

Abstract
Psychosocial stress alters susceptibility to infectious and systemic illnesses and may enhance airway inflammation in asthma by modulating immune cell function through neural and hormonal pathways. Stress activates the hypothalamic-pituitary-adrenal (HPA) axis. Release of endogenous glucocorticoids, as a consequence, may play a prominent role in altering the airway immune homeostasis. Despite substantial corticosteroid and catecholamine plasma levels, chronic psychosocial stress evokes asthma exacerbations.
Animal studies suggest that social stress induces corticosteroid insensitivity that in part may be due to impaired glucocorticoid receptor (GR) expression and/or function. Such mechanisms likely promote and amplify airway inflammation in response to infections, allergen or irritant exposure. This review discusses evidence of an altered corticosteroid responsive state as a consequence of chronic psychosocial stress. Elucidation of the mechanisms of stress-induced impairment of glucocorticoid responsiveness and immune homeostasis may identify novel therapeutic targets that could improve asthma management.
Keywords: Airway inflammation, psychosocial stress, corticosteroids, innate immune system
“…the regulation of gene expression by social factors makes all bodily functions…susceptible to social influences…” Eric Kandel, Nobel Laureate1
Introduction
Ethnic disparity in asthma prevalence, morbidity and mortality is highly correlated with low socioeconomic status and urban living, both of which are predisposing factors to psychosocial stress. Perception of stress in epidemiological studies associates with socioeconomic status and health2. Social stress was identified next to air pollution and urban living as one of the most significant contributors to asthma exacerbations3, 4. Predisposing factors to psychosocial stress such as low socioeconomic status also correlate with health care disparity in asthma prevalence, morbidity and mortality5. Both physical and social factors induce stress3, 4.
Evidence suggests that stressful experiences evoke asthma exacerbations6-12. Asthma patients respond with increased bronchial tone to distressful experiences13, listening to stressful interactions14 or exposure to asthma-related visual brain stimulation15. Further, school examinations induce asthma exacerbations in children16. The mechanisms of bronchoconstriction in response to such acute stress may include alterations in adrenergic (sympathetic) and cholinergic (parasympathetic) neural control. Asthma exacerbations were also noted in patients suffering from chronic depression and anxiety as well as in adults17 and children18 who had inadequate social support. For instance, greater caregiver-perceived stress was independently associated with subsequent risk of recurrent wheeze in early childhood and high levels of caregiver stress predicted increased wheezing in the index children19. Severely negative life events in another study increased the risk of asthma attacks in children, and this risk was magnified if the child's life situation was characterized by multiple chronic stressors20. Chronic caregiver stress in early childhood was also associated with the development of atopy while increased stress in the home predicted higher levels of IgE expression, enhanced allergen-specific lymphocyte proliferation, and differential cytokine expression in children21. The increase in asthma symptoms during chronic stress may be due to the combined effects of hormones, neurotransmitters, and neuropeptides involved in the autonomic control and inflammation of the airways. While stress may alter the magnitude of the airway inflammatory response elicited by allergens, irritants and/or infections in persons with asthma22, the precise mechanisms are unknown. This review discusses evidence that highlights the role of altered corticosteroid function in modulating airway inflammation due to social stress.
What is stress?
Hans (János) Selye defined stress as a state of altered homeostasis resulting from either an internal or external stimulus. Stressors can be physical or psychological in nature. All organisms experience stress, which by itself, is necessary for biological and social adaptation to the environment and for health23. The physiological response to stress involves the development of adaptive neuroendocrine mechanisms 24-26. These mechanisms are designed to counteract stress and to restore homeostasis. Stress, however, also evokes physiological changes that may potentially promote disease. Selye called this kind of stress “distress”: The reaction to noxious physical or psychological stimuli involves a short-term activation of the neuroendocrine and autonomic nervous systems that promotes adaptation and survival during the period of challenge. Such mechanisms, termed allostasis, literally mean “re-establishing stability through change”26. During allostasis, adaptive mechanisms operate at a higher or lower level than during “normal” homeostasis. For example, emotional distress elevates heart rate and blood pressure as well as elevating glucocorticoid levels and suppressing expression of inflammatory cytokines. Assuming allostatic responses cease when no longer needed, the organism adapts to and survives the immediate challenge without suffering long-term consequences. If, however, the same responses are repeatedly activated or persist over a longer period of time, receptor alterations and tissue damage occur that precipitate or exacerbate disease processes. This has been termed allostatic load26.
