

Abstract
In the present study, mice lacking gp91phox gene were used to address the role of NADPH oxidase in hyperhomocysteinemia-induced podocyte injury. It was found that the folate-free diet increased plasma homocysteine levels, but failed to increase O2.− production in the glomeruli from gp91phox gene knockout (gp91−/−) mice, compared with wild-type (gp91+/+) mice. Proteinuria and glomerular damage index (GDI) were significantly lower, while the glomerular filtration rate (GFR) was higher in gp91−/− than gp91+/+ mice when they were on the folate-free diet (Urine albumin excretion: 21.23±1.88 vs. 32.86±4.03 μg/24 h; GDI: 1.17±0.18 vs. 2.59 ±0.49; and GFR: 53.01±4.69 vs. 40.98±1.44 μL/min). Hyperhomocysteinemia-induced decrease in nephrin expression and increase in desmin expression in gp91+/+ mice were not observed in gp91−/− mice. Morphologically, foot process effacement and podocyte loss due to hyperhomocysteinemia were significantly attenuated in gp91−/− mice. In in vitro studies of podocytes, homocysteine was found to increase gp91phox expression and O2.− generation, which was substantially inhibited by gp91phox siRNA. Functionally, homocysteine-induced decrease in vascular endothelial growth factor-A (VEGF-A) production was abolished by gp91phox siRNA or diphenyleneiodonium, a NADPH oxidase inhibitor. These results suggest that the functional integrity of NADPH oxidase is essential for hyperhomocysteinemia-induced podocyte injury and glomerulosclerosis.
Keywords: homocysteinemia, glomerulosclerosis, glomerulus, redox signaling, free radicals
Introduction
Hyperhomocysteinemia (hHcys) has been known as a critical pathogenic factor both in the progression of end-stage renal disease (ESRD) and in the development of cardiovascular complications related to ESRD [1-3]. It has been reported that sustained hHcys may induce extracellular matrix accumulation and inhibit their degradation in the kidney, which ultimately leads to glomerulosclerosis [4]. Although many downstream signaling pathways are found to be involved in hHcys-induced glomerulosclerosis, reactive oxygen species (ROS) are currently regarded as one of the most important mediators in the initiation of hHcys-induced kidney damage. Among ROS, superoxide (O2.−) is a main source for other oxygen-centered radicals, such as H2O2 and .OH, which participate in lipid peroxidation and induce cellular membrane damage. Emerging evidence has highlighted the important role for the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent system as a major source of O2.− in the kidney [5]. Recently, we and others have shown that NADPH oxidase activation and subsequent O2.− generation are important mechanisms in the development of hHcys-induced glomerulosclerosis [6, 7].
NADPH oxidase comprises the membrane-bound cytochrome b558 (formed by gp91phox and p22phox subunits) and the cytosolic proteins p40phox, p47phox, p67phox, and Rac1/2. The catalytic subunits of this enzyme are termed NOX proteins, which include NOX1, NOX2 (i.e. gp91phox), NOX3, NOX4, NOX5, DUOX1, and DUOX2 [8]. It has been reported that almost all components of the NADPH oxidase complex, including gp91phox, p22phox, p47phox, p67phox, and Rac1/2 are expressed in the kidney; and that these NADPH oxidase components could be detected in renal fibroblasts, endothelial cells, vascular smooth muscle cells, mesangial cells, and tubular cells [9]. Among these, the catalytic subunit gp91phox was shown to be essential for oxidative stress in the kidney. Previous studies using gp91phox knockout (KO) mice have demonstrated that this enzyme plays important roles in the maintenance of renal vascular tone [10], as well as in the regulation of renal hemodynamics and excretory function under a condition of nitric oxide deficiency [11]. Recently, several NADPH oxidase subunits, gp91phox, p22phox, p67phox, and p47phox, have been also demonstrated to be present in cultured podocytes [12]. Further studies showed that NADPH oxidase activation and subsequent O2.− production play a key role in high glucose-, angiotensin II-, and adriamycin-induced podocyte injury [13-15]. However, it remains unknown whether activation of NADPH oxidase is involved in hHcys-induced podocyte injury and related pathological changes.
Podocytes are terminally differentiated epithelial cells, which cover the outmost layer of glomerular filtration barrier and constitute the last and the most important barrier in preventing protein leakage from the plasma. Although podocyte injury has been considered as the most important early event in initiating glomerulosclerosis and thereby resulting in ESRD in different animal models and humans [16-18], its involvement in hHcys-induced glomerular damage and sclerosis is still poorly understood. The present study hypothesized that homocysteine (Hcys) may directly cause podocyte injury through NADPH oxidase-mediated O2.− production and that knockout or deletion of its functional subunit, gp91phox, will protect podocytes from hHcys-induced injury. To test this hypothesis, we determined the possible injurious response of podocytes to hHcys in mice lacking gp91phox gene and compared related pathological changes with their genetic background strain C57BL/6 mice. We further used cultured murine podocytes to examine the direct effects of gp91phox gene deletion on Hcys-induced functional changes in podocyte and explored the underlying mechanisms.
Materials and Methods
Animal procedures
C57BL/6J wild-type (WT, 6 weeks old, male) and gp91phox knockout (KO) mice (6 weeks old, male, The Jackson Laboratory, Bar Harbor, ME) were used. All protocols were approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University. Given the difficulty for the development of hyperhomocysteinemia in mice with both kidneys due to treatment that requires a long period of time, all gp91phox KO mice and WT mice were subjected to a uninephrectomy which helped accelerate the progression of hHcys-induced glomerular injury as we reported previously [6]. This model was demonstrated to induce glomerular damage unrelated to the uninephrectomy and arterial blood pressure, but specific to hHcys. After a 1-week recovery period from the uninephrectomy, KO and WT mice were fed a normal diet or a folate-free (FF) diet (Dyets Inc, Bethlehem, PA) for 4 weeks to induce hHcys. One day before these mice were sacrificed, 24-hour urine samples were collected using mouse metabolic cages. After blood samples were collected, the mice were sacrificed and renal tissues were harvested for biochemical and molecular analysis as well as morphological examinations.
