblasts. Data are the average of two measurements.
Fig. 3.

The influence of propargylglycine and sulfasalazine on thiol metabolites in normal and cystinotic fetal human lung fibroblasts. Data are the average of two measurements.
Both inhibitors had similar effects in both normal and cystinotic fibroblasts. In Figs. 2 and 3, the effect of these inhibitors is shown on the intracellular concentrations of cystine, cysteine (Cys), GSH, and the GSH/GSSG ratio. In cystinotic and normal skin fibroblasts, both propargylglycine and sulfasalazine resulted in decreased cysteine (Cys) and GSH levels (Fig. 2). In fetal lung cells, only propargylglycine caused a decrease in both cysteine and GSH levels (Fig. 3), whereas SAS caused a decrease only in cysteine. Interestingly, proparglycine resulted in a decrease in cystine levels in fetal lung but not in skin fibroblasts. The reason for this difference is not known and may reflect differences in the quantitative importance of the transsulfuration pathway for cysteine generation in fetal versus non-fetal cells. The [GSH]/[GSSG] ratio was not significantly affected by propargylglycine or sulfasalazine in either skin or lung fibroblasts. The effects were statistically insignificant between cystinotic and normal cells for all measured parameters, hence these results confirm that perturbations in GSH-linked intracellular redox homeostasis are not present in lung or skin fibroblasts derived from patients with nephropathic cystinosis.
Based on the similar sensitivity of the GSH pool size to inhibition of the transsulfuration pathway or to extracellular cystine import (Figs. 2–3) and because they exhibit comparable [GSH]/[GSSG] ratios, we conclude that cystinotic cells do not exhibit signs of impaired cytoplasmic redox metabolism. Note that if increased lysosomal cystine pools caused the ambient cytosolic redox potential to be more oxidizing, a decrease rather than an increase as seen in line GM00008 in the [GSH]/[GSSG] ratio (Table 2), would be expected.
A priori, an alteration in cytosolic redox is not expected from lysosomal cystine storage in cystinotic cells, based on the compartmentalization of cystine in the lysosome, and on the magnitude of the stored cystine pool size compared to cytosolic GSH levels. Release of the entire lysosomal cystine store into the cytosol would lead to an increase in intracellular cysteine concentration due to rapid conversion of cystine to cysteine in the reducing milieu of the cytoplasm. This redox change should be readily buffered given the 10-fold higher concentration of GSH measured in fibroblasts and the high [GSH]/[GSSG] ratio ranging from ~60 in skin to ~140 in fetal lung fibroblasts (Figs. 2–3). Alternatively, if the lysosomal cystine pool in normal cells is an important feeder of the cytoplasmic cysteine pool, then an increased sensitivity to pharmacological inhibition of the transsulfuration pathway for cysteine synthesis or of the cytoplasmic xc transporter, might have been expected in cystinotic cells, however; this was not observed.
In contrast to the data reported in this study, a significantly lower [GSH]/[GSSG] ratio has been previously reported in cystinotic fibroblasts and in polymorphonuclear cells (7). In that study, the cysteine pool was reported to be 1.5-to 1.8-fold lower than the total GSH pool, which represents a significant underestimation of the GSH concentration, which is typically in the 1 mM range, while intracellular cysteine is in the 50–100 μM range in fibroblasts. We found that the cysteine pool is ~13-fold and ~8-fold lower than GSH concentrations in the skin and fetal lung fibroblasts used in this study. Thiol metabolites are unstable in air, notoriously difficult to quantify accurately and therefore easily underestimated, while the disulfide pools are overestimated. In the prior study (7) the relatively low levels of total GSH in comparison to cysteine and the wide range in the percentage of GSSG of the total GSH pool, which varied over a 4-and 7-fold range in cystinotic and control fibroblast cells respectively, raise questions about the accuracy of redox metabolite determinations. Higher GSSG levels were also reported in immortalized proximal tubular cystinotic cells compared to controls (8). Remarkably, GSH levels, which are in the millimolar range, were reported to be below the detection limit in half the control and cystinotic cell lines, also suggesting technical problems associated with redox metabolite measurements.
In summary, our study demonstrates that the steady-state cytosolic redox status in fetal and non-fetal skin and lung-derived cystinotic fibroblasts compared to origin and age-matched normal control fibroblasts are comparable. However, the question still remains as to how the lysosomal cystine pool elicits the cystinotic phenotype. Recently, an alternative splice site in the CTNS (17p13) gene encoding the lysosomal cystine transporter, cystinosin was reported, which leads to an isoform that localizes in the plasma membrane in addition to the lysosome, endoplasmic reticulum and the Golgi apparatus (15). It is possible that mutations affecting the relative distribution of the splice site variants could alter cystine dynamics by a mechanism that awaits elucidation. Alternatively, increased apoptosis has been reported in cystinotic fibroblasts and cultured renal tubule epithelial cells, and a mechanism for development of the cystinotic phenotype has been proposed (16–20). Lysosomal permeability, which occurs early in the apoptosis cascade, could provide a route by which lysosomal cystine could reach the cystosol and produce deleterious effects.