Acute stress, the “flight-or-fight response” (positive stress) is a response to a perceived threat or challenge that requires a short period of time in which to successfully adapt. Such stress evokes an immediate activation of the hypothalamic-pituitary-adrenal (HPA) axis. Hypothalamic corticotrophin-releasing hormone (CRH) stimulates adrenocorticotropic hormone (ACTH) secretion from the pituitary that in turn promotes the release of corticosteroids from the adrenal cortex. The HPA axis is one of the central mechanisms by which stress, detected by the brain, promotes adaptive physiological responses. Acute stressors, through the stimulation of the sympathetic nervous system, prepare the body for an immediate response and are beneficial. The HPA axis modulates immune/inflammatory responses to prevent such responses from becoming unchecked.
The aforementioned adaptive responses to acute stress stand in distinction from those of chronic stress, where the presence of prolonged stress induces maladaptive responses that promote disease. Traditionally, stress in clinical studies suggests chronic psychological (maladaptive) stimuli. Reduced social status, loss of a job, death of a loved one, long-term anxiety, repeated exposure to violence and depression4, 23, 27 are examples of such stressors. The clinical studies linking psychosocial factors with asthma severity are observational and cannot determine causality. Animal models may offer novel insight into the mechanisms relating psychosocial risk factors to physical health27.
Animal models of stress and asthma
To date, few studies have investigated the effects of stress on allergen-induced airway inflammation. For example, in a rat model of unpredictable changes in daily living conditions, increased edema and cellular influx were observed in inflamed paws and airways28. Repeated ultrasound-induced stress in ovalbumin-sensitized mice increased inflammatory cell number, IL-4, IL-529, TNFα30 and eotaxin31 in the airways. In pregnant BALB/c mice, maternal stress increased susceptibility of the offspring to allergic sensitization to ovalbumin 32.
A model of social disruption stressor (SDR) has been used to study the effects on allergic airway inflammation in our laboratory. Since this platform mimics human psychological stress, such an approach may be more relevant than exposure to sound, ultrasound or physical stressors. In SDR, the social hierarchy of a cohort of young male mice is disrupted by an older, aggressive intruder, and a stress response is elicited by repeated experience of defeat and loss of social status33-38. Exposure to psychological defeat as a consequence of confrontation is a relatively common experience in industrialized societies. Accordingly, epidemiological and psychosocial studies show that younger age, lower educational level, subordinate socioeconomic status or rank all lead to repeated experience of defeat and contribute to stress39-41. Social stressors may also modulate immune responses and predispose to asthma morbidity in humans4, 5, 27. Using the SDR model, we showed that combined exposure to allergen and repeated stress prolonged and enhanced the allergic airway response and reduced the glucocorticoid sensitivity of Th2 cytokine release by immune cells in vitro 42, 43. Recently, stress-related glucocorticoid resistance of Th2 cytokine production by T cells was also reported in asthmatic children who reported inadequate social support18.
The molecular mechanisms by which SDR enhances allergic airway inflammation remain unclear. Earlier findings in rodent models of stress revealed acute atrophy of the immune organs that increased susceptibility to viral or bacterial infections24, 25. Stress in the SDR model however was not immunosuppressive. We found increased spleen mass and enhanced p65 expression in the nuclear extract of the SDR mice, suggesting activation (not suppression) of the innate immune cells and Th2-type immune mediators. There are indications that both the duration (acute vs. chronic or repeated) and type of stressors can alter the effects of corticosteroids44. Opposing effects of short- and long-term stress on the immune system were suggested in models of restraint44 and the forced swim test45. In restraint stress mice are placed in ventilated Plexiglas restrainers [11.5 cm×3 cm (diameter) with 8–0.5 cm diameter air holes for 2 hours. In the forced swim stress model, mice are individually placed in 10 cm deep water (23-24°C) in an uncovered, cylindrical Plexiglas container, where they are required to swim or float for 2 minutes. When applied chronically (daily for 25 days), both the restraint and forced swim stress models enhanced inflammatory responses 45. Chida and colleagues showed that repeated psychological stress exacerbated allergic airway responses, and pretreatment of mice with the glucocorticoid receptor antagonists RU486 before allergen challenge, abolished the stress effects. On the other hand, in acute/short-term stress, airway inflammation46 and bone marrow eosinophilia in allergen-sensitized animals were inhibited, and this inhibition was abrogated by RU486 or metirapon47, suggesting that endogenous corticosteroids effectively suppress allergic inflammation. Accordingly, corticotropin-releasing hormone (CRH)-deficient mice experienced significantly enhanced cellular inflammation to allergic sensitization as compared with that of wild-type mice48.