High-performance liquid chromatography (HPLC) analysis
Plasma total Hcys levels were measured by HPLC method as we described previously [19]. Briefly, blood samples were centrifuged at 1000 g for 10 min at 4°C to isolate the plasma. A 100 μL plasma, or standard solution mixed with 10 μL of internal standard, thionglycolic acid (2.0 mmol/L) solution, was treated with 10 μL of 10% tri-n-butylphosphine (TBP) solution in dimethylformamide at 4°C for 30 minutes. Then, 80 μL of ice-cold 10% trichloroacetic acid (TCA) in 1 mmol/L EDTA was added and centrifuged to remove proteins in the sample. 100 μL of the supernatant was transferred into the mixture of 20 μL of 1.55 mol/L sodium hydroxide, 250 μL of 0.125 mol/L borate buffer (pH 9.5), and 100 μL of 1.0 mg/mL ABD-F solution. The resulting mixture was incubated at 60°C for 30 minutes to accomplish derivatization of plasma thiols. HPLC was performed with a HP 1100 series equipped with a binary pump, a vacuum degasser, a thermostated column compartment, and an autosampler (Agilent Technologies, Waldbronn, Germany). Separation was carried out at an ambient temperature on an analytical column, Supelco LC-18-DB (1504.6 mm ID, 5 m) with a Supelcosil LC-18 guard column (204.6 mm ID, 5 m). Fluorescence intensities were measured with an excitation wavelength of 385 nm and emission wavelength of 515 nm by a Hewlett-Packard Model 1046A fluorescence spectrophotometer. The peak area of the chromatographs was quantified with a Hewlett-Packard 3392 integrator. The analytical column was eluted with 0.1 mol/L potassium dihydrogenphosphate buffer (pH 2.1) containing 6% acetonitrile (v/v) as the mobile phase with a flow rate of 2.0 mL/min.
Monitoring of arterial blood pressure in conscious mice
Mean arterial pressure (MAP) was measured 4 weeks after mice were treated with the normal or FF diet as we described previously [20]. In brief, mice were anesthetized by inhalation of isoflurane, and then a catheter connected to a telemetry transmitter was implanted into the carotid artery and the transmitter was placed subcutaneously. The arterial blood pressure signal from the transmitter was received by a remote receiver and then recorded by a computer program (Data Sciences International, St. Paul, MN). Arterial blood pressure was continuously measured for one week after an equilibration period.
Electromagnetic spin resonance (ESR) analysis of O2.− production
For detection of NADPH oxidase-dependent O2.− production, proteins from the isolated glomeruli of mice or cultured podocytes were extracted using sucrose buffer and resuspended with modified Kreb's-Hepes buffer containing deferoximine (100 μmol/L, Sigma, St. Louis, MO) and diethyldithiocarbamate (5 μmol/L, Sigma, St. Louis, MO). NADPH oxidase-dependent O2.− production was examined in 50 μg of protein in the presence or absence of SOD (200 U/ml, Sigma, St. Louis, MO). The mixture was loaded in glass capillaries and immediately analyzed for O2.− production kinetically for 10 min in a Miniscope MS200 ESR spectrometer (Magnettech Ltd, Berlin, Germany) as we described in other studies [21, 22]. The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; 4,096 points of resolution; receiver gain, 20 for tissue and 50 for cells. The results were expressed as the fold changes of control.
Measurement of creatinine clearance (Ccr)
Plasma and urinary creatinine concentrations (Pcr and Ucr) were measured as described before [23]. In brief, 24-hour urine samples were collected using metabolic cages. Blood samples for serum creatinine were collected at the time the mice were sacrificed. Serum and urinary creatinine concentrations were measured by a QuantiChrom™ Creatinine Assay kit (BioAssay Systems, Hayward, CA). Ccr was calculated using the following formula: Ccr (mL/min) = (Ucr/Pcr)×urine volume (mL/min).
Urinary total protein and albumin excretion measurement
The 24-hour urine samples were collected using metabolic cages and subjected to total protein and albumin excretion measurement, respectively. Total protein content in the urine was detected by Bradford method using a UV spectrometer. Urine albumin was detected using a commercially available mouse albumin ELISA kit (Bethyl Laboratories, Montgomery, TX).
Morphological examination
For observation of renal morphology using light microscope, renal tissues were fixed with 10% formalin solution, paraffin-embedded, and stained with periodic acid-Schiff (PAS). Glomerular sclerosis was assessed by a standard semi-quantitative analysis and expressed as glomerular damage index (GDI) [24]. Fifty glomeruli per slide were counted and graded as 0, 1, 2, 3, or 4, according to 0, <25, 25–50, 51–75, or >75% sclerotic changes cross a longitudinal kidney section, respectively. The GDI for each mouse was calculated by the formula: (N1 × 1 + N2 × 2 + N3 × 3 + N4 × 4)/n, where N1, N2, N3, and N4 represent the numbers of glomeruli exhibiting grades 1, 2, 3, and 4, respectively; and n is the total number of glomeruli graded.
For transmission electron microscopic (TEM) observation of ultrastructural changes in podocytes, the kidneys were perfused with a fixative containing 3% glutaraldehyde and 4% paraformaldehyde in 0.1M phosphate buffer. The kidneys were sliced longitudinally and cortical strips were cut into 1 mm3 tissue blocks, which were further fixed with 3% glutaraldehyde for 12 h. After dehydration with ethanol, the samples were embedded in Durcupan resin for ultra-thin sectioning and TEM examinations by VCU electron microscopy core facility.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from isolated mouse glomeruli or cultured podocytes were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the protocol as described by the manufacturer. RNA samples were quantified by measurement of optic absorbance at 260 nm and 280 nm in a spectrophotometer. The concentrations of RNA were calculated according to A260. Aliquots of total RNA (1 μg) from each sample were reverse-transcribed into cDNA according to the instructions of the first strand cDNA synthesis kit manufacturer (Bio-Rad, Hercules, CA). Equal amounts of the reverse transcriptional products were subjected to PCR amplification using SYBR Green as the fluorescence indicator on a Bio-Rad iCycler system (Bio-Rad, Hercules, CA). The mRNA levels of target genes were normalized to the β-actin mRNA levels. The primers used in this study were synthesized by Operon (Huntsville, AL) and the sequences were: gp91phox sense TGGCACATCGATCCCTCACTGAAA, antisense GGTCACTGCATCTAAGGCAACCT; vascular endothelial growth factor-A (VEGF-A) sense CAATGATGAAGCCCTGGAG, antisense TCTCCTATGTGCTGGCTTTG; β-actin sense TCGCTGCGCTGGTCGTC, antisense GGCCTCGTCACCCACATAGGA.