We find that the steady-state redox status as revealed by the [GSH]/[GSSG] ratio, an indicator of the intracellular redox poise, is unperturbed in cystinotic cells. Furthermore, the dependence of the intracellular GSH and cysteine pool sizes and the [GSH]/[GSSG] ratio are similarly dependent on the two major sources of cysteine, i.e. the transsulfuration pathway and the plasma membrane cystine transporter, xc-, in both cystinotic and control cells, and that the presence of lysosomal cystine has no measurable effect on the redox status of these cells. Hence, mechanisms other than cytosolic redox perturbations are involved in the etiology of nephropathic cystinosis.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (DK64959 to R.B.) and the Cystinosis Research Foundation (to JGT)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1).Gahl WA, Thoene J, Schneider J. Cystinosis: A disorder of lysosomal membrane transport. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edition McGraw Hill; 2001. pp. 5085–5108. [Google Scholar]
- 2).Town M, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van't Hoff W, Antignac C. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nature Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- 3).Thoene J, Oshima R, Crawhall J, Olson D, Schneider J. Cystinosis: Intracellular Cystine Depletion by Aminothiols in Vitro and in Vivo. J. Clin. Invest. 1976;58:180–189. doi: 10.1172/JCI108448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Oshima R, Rhead W, Thoene J, Schneider J. Cystine Metabolism in Human Fibroblasts. J. Biol. Chem. 1976;251:4287–4293. [PubMed] [Google Scholar]
- 5).Patrick A. Deficiences of -SH-dependent enzymes in cystinosis. J.Clin.Path. 1965;28:427–443. [PubMed] [Google Scholar]
- 6).Baum M. The Fanconi syndrome of cystinosis: insights into the pathophysiology. Pediatr Nephrol. 1998;12:492–497. doi: 10.1007/s004670050495. [DOI] [PubMed] [Google Scholar]
- 7).Levtchenko E, de Graaf-Hess A, Wilmer M, van den Heuvel L, Monnens L, Blom H. Altered status of glutathione and its metabolites in cystinotic cells. Nephrol Dial Transplant. 2005;20:18281832. doi: 10.1093/ndt/gfh932. [DOI] [PubMed] [Google Scholar]
- 8).Wilmer MJ, de Graaf-Hess A, Blom HJ, Dijkman HB, Monnens LA, van den Heuvel LP, Levtchenko EN. Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun. 2005;337(2):610–4. doi: 10.1016/j.bbrc.2005.09.094. [DOI] [PubMed] [Google Scholar]
- 9).Chol M, Nevo N, Cherqui S, Antignac C, Rustin P. Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem Biophys Res Commun. 2004;324(1):231–5. doi: 10.1016/j.bbrc.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 10.Mannucci L, Pastore A, Rizzo C, Piemonte F, Rizzoni G, Emma F. Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr Res. 2006;59:332–5. doi: 10.1203/01.pdr.0000196370.57200.da. [DOI] [PubMed] [Google Scholar]
- 11.J Steegborn C, Clausen T, Sondermann P, Jacob U, Worbs M, Marinkovic S, Huber R, Wahl MC. Kinetics and inhibition of recombinant human cystathionine gamma-lyase. Toward the rational control of transsulfuration. J Biol Chem. 1999;274:12675–84. doi: 10.1074/jbc.274.18.12675. [DOI] [PubMed] [Google Scholar]
- 12).Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)-cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–40. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 13).Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–11. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 14).Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem. 2006;281:3578593. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- 15).Taranta A, Petrini S, Palma A, Mannucci L, Wilmer MJ, De Luca V, Diomedi-Camassei F, Corallini S, Bellomo F, van den Heuvel LP, Levtchenko EN, Emma F. Identification and subcellular localization of a new cystinosin isoform. Am J Physiol Renal Physiol. 2008;294(5):F1101–8. doi: 10.1152/ajprenal.00413.2007. [DOI] [PubMed] [Google Scholar]
- 16).Park MA, Helip-Wooley A, Thoene JG. Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol. 2002;13:2878–2887. doi: 10.1097/01.asn.0000036867.49866.59. [DOI] [PubMed] [Google Scholar]
- 17).Park MA, Thoene JG. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol. 2005;20:441–446. doi: 10.1007/s00467-004-1712-9. [DOI] [PubMed] [Google Scholar]
- 18).Park M, Pejovic V, Kerisit K, Junius S, Thoene J. Increased Apoptosis in Cystinotic Fibroblasts and Renal Proximal Tubule Epithelial Cells Results from Cysteinylation of PKCδ. J Am Soc Neph. 2006;17:3167–3175. doi: 10.1681/ASN.2006050474. [DOI] [PubMed] [Google Scholar]
- 19).Laube G, Shah V, Stewart V, Hargreaves I, Haq M, Heales S, van't Hoff W. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol. 2006;21:503–509. doi: 10.1007/s00467-006-0005-x. [DOI] [PubMed] [Google Scholar]
- 20).Thoene JG. A review of the role of enhanced apoptosis in the pathophysiology of cystinosis. Mol Genet Metab. 2007;92:292–8. doi: 10.1016/j.ymgme.2007.07.008. [DOI] [PubMed] [Google Scholar]