Direct comparison of the SDR stressor with a repeated restraint stress paradigm also suggests differential effects on the immune function 49, 50. While the HPA axis and the sympathetic nervous systems were similarly activated by both stressors, repeated restraint disrupted the circadian rhythm of the HPA axis, and a prolonged elevation of circulating corticosterone levels with splenic hypotrophy, suppressed mononuclear cell function and cytokine production, 49 and these responses were restored with RU486 51. In comparison, repeated cycles of SDR elevated circulating corticosterone levels but did not disrupt the circadian rhythm of the HPA axis, induce splenic hypotrophy or suppress mononuclear cell activation 52. Further, development of glucocorticoid insensitivity of cytokine production by splenocytes 53 was observed in SDR but not the restraint stress model 54. Taken together, exposure to sound 29-32 or psychological stressors 55 enhances an allergic immune response while restraint stress suppresses it. Further, while acute stress evokes immunosuppression, after prolonged or repeated stress glucocorticoid insensitivity may develop. Although paradoxical, increases in circulating corticosterone due to repeated psychological stress may promote a loss of the anti-inflammatory properties of corticosteroids and in fact perpetuate airway inflammation55. Endogenous glucocorticoid levels can regulate the development, distribution and function of immune and structural cells 56-58 but the effects of stress-induced glucocorticoid release on airway immune function require further study.
Mechanisms of stress-induced glucocorticoid insensitivity: Role of the GR
Our studies42 showed that social stress prolonged the airway inflammation to allergen in mice including hyperresponsiveness to methacholine challenge, increased BAL eosinophil and lymphocyte numbers, and elevated levels of cytokines, chemokines and immunoglobulins, reflecting activation of both the innate and adaptive immune system. Importantly, heightened inflammation occurred in the presence of elevated serum corticosterone in mice exposed to social stress and allergen suggesting a diminished inhibitory action of endogenous corticosteroids42. Conceivably, repeated exposure to social stress skews the immune status towards a Th2 phenotype in the airways inducing a persistent and amplified inflammation after allergen challenge. Such changes, in part, may be due to endogenous corticosteroid insensitivity, mediated by decreased function and expression of the GR. In fact, in vitro splenocyte studies suggest that stress and allergen are synergistic in reversing the immunosuppressive action of corticosteroids on pro-inflammatory cytokine production by immune cells43. The combination of stress and allergen exposure also profoundly altered the in vitro corticosteroid responsiveness of LPS-stimulated splenocytes42. Investigators previously showed that stress-induced disruption of the glucocorticoid action was associated with a failure of the GR to translocate to the nucleus 53, 59, and GR binding to glucocorticoid response elements in the nuclear fractions of splenocytes was attenuated42. Stress alone significantly inhibited GR binding to glucocorticoid response elements 53, 59 but additional Af exposure of the stressed mice did not increase this effect42. Thus, the mechanism by which the synergistic reversal of corticosteroid action occurs in LPS-stimulated cells from the allergen- and stress-exposed mice remains unclear.