Immunofluorescent staining and immunohistochemistry
Indirect-immunofluorescent staining was performed using frozen slides of mouse kidneys. After fixation with acetone, the slides were incubated with rabbit anti-nephrin 1: 100 (Abcam, Cambridge, MA), rabbit anti-podocin 1: 200 (Sigma, St. Louis, MO) or rabbit anti-desmin 1: 100 (BD Biosciences, San Jose, CA), overnight at 4 °C. Then, the slides were washed and incubated with corresponding FITC-labeled secondary antibodies. Finally, the slides were washed, mounted, and subjected to fluorescent microscopy examination. The images were captured with a Spot CCD camera (Diagnostic Instruments Inc., Sterling Heights, MI., USA). All exposure settings were kept constant for each group of kidneys.
Since WT1 (Wilms' tumor suppressor gene) is exclusively expressed by podocytes but not by other cell types in the glomeruli, podocyte numbers were counted in mouse glomeruli using immunohistochemistry staining for WT1 as reported previously[25]. Briefly, kidney sections from paraffin-embedded tissues were prepared at 4 μm thickness and stained by an antibody to WT1 (1:50 dilution; Abcam, Cambridge, MA) using a routine immunohistochemistry procedure. After staining, 50 consecutive glomerular cross-sections per mouse were randomly chosen and the positively-stained podocytes were counted by an observer who was blind to the sample identity. Six mice were analyzed for each group. The number of podocytes per glomerulus was calculated from podocyte density multiplied by the glomerular volume as described before [26].
Cell culture
Conditionally immortalized mouse podocyte cell line, kindly provided by Dr. Klotman PE (Division of Nephrology, Department of Medicine, Mount Sinai School of Medicine, New York, NY, USA), were cultured on collagen I-coated flasks or plates in RPMI 1640 medium supplemented with recombinant mouse interferon–γ at 33°C. After differentiated at 37°C for 10-14 days without interferon–γ, podocytes were used for the proposed experiments. In the present study, preparation of L-Hcys (a pathogenic form of Hcys), the concentration and the incubation time of L-Hcys treatment were chosen based on our previous studies in mesangial cells [27] and our preliminary experiments on podocytes.
Gp91phox siRNA transfection
Gp91phox siRNA was purchased from Qiagen, which was confirmed to be effective in silencing gp91phox gene in different cells by the company. The scrambled RNA (sRNA, Qiagen, Valencia, CA) was confirmed as non-silencing double-stranded RNA and used as the control in the present study. Podocytes were serum-starved for 12 h and then transfected with gp91phox siRNA or scrambled sRNA using siLentFect Lipid Reagent (Bio-rad, Hercules, CA). After 18 h of incubation at 37 °C, the medium was changed and L-Hcys (80 μmol/L) added into the medium for the indicated time span.
ELISA for VEGF-A secretion in podocytes
After transfected with gp91phox siRNA, scrambled sRNA, or pretreated with diphenyleneiodonium (DPI, a NADPH oxidase inhibitor), podocytes were incubated with L-Hcys (80 μmol/L) for 24 h. A relatively specific podocyte injury compound, puromycin aminonucleoside (PAN, 100 μg/ml) was used to treat cells for 24 h to serve as a positive control. The supernatant was collected for ELISA assay of VEGF-A using a commercially available kit (R&D system, Minneapolis, MN).
Statistical analysis
All of the values are expressed as mean ± SEM. Significant differences among multiple groups were examined using ANOVA followed by a Student-Newman-Keuls test. χ2 test was used to access the significance of ratio and percentage data. P<0.05 was considered statistically significant.
Results
Reduction of hHcys-induced O2.− production in gp91phox KO mice
As shown by HPLC analysis, plasma Hcys levels were similar in WT and gp91phox KO mice on the normal diet. FF diet treatment significantly increased plasma Hcys levels in both WT and gp91phox KO mice (Figure 1A). ESR analysis showed that glomerular O2.− production was lower in KO mice than WT mice on the normal diet. On the FF diet, glomerular O2.− production increased by 2.4 folds in WT mice. However, FF diet-induced O2.− production was much less in KO mice as compared with WT mice (Figure 1B and 1C).
Figure 1. Effects of normal and FF diets on plasma Hcys levels and glomerular O2.− production in gp91phox KO and WT mice.

A. Plasma Hcys levels measured by HPLC in 4 groups of mice (n=6). B. Representative ESR spectra traces for O2.− production in gp91phox KO and WT mice (n=5). C. Summarized data show the fold changes of O2.− production, which are normalized to WT mice on the normal diet (n=5). * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet; & p<0.05 vs. KO mice on the normal diet.
Alleviation of glomerular injury in gp91phox KO mice on the FF diet
In parallel to the elevation of plasma Hcys levels, the 24-hour urinary total protein and albumin excretions significantly increased in WT mice on the FF diet. In gp91phox KO mice, however, a similar level of plasma Hcys caused much less urinary total protein and albumin excretion (Figure 2A and 2B). By PAS staining, we observed a typical pathological change in glomerular sclerotic damage in WT mice on the FF diet (Figure 3A). The average GDI was substantially higher in WT mice on the FF diet, but much less in gp91phox KO mice on the same diet (p<0.05, Figure 3B). To evaluate the glomerular filtration rate (GFR) of these mice, creatinine clearance (Ccr) was measured. It was found that FF diet treatment induced a decline of Ccr in WT mice, while the Ccr was mostly preserved in gp91phox KO mice on the same diet (Figure 4A). To explore whether these renal functional and pathological changes were associated with elevation of arterial blood pressure, mean arterial pressure (MAP) was measured in conscious mice using telemetry. FF diet treatment had no effect on MAP in both WT and KO mice (Figure 4B).