Since constitutive GR expression is essential for an adequate glucocorticoid action, we also investigated GR expression both in the spleen and the lung of mice exposed to stress, allergen or the combination of the two. GR mRNA and protein expression was significantly inhibited in the allergen- and stress-exposed group in comparison with the allergen-sensitized control mice42. We speculate that an impaired nuclear translocation and DNA binding together with reduced GR mRNA and protein expression may in part explain the observed alterations in corticosteroid responsiveness. Although evidence suggests that stress modulates AHR and airway inflammation in mice 29, 30, 32, 44, 46-48, 55, 60, our recent study is the first demonstration that social stress enhanced the allergic airway response in association with innate immune cell corticosteroid insensitivity and impaired GR translocation and expression. GR expression is regulated by complex transcriptional and post translational mechanisms that can be significantly modified by inflammatory processes during airway inflammation61. Indeed reduced GR expression was reported in asthmatic and COPD patients with insensitivity to corticosteroid treatment62, 63. While these findings may point to potential mechanisms of asthma exacerbation and diminished effectiveness of corticosteroid therapies a number of questions remain unanswered. For instance, what is the mechanism of reduced GR expression in the lung? While GR expression is likely to be reduced by homologous ligand down-regulation (by GR agonists) during the combination of stress and allergic inflammation, other pathways may be also operational. Specifically, the human GRα isoform can be transrepressed by NF-κB, a mechanism that could mediate steroid insensitivity in the inflamed tissue62, 64. Interestingly, ligand-induced GR down regulation is a significant mechanism in various tissues and cell types but not in T lymphocytes64. Thus, innate immune cells may be important. The exact cell types that are ultimately responsible for mediating the effects of corticosteroid insensitivity in the lung however still need to be identified. It is also unclear whether low levels of GR mRNA are due to suppression of promoter activation, decreased mRNA stability, or both64. Further, the expression and function of the human GR are distinct from that of the mouse. For example, it is unclear whether transrepression of the GRα by NF- κB plays a role in corticosteroid resistance in mice. Existence of the dominant negative GRβ isoform (a genetic variant of the GR in humans that was also implied in glucocorticoid resistance61, 64, 65) could not be demonstrated in mice either.
GR signaling and glucocorticoid resistance in airway inflammation
In the canonical signaling pathway, glucocorticoid binding to the intracellular receptor (GR) induces molecular rearrangement of the GR-HSP90 heterocomplex and promotes GR nuclear localization, homodimerization, and DNA-binding. The activated GR homodimer exerts its genomic effect by interacting with specific DNA elements, termed glucocorticoid response elements (GRE), located within regulatory regions of glucocorticoid-responsive genes (Figure 1). After recruitment of co-activators or co-repressors, the GR modulates the rate of gene transcription in the nucleus by transactivation or transrepression. Transactivation is an increased rate of gene expression triggered by GRE which acts in “trans”, i.e. intermolecularly (this may be considered the opposite of “cis”-acting i.e., intramolecular). On the other hand, transrepression is a process whereby one protein represses (i.e., inhibits) the activity of a second protein through a protein-protein interaction66. The protein that is repressed is usually a transcription factor whose function is to up-regulate the rate of gene transcription. Hence the net result of transrepression is down regulation of gene transcription67. The phenomenon of transrepression was first observed in the ability of the glucocorticoid receptor to inhibit the transcriptional promoting activity of the AP-1 and NF-κB transcription factors. Both transactivation and transrepression are important in mediating the anti-inflammatory effects of glucocorticoids.
Figure 1.
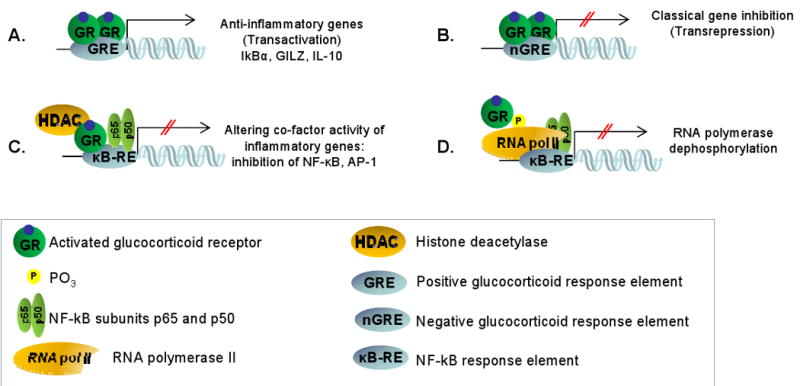
Mechanisms of glucocorticoid action. The activated glucocorticoid receptor (GR) homodimer binds to glucocorticoid response elements (GRE), located within regulatory regions of glucocorticoid-responsive genes in the nucleus. (A): Positive GRE mediates transcriptional up-regulation of anti-inflammatory genes such as IκBα, the glucocorticoid-inducible leucine zipper (GILZ) or IL-10. B: Negative GRE mediates transcriptional down-regulation. C-D: In monomeric form the activated GR may interact with other transcription factors such as nuclear factor κB (NFκB). (C): This interaction can occur indirectly through transcriptional co-factor (HDAC) binding or (D): by RNA polymerase dephosphorylation.