Figure 2. Proteinuria was attenuated in gp91phox KO mice on the FF diet.

A. Urinary total protein levels in 4 groups of mice (n=6). B. Urinary albumin excretion in 4 different groups of mice as indicated (n=6). * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet; & p<0.05 vs. KO mice on the normal diet.
Figure 3. Glomerular damage was alleviated in gp91phox KO mice on the FF diet.

A. PAS staining shows glomerular morphological changes (original magnification, ×400). B. Summarized data of glomerular damage index (GDI) by semi-quantitation of scores in 4 different groups of mice (for each group, n=6). For each kidney section, 50 glomeruli were randomly chosen for the calculation of GDI. * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet.
Figure 4. Creatinine clearance (Ccr) and arterial blood pressure in gp91phox KO and WT mice.

A. Creatinine clearance in WT and KO mice on a normal or FF diet (n=7). B. Mean arterial pressure (MAP) in WT and KO mice with or without FF diet (n=5). * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet.
Expression of nephrin, podocin, and desmin in gp91phox KO and WT mice
Next we determined whether the protective effect of gp91phox gene knockout is associated with the protection of podocytes from hHcys-induced injury. Real-time RT-PCR demonstrated that the mRNA levels of two important slit diaphragm molecules, nephrin and podocin, were significantly decreased by the FF diet in WT mice, which was much less in KO mice on the same diet. However, the expression of desmin, a podocyte injury marker, was significantly increased in WT mice on the FF diet as compared with gp91phox KO mice (p<0.05, Figure 5A). As shown by indirect immunofluorescent staining in Figure 5B, nephrin and podocin staining was shown as a fine linear-like pattern along the glomerular capillary loop in mice on a normal diet. Both nephrin and podocin expression showed a dramatic decrease in WT mice on the FF diet, which was less obvious in the gp91phox KO mice on the same diet. In contrast, the expression of desmin was less increased in gp91phox KO mice on the FF diet as compared with WT mice.
Figure 5. Expressions of nephrin, podocin, and desmin in gp91phox KO and WT mice.

A. Real-time RT-PCR analysis of the expressions of nephrin, podocin, and desmin in the glomeruli of gp91 KO and WT mice (n=6). B. Immunofluorescent staining of nephrin, podocin, and desmin in the glomeruli of 4 groups of mice (n=4). * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet; & p<0.05 vs. KO mice on the normal diet.
Attenuation of hHcys-induced foot process effacement and podocyte loss in gp91phox KO mice
To observe ultrastructural changes in podocytes, TEM was conducted. Compared with the distinct brush-like structures of podocyte foot processes shown in mice on the normal diet, evident foot process effacement was observed in WT mice 4 weeks after FF diet treatment. In contrast, podocytes of gp91phox KO mice on the FF diet had relatively normal ultrastructures (Figures 6A). To investigate whether gp91phox gene knockout helps to maintain podocyte numbers, we counted the number of podocytes per glomerular cross-section in WT and KO mice. Podocytes were identified by staining with an antibody against the podocyte specific marker, WT1. It was found that there were similar numbers of WT1-stained podocytes in WT and KO mice fed a normal diet. FF diet treatment induced a significant decrease in podocyte numbers in WT mice, but not in KO mice (Figure 6B, 6C).
Figure 6. Attenuation of foot process effacement and podocyte loss in the glomeruli of gp91phox KO mice.
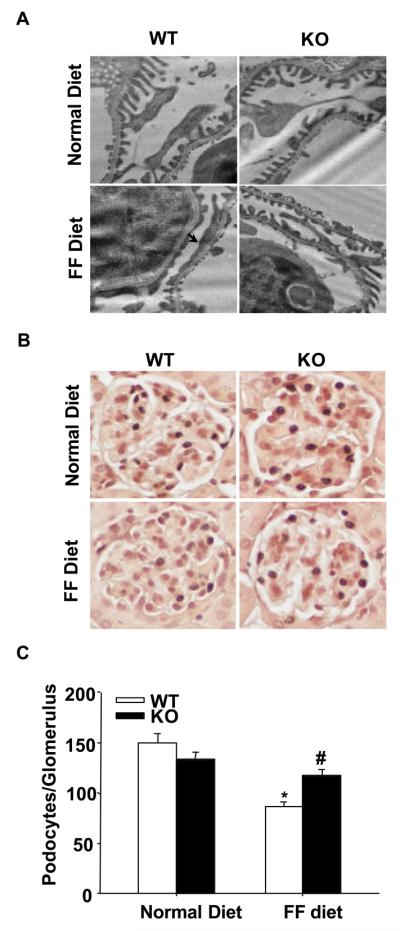
A. gp91phox gene deletion improved podocyte ultrastructure in FF diet-treated mice. Arrow denotes the area of foot process effacement in WT mice on the FF diet. Images are representative of 6 TEM images per kidney from 3 mice per group. Original magnification: ×8,000. B. Typical images of WT1-stained glomeruli from 4 groups of mice (Original magnification, ×400). C. Summarized data showing podocyte numbers per glomerulus in each group (n=6). * p<0.05 vs. WT mice on the normal diet; # p<0.05 vs. WT mice on the FF diet.
Blockade of Hcys-induced O2.− production by gp91phox gene silencing in cultured podocytes
Although we have demonstrated in whole animal experiments that the glomeruli of gp91phox KO mice were protected from hHcys-induced renal injury, it has yet to be determined whether such protective action is due to the direct protection of podocytes. To address this issue and to explore potential underlying mechanisms, we used in vitro cultured podocytes to examine the role of gp91phox-containing NADPH oxidase in Hcys-induced podocyte injury. Real-time RT-PCR showed that L-Hcys stimulated the expression of gp91phox both at mRNA and protein levels (p<0.05, Figure 7A). This increased gp91phox expression was accompanied by an elevated NADPH oxidase-dependent O2.− production (Figure 7B). When podocytes were transfected with gp91phox siRNA, Hcys-induced O2.− production was significantly inhibited, which was comparable to DPI, a NADPH oxidase inhibitor (p<0.05, Figure 7C).
Figure 7. L-Hcys increases gp91phox expression and NADPH oxidase-dependent O2.− production in cultured podocytes.

A. L-Hcys stimulation for 12 h caused elevated gp91phox mRNA expression in podocytes as determined by real-time RT-PCR (n=4). B. ESR analysis shows that Hcys induces O2.− production in a concentration-dependent manner in podocytes (n=5). C. ESR analysis shows that gp91phox siRNA transfection or NADPH oxidase inhibition by DPI reduces O2.− production in podocytes. Ctrl: Control; Vehl: Vehicle; Scra: scrambled sRNA; siRNA: gp91phox siRNA. (n=6). * p<0.05 vs.ctrl, # p<0.05 vs. Hcys.
Blockade of Hcys-induced decrease in VEGF-A secretion in podocytes by gp91phox siRNA gene silencing
As a functional parameter of podocytes, VEGF-A production was detected in podocytes under different conditions. Figure 8A showed that the mRNA expression of VEGF-A was decreased by L-Hcys in a concentration-dependent manner. As shown in Figure 8B and 8C, VEGF-A production and secretion were dramatically reduced by the treatment with L-Hcys (p<0.05), which was similar to the designated positive control, PAN. When gp91phox siRNA was transfected to inhibit O2.− production via NADPH oxidase, Hcys-induced decrease of VEGF-A secretion was significantly restored (p<0.05). Similarly, inhibition of NADPH oxidase activity using DPI also recovered VEGF secretion in podocytes.
Figure 8. Effects of gp91phox gene silencing on L-Hcys-induced decrease in VEGF-A production and secretion in podocytes.

A. Real-time RT-PCR analysis shows that Hcys stimulation for 12 h decreases VEGF-A mRNA levels in a concentration-dependent manner (n=4). B. Real-time RT-PCR analysis shows the expression of VEGF-A mRNA challenged by Hcys (80 μmol/L) for 12 h with different pretreatments. PAN was used as a positive control (n=4). C. VEGF-A secretion in the culture medium detected by ELISA after pretreatment with different inhibitors and Hcys for 24 h. Ctrl: Control; Vehl: Vehicle; Scra: scrambled sRNA; siRNA: gp91phox siRNA. (n=5). * p<0.05 vs. ctrl. # p<0.05 vs. Hcys.
Discussion
The goals of this study was to determine whether podocytes are a direct target of hHcys and to investigate the role of NADPH oxidase-dependent O2.− production in hHcys-induced podocyte injury. Using a FF diet-induced hHcys animal model in gp91phox gene deficient mice and in vitro cultured podocytes, we clearly demonstrated that hHcys directly induces podocyte injury and depletion through gp91phox-containing NADPH oxidase activation and O2.− production.
There is considerable evidence supporting the critical role of ROS, particularly O2.−, in the pathogenesis of kidney diseases, such as diabetic nephropathy [28] and hypertension-related nephropathy [29]. Among various enzymes involved in the generation of ROS, NADPH oxidase is regarded as the main source of O2.− production. Gp91phox, also known as NOX2, is the catalytic subunit of NADPH oxidase. Although there are some reports indicating that NOX1 and NOX4 are also expressed in the kidney [30, 31], data from our laboratory and by other groups have strongly suggested that gp91phox-containing NADPH oxidase system is more critical in mediating O2.− production in the kidney [4, 10, 11, 32]. Therefore, we used gp91phox deficient mice to explore the role of NADPH oxidase-mediated O2.− production in hHcys-induced podocyte injury. It was found that the FF diet treatment increased plasma Hcys levels in both WT and KO mice, which indicates a successful establishment of hHcys model and also suggests that gp91phox itself is not involved in the metabolism of Hcys. Gp91phox KO mice on the FF diet exhibited a lower level of renal O2.− production compared with WT mice, implying that gp91phox gene deficiency prevents hHcys-induced local O2.− production in the kidney. In previous studies, gp91phox KO mice have also been reported to have lowered O2.− production in many organs, such as the heart, lungs, and brain, which protect these mice from different pathophysiological conditions such as cardiac hypertrophy [8], hypoxic pulmonary hypertension [33], and surgically-induced brain injury [34]. In accordance with lowered O2.− production in gp91phox KO mice on the FF diet, urine albumin excretion in these mice was significantly decreased as compared with WT mice on the same diet, suggesting that ROS-associated renal injury during hHcys is alleviated in these KO mice. Pathological studies using PAS staining showed that there was a significant improvement of hHcys-induced glomerulosclerosis in these KO mice as compared with WT mice. In consistent with these pathological changes, GFR was also preserved in these KO mice on the FF diet. In addition, a 4-week FF diet treatment had no influence on arterial blood pressure of WT and gp91phox KO mice, indicating that the preservation of podocyte function and structure in these KO mice is not associated with any change in blood pressure. All these results support the view that gp91phox-containing NADPH oxidase mediates O2.− production in the glomeruli and thereby plays a critical role in hHcys-induced renal injury.