Transactivation GRE up-regulates anti-inflammatory genes such as the NF-κB inhibitor IκBα, the AP-1 inhibitor glucocorticoid-inducible leucine zipper (GILZ) or IL-10 (Figure 1A). Transrepression GRE mediates transcriptional down-regulation (Figure 1B). In its monomeric form, the activated GR can also modulate transcription independent of the canonical GRE pathway68. For example, GR physically interacts with other transcription factors (a mechanism called “tethering”), such as nuclear factor κB (NF-κB), activator protein-1 (AP-1), signal transducers and activators of transcription (STAT) or CAAT Enhancer Binding Protein (C/EBP), and modulates activation of target genes69, 70. Figure 1C illustrates that the activated monomeric GR with the help of HDAC (histone deacetylase), can interfere with the p65 and p50 NF-κB heterodimer activation of the κB responsive element (κB-RE). Although the main function of HDACs in the context of regulating gene transcription is to modify histones and chromatin structure, different HDAC isoforms were identified to have various cytoplasmic and nuclear regulatory functions. HDAC1 for example is considered to be a transcriptional co-activator71 and impaired HDAC2 function has been implicated in corticosteroid resistance of asthmatic and COPD patients72. Phosphorylation-dephosphorylation is an important mechanism in the function of the transcription regulator enzyme, RNA polymerase II. Figure 1D shows that the GR may interfere with transcription activation through dephosphorylating RNA polymerase II70.
GR expression and function are regulated by transcriptional and post translational mechanisms64 such as kinase-dependent phosphorylation as well as by homologous ligand down-regulation (by GR agonists) that can be significantly modified by stress and increased NF-κB expression61. Our studies42 showed that SDR enhanced expression of NF-κB in the nuclear fraction of immune cells parallel with an impairment of GR nuclear translocation, DNA binding and a decrease in the expression of GR. A mutual transrepression between the GR and NF-κB maybe responsible for mediating steroid insensitivity in the inflamed airways in asthma62-64. Mechanisms by which NF-kB can affect glucocorticoid responsiveness are illustrated in Figure 2. Glucocorticoid binding to the GR induces molecular rearrangement of the GR-HSP90 heterocomplex and promotes GR nuclear translocation, homodimerization, and DNA-binding. These processes can be modulated by GR/NF-κB interactions. NF-kB for example can directly interefere with GRE-mediated transactivation as well as transrepression (Figure 2A)53. Indirect inhibition of the GR function maybe achieved by transcription factor “tethering” (Figure 2B). Thus, GR/NF-κB interactions regulate GR expression, modulate nuclear translocation of the GR and affect GRE-mediated function as well as transcription factor “tethering” in the inflamed airways. It is important to note that while the GR is ubiquitously expressed, regulation of the GR gene leads to differences in the expression of GR transcript and protein in individual cell types73, 74. Cell type-specific increase in NF-κB for example in dendritic cells, may significantly inhibit GR expression and modulate allergic airway inflammation during stress.
Figure 2.
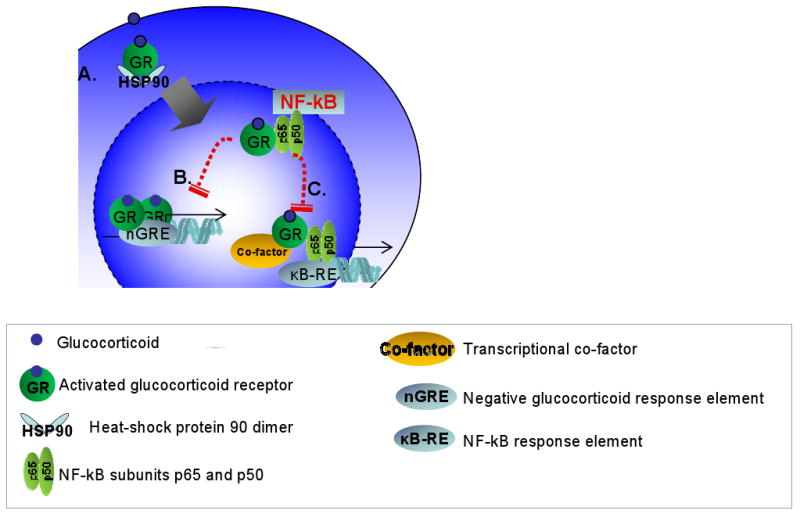
Hypothesized mechanisms by which glucocorticoid resistance could occur. (A): Glucocorticoid binding to the GR induces molecular rearrangement of the GR-HSP90 heterocomplex and promotes GR nuclear translocation, homodimerization, and DNA-binding. (B): This process can be modulated by GR/NF-κB interactions, directly by affecting transactivation (GRE-mediated) as well as transrepression of GR functions. Indirect inhibition of the GR function maybe achieved by transcription factor “tethering”.