One of the most important findings of the present study is that gp91phox gene deficiency may protect podocytes from hHcys-induced injury. Podocytes serve as the final defense mechanism against urinary protein loss in the normal glomerulus [35]. Damages of podocytes and their slit diaphragm are intimately associated with proteinuria and glomerulosclerosis in many kidney diseases, such as focal segmental glomerulosclerosis, diabetic nephropathy, and hypertension-associated nephropathy [36, 37]. In recent studies, we have reported that foot process effacement was presented in a methionine-induced animal model of hHcys [23], but the mechanism has not yet been explored. Here, we demonstrated for the first time that hHcys could induce podocyte injury through NADPH oxidase-mediated O2.− production, which was supported by analysis of the levels of some slit diaphragm molecules such as nephrin and podocin and by measuring the podocyte damage marker, desmin. As it is well known, nephrin and podocin are two important components of the slit diaphragm complex, which play an essential role in maintaining the normal structure of the glomerular filtration barrier. Mutations or down-regulations of these proteins are associated with a defective filtration barrier causing proteinuria [38]. In addition, nephrin affords important signaling cascades through its interaction with podocin and CD2-associated protein (CD2AP) for maintaining the normal growth of podocytes and preventing their apoptosis [38]. So, the decreased expression of nephrin is not only an indicator of slit-diaphragm damage, but also a sign of disturbed intracellular signaling pathway, which is essential for podocyte living. The results from the present study showed that hHcys induced a dramatic decrease in nephrin and podocin expression in WT mice, but not in gp91phox KO mice. It appears that hHcys may disrupt the normal structure of the slit diaphragm complex and the related intracellular signaling events through NADPH oxidase-mediated O2.− production. This Hcys-induced podocyte injury and its attenuation in gp91phox KO mice were also demonstrated by the increase of desmin expression. Desmin is an intermediate filament protein, which has long been suggested as a specific and sensitive podocyte injury indicator [7] and its expression is often upregulated in various glomerular diseases in which podocyte damage is involved [18, 39]. In the present study, we showed that hHcys induced by the FF diet significantly increased desmin expression in the glomeruli of WT mice, but much less in gp91phox gene KO mice, further suggesting that podocyte injury is significantly attenuated due to reduced O2.− generation in the kidney.
Among various morphologic characteristics, foot process effacement and consequent podocyte depletion are strong predictors for the progression of glomerulosclerosis. It has been proposed that the loss of <20% of podocytes triggers mesangial expansion, loss of 20-40% produces small patches of podocyte depletion and synechia formation, and >40% podocyte depletion produces the full picture of FSGS with segmental and eventually global sclerosis [40]. Although these observations identify podocyte depletion as one of the earliest cellular features of many types of kidney diseases, the mechanisms underlying the loss of podocytes remain poorly understood. Recently, it has been demonstrated ROS activation is a key mechanism leading to podocyte loss in animal models of diabetic nephropathy [13] and PAN-induced nephropathy [41]. In the present study, the TEM data showed that FF diet treatment caused podocyte foot process effacement in WT mice, and such damage was dramatically attenuated in gp91phox KO mice on the same diet. The fact that gp91phox gene deletion provided protection from podocyte foot process effacement indicates that this podocyte injury is a direct action of hHcys on podocytes and it is through NADPH oxidase-mediated ROS toxicity. Using WT1 staining to calculate podocyte numbers and evaluate podocyte loss in the glomeruli, we found that hHcys significantly decreased podocyte numbers in the glomeruli of WT mice, indicating a podocyte loss under such pathological conditions. Interestingly, gp91phox gene deletion prevented such podocyte loss induced by hHcys. These results suggested that the activation of NADPH oxidase and subsequent O2.− production is an important mechanism in mediating Hcys-induced podocyte injury and death. In harmony with our results, Liu et al recently provided evidence showing that up-regulation of NADPH oxidase activity plays a pivotal role in mediating proximal tubular cell apoptosis in transgenic mice over-expressing angiotensinogen [42]. It should be noted that some previous studies reported the effects of hHcys in inducing renal cell proliferation through the activation of NADPH oxidase in other cell types such as mesangial cells [27]. It seems that the action of NADPH oxidase in causing cell proliferation or death may be cell-type specific. Although the function of NADPH oxidase-derived ROS in mediating renal cell proliferation and death seems to be paradoxical, these actions may eventually converge at the functional level, in that both processes may be detrimental (for example, proliferation of mesangial cells and depletion of podocytes) to normal kidney functions [43].
To address whether this protective effect of gp91phox gene deletion is directly associated with counteracting Hcys-induced injury to podocytes, we used cultured murine podocytes to examine the direct effects of gp91phox gene silencing on Hcys-induced functional changes. It was found that Hcys induced an increase in the expression of gp91phox and a dramatic enhancement of O2.− production in a concentration-dependent manner, which was blocked when podocytes were transfected with gp91phox siRNA. These results provide evidence that Hcys is able to directly act on podocytes, initiating local oxidative stress and resulting in podocyte injury via activation of NADPH oxidase. In accordance with our findings, Eid AA et al demonstrated that NADPH oxidase-dependent O2.− generation also mediates high glucose-induced podocyte injury using the same cell line as we used [13]. There is also another study indicating that NADPH oxidase-mediated O2.− generation serves as the main mediator of PAN-induced podocyte damage [44]. It seems that NADPH oxidase-mediated O2.− production is an important detrimental factor in podocyte injury, which may initiate and promote glomerulosclerosis.
To further investigate the functional abnormality of Hcys-induced podocyte injury via NADPH oxidase activation, we examined the production of VEGF-A in cultured podocytes. In the kidney, VEGF-A is mainly derived from podocytes, which regulates the cytoskeletal organization and migration of glomerular endothelial cells facilitating the formation of a new vascular lumen [45]. A recent report has demonstrated that podocytes have a functional autocrine VEGF-A system, which is regulated by differentiation and ligand availability [46]. The function of VEGF-A in podocytes includes promoting cell survival through VEGFR2, inducing podocin upregulation and increasing nephrin/CD2AP interaction [46]. VEGF-A may also serve as a crucial growth factor in maintaining the normal function of podocytes by preventing their apoptosis through the interaction with nephrin and the activation of AKT signaling pathway [47]. Podocyte-derived VEGF-A is found to be decreased in sclerotic glomeruli [48], while treatment with exogenous VEGF-A decreases renal sclerotic injuries and restores glomerular capillaries [49]. In the present study, we found that Hcys treatment significantly decreased the production of VEGF-A in podocytes, which was restored by silencing of gp91phox gene or by inhibition of NADPH oxidase activity. It appears that elevated O2.− production induced by Hcys damages the normal function of podocytes to secrete this important growth and permeability factor in the glomeruli. Given the important role of VEGF in maintaining the normal function of podocytes and preventing their apoptosis, it is plausible that decreased nephrin expression, reduced VEGF excretion and subsequent disturbed VEGF-nephrin interaction induced by hHcys may contribute importantly to Hcys-induced podocyte depletion.
In summary, this is the first report showing that Hcys may directly cause podocyte injury and depletion, which are important events leading to glomerulosclerosis associated with hHcys. Using a gp91phox gene KO mouse model, we found that all hHcys-induced structural and functional changes reflecting podocyte injury were alleviated, such as proteinuria, reduced expression of podocin and nephrin, increased desmin level, foot process effacement, and podocyte loss. In cultured murine podocytes, we further demonstrated that silencing of gp91phox gene inhibited O2.− production induced by Hcys and recovered Hcys-induced decrease in secretion of VEGF-A. Collectively, these results demonstrate that a direct injurious effect of hHcys on podocytes is mainly mediated by a mechanism that involves generation of O2.− by the gp91phox-containing NADPH oxidase, which may represent a specific early mechanism mediating hHcys-induced glomerulosclerosis.
Acknowledgements
This study was supported by grants DK54927, HL075316, and HL57244 from National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.Robinson K, Gupta A, Dennis V, Arheart K, Chaudhary D, Green R, Vigo P, Mayer EL, Selhub J, Kutner M, Jacobsen DW. Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation. 1996;94:2743–2748. doi: 10.1161/01.cir.94.11.2743. [DOI] [PubMed] [Google Scholar]
- 2.Moustapha A, Gupta A, Robinson K, Arheart K, Jacobsen DW, Schreiber MJ, Dennis VW. Prevalence and determinants of hyperhomocysteinemia in hemodialysis and peritoneal dialysis. Kidney Int. 1999;55:1470–1475. doi: 10.1046/j.1523-1755.1999.00378.x. [DOI] [PubMed] [Google Scholar]
- 3.Ducloux D, Motte G, Challier B, Gibey R, Chalopin JM. Serum total homocysteine and cardiovascular disease occurrence in chronic, stable renal transplant recipients: a prospective study. J Am Soc Nephrol. 2000;11:134–137. doi: 10.1681/ASN.V111134. [DOI] [PubMed] [Google Scholar]
- 4.Yi F, Li PL. Mechanisms of homocysteine-induced glomerular injury and sclerosis. Am J Nephrol. 2008;28:254–264. doi: 10.1159/000110876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 6.Yi F, Zhang AY, Li N, Muh RW, Fillet M, Renert AF, Li PL. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006;70:88–96. doi: 10.1038/sj.ki.5001517. [DOI] [PubMed] [Google Scholar]
- 7.Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, Shah KS, Passmore JC, Tyagi SC. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297:F410–419. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L. Gp91phox-containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol Med. 2007;42:466–473. doi: 10.1016/j.freeradbiomed.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 10.Haque MZ, Majid DS. Assessment of renal functional phenotype in mice lacking gp91PHOX subunit of NAD(P)H oxidase. Hypertension. 2004;43:335–340. doi: 10.1161/01.HYP.0000111137.15873.4a. [DOI] [PubMed] [Google Scholar]
- 11.Haque MZ, Majid DS. Reduced renal responses to nitric oxide synthase inhibition in mice lacking the gene for gp91phox subunit of NAD(P)H oxidase. Am J Physiol Renal Physiol. 2008;295:F758–764. doi: 10.1152/ajprenal.90291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greiber S, Munzel T, Kastner S, Muller B, Schollmeyer P, Pavenstadt H. NAD(P)H oxidase activity in cultured human podocytes: effects of adenosine triphosphate. Kidney Int. 1998;53:654–663. doi: 10.1046/j.1523-1755.1998.00796.x. [DOI] [PubMed] [Google Scholar]
- 13.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whaley-Connell AT, Chowdhury NA, Hayden MR, Stump CS, Habibi J, Wiedmeyer CE, Gallagher PE, Tallant EA, Cooper SA, Link CD, Ferrario C, Sowers JR. Oxidative stress and glomerular filtration barrier injury: role of the reninangiotensin system in the Ren2 transgenic rat. Am J Physiol Renal Physiol. 2006;291:F1308–1314. doi: 10.1152/ajprenal.00167.2006. [DOI] [PubMed] [Google Scholar]
- 15.Guo J, Ananthakrishnan R, Qu W, Lu Y, Reiniger N, Zeng S, Ma W, Rosario R, Yan SF, Ramasamy R, D'Agati V, Schmidt AM. RAGE mediates podocyte injury in adriamycin-induced glomerulosclerosis. J Am Soc Nephrol. 2008;19:961–972. doi: 10.1681/ASN.2007101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagase M, Yoshida S, Shibata S, Nagase T, Gotoda T, Ando K, Fujita T. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–3446. doi: 10.1681/ASN.2006080944. [DOI] [PubMed] [Google Scholar]
- 17.Matsui I, Hamano T, Tomida K, Inoue K, Takabatake Y, Nagasawa Y, Kawada N, Ito T, Kawachi H, Rakugi H, Imai E, Isaka Y. Active vitamin D and its analogue, 22-oxacalcitriol, ameliorate puromycin aminonucleoside-induced nephrosis in rats. Nephrol Dial Transplant. 2009 doi: 10.1093/ndt/gfp117. [DOI] [PubMed] [Google Scholar]
- 18.Zou J, Yaoita E, Watanabe Y, Yoshida Y, Nameta M, Li H, Qu Z, Yamamoto T. Upregulation of nestin, vimentin, and desmin in rat podocytes in response to injury. Virchows Arch. 2006;448:485–492. doi: 10.1007/s00428-005-0134-9. [DOI] [PubMed] [Google Scholar]
- 19.Chen YF, Li PL, Zou AP. Effect of hyperhomocysteinemia on plasma or tissue adenosine levels and renal function. Circulation. 2002;106:1275–1281. doi: 10.1161/01.cir.0000027586.64231.1b. [DOI] [PubMed] [Google Scholar]
- 20.Li N, Chen L, Yi F, Xia M, Li PL. Salt-sensitive hypertension induced by decoy of transcription factor hypoxia-inducible factor-1alpha in the renal medulla. Circ Res. 2008;102:1101–1108. doi: 10.1161/CIRCRESAHA.107.169201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang G, Zhang F, Muh R, Yi F, Chalupsky K, Cai H, Li PL. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2007;292:H483–495. doi: 10.1152/ajpheart.00632.2006. [DOI] [PubMed] [Google Scholar]
- 22.Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, Li PL. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal. 2007;9:817–828. doi: 10.1089/ars.2007.1509. [DOI] [PubMed] [Google Scholar]
- 23.Yi F, dos Santos EA, Xia M, Chen QZ, Li PL, Li N. Podocyte injury and glomerulosclerosis in hyperhomocysteinemic rats. Am J Nephrol. 2007;27:262–268. doi: 10.1159/000101471. [DOI] [PubMed] [Google Scholar]
- 24.Iacobini C, Menini S, Oddi G, Ricci C, Amadio L, Pricci F, Olivieri A, Sorcini M, Di Mario U, Pesce C, Pugliese G. Galectin-3/AGE-receptor 3 knockout mice show accelerated AGE-induced glomerular injury: evidence for a protective role of galectin-3 as an AGE receptor. FASEB J. 2004;18:1773–1775. doi: 10.1096/fj.04-2031fje. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Zheng S, Epstein PN. Metallothionein over-expression in podocytes reduces adriamycin nephrotoxicity. Free Radic Res. 2009;43:174–182. doi: 10.1080/10715760802657308. [DOI] [PubMed] [Google Scholar]
- 26.Zheng S, Carlson EC, Yang L, Kralik PM, Huang Y, Epstein PN. Podocyte-specific overexpression of the antioxidant metallothionein reduces diabetic nephropathy. J Am Soc Nephrol. 2008;19:2077–2085. doi: 10.1681/ASN.2007080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang ZZ, Zou AP. Homocysteine enhances TIMP-1 expression and cell proliferation associated with NADH oxidase in rat mesangial cells. Kidney Int. 2003;63:1012–1020. doi: 10.1046/j.1523-1755.2003.00825.x. [DOI] [PubMed] [Google Scholar]
- 28.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 29.Manning RD, Jr., Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol. 2005;25:311–317. doi: 10.1159/000086411. [DOI] [PubMed] [Google Scholar]
- 30.Fujii M, Inoguchi T, Maeda Y, Sasaki S, Sawada F, Saito R, Kobayashi K, Sumimoto H, Takayanagi R. Pitavastatin ameliorates albuminuria and renal mesangial expansion by downregulating NOX4 in db/db mice. Kidney Int. 2007;72:473–480. doi: 10.1038/sj.ki.5002366. [DOI] [PubMed] [Google Scholar]
- 31.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 32.Yi F, Zhang AY, Janscha JL, Li PL, Zou AP. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int. 2004;66:1977–1987. doi: 10.1111/j.1523-1755.2004.00968.x. [DOI] [PubMed] [Google Scholar]
- 33.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 34.Lo W, Bravo T, Jadhav V, Titova E, Zhang JH, Tang J. NADPH oxidase inhibition improves neurological outcomes in surgically-induced brain injury. Neurosci Lett. 2007;414:228–232. doi: 10.1016/j.neulet.2006.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 36.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 37.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Kawachi H, Miyauchi N, Suzuki K, Han GD, Orikasa M, Shimizu F. Role of podocyte slit diaphragm as a filtration barrier. Nephrology (Carlton) 2006;11:274–281. doi: 10.1111/j.1440-1797.2006.00583.x. [DOI] [PubMed] [Google Scholar]
- 39.Lenz O, Elliot SJ, Stetler-Stevenson WG. Matrix metalloproteinases in renal development and disease. J Am Soc Nephrol. 2000;11:574–581. doi: 10.1681/ASN.V113574. [DOI] [PubMed] [Google Scholar]
- 40.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 41.Xiao H, Shi W, Liu S, Wang W, Zhang B, Zhang Y, Xu L, Liang X, Liang Y. 1,25-Dihydroxyvitamin D(3) Prevents Puromycin Aminonucleoside-Induced Apoptosis of Glomerular Podocytes by Activating the Phosphatidylinositol 3-Kinase/Akt-Signaling Pathway. Am J Nephrol. 2009;30:34–43. doi: 10.1159/000200769. [DOI] [PubMed] [Google Scholar]
- 42.Liu F, Wei CC, Wu SJ, Chenier I, Zhang SL, Filep JG, Ingelfinger JR, Chan JS. Apocynin attenuates tubular apoptosis and tubulointerstitial fibrosis in transgenic mice independent of hypertension. Kidney Int. 2009;75:156–166. doi: 10.1038/ki.2008.509. [DOI] [PubMed] [Google Scholar]
- 43.Jiang F. NADPH oxidase in the kidney: a Janus in determining cell fate. Kidney Int. 2009;75:135–137. doi: 10.1038/ki.2008.478. [DOI] [PubMed] [Google Scholar]
- 44.Marshall CB, Pippin JW, Krofft RD, Shankland SJ. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int. 2006;70:1962–1973. doi: 10.1038/sj.ki.5001965. [DOI] [PubMed] [Google Scholar]
- 45.Munoz-Chapuli R, Quesada AR, Angel Medina M. Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci. 2004;61:2224–2243. doi: 10.1007/s00018-004-4070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guan F, Villegas G, Teichman J, Mundel P, Tufro A. Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol. 2006;291:F422–428. doi: 10.1152/ajprenal.00448.2005. [DOI] [PubMed] [Google Scholar]
- 47.Foster RR, Saleem MA, Mathieson PW, Bates DO, Harper SJ. Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol. 2005;288:F48–57. doi: 10.1152/ajprenal.00146.2004. [DOI] [PubMed] [Google Scholar]
- 48.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang DH, Johnson RJ. Vascular endothelial growth factor: a new player in the pathogenesis of renal fibrosis. Curr Opin Nephrol Hypertens. 2003;12:43–49. doi: 10.1097/00041552-200301000-00008. [DOI] [PubMed] [Google Scholar]