Role of the innate and adaptive immune system in stress and the asthmatic response
Altered innate immune cell-T cell interactions may play a role in stress-induced enhancement of immune responses. After interaction with the antigen-presenting cells and recognition of the antigen, naive CD4 T cells rapidly undergo a differentiation process that promotes development of four functionally distinct cell subsets characterized by a mutually exclusive pattern of transcription factor and cytokine secretion. Th1 cells express Tbet and secrete IFNγ while Th2 cells are GATA3 positive, produce IL-4, IL-5, IL-9, and IL-1375-77. The proinflammatory Th17 cells express Rorγt and produce IL-17 while the Foxp3 positive regulatory (Treg) T cells release IL-10 and TGFβ and exert profound immunosuppressive activities. A critical role of CD4 T cells in the immune response to allergens has been supported by evidence including previous studies on asthmatic patients and animal models76-79. We have shown that T cell activation in asthmatic patients 80, 81 and in a Brown-Norway rat model of allergic airway inflammation82, 83 was associated with production of IL-4 and IL-5, but not IFNγ82-84 and that an asthmatic phenotype was transferable from sensitized to naïve animals by CD4 T cells85. Our studies also demonstrated that deficiency of either IL-4 or IL-5 abolished allergic airway inflammation86 while overexpression of IL-587 or IL-988, 89 was responsible for exaggerated eosinophilia, mucus production and airway hyperresponsiveness.
Cell-cell interactions provided by dendritic cells (through MHC-II and CD86) and paracrine secretion of proinflammatory cytokines90 are the main factors influencing CD4 T cell differentiation that determine the kinetics and magnitude of airway inflammation. Dendritic cells within the bronchial and alveolar wall cannot present antigen or stimulate T cells in their resting state. In response to allergen exposure however, proinflammatory, maturing dendritic cell precursors migrate from the bone marrow to the airway epithelium and evoke Th2-cell activation. These CD11b/MHC-II/CD86 positive myeloid dendritic cells have central importance in promoting airway inflammation and can be distinguished from the more tolerogenic plasmacytoid dendritic cells by membrane markers and intracellular mediators.
Although stress may impair asthma control 6-12, 15, 91, few studies address whether stress directly modulates immune function in disease. Our studies showed that levels of the immunoglobulin IgG1 and TARC (CCL17), a chemokine produced by dendritic cells and macrophages and responsible for attracting Th2 lymphocytes and activated dendritic cells 92, were enhanced in mice exposed to stress alone. Both IgG1 93 and TARC 92 are regulated by IL-4/IL-13 and are part of the Th2 immune response. Since these cytokine/chemokine and immunoglobulin changes occurred without the presence of antigenic stimulation, we hypothesized that alterations in innate immune cell function played an important role in mediating stress effects on airway inflammation. TNFα and IL-6 promote allergen-induced inflammation but also LPS-stimulated innate immune response. Therefore levels of TNFα and IL-6 are biomarkers of the activation state of innate immune cells both in the lung (in vivo) and in the spleen (in vitro). Activation of dendritic cells and macrophages 94 also contributes to elevating TNFα and IL-6 in mice that received Af+SDR.
Because TARC and IgG1 can also characterize Th2-type immune responses, we also raised the question whether animals exposed to repeated social stress would experience an increased susceptibility to developing allergic airway inflammation 95-98. Combined exposure to allergen and stress significantly enhanced the numbers of eosinophils and lymphocytes in the airways. These cells will interact during the allergen-induced airway inflammation through the T lymphocyte-derived IL-4, IL-5 and GM-CSF, which are essential for promoting allergen-induced tissue eosinophilia. The genes for these cytokines are located in a cluster of chromosome 5q and are sensitive to corticosteroid regulation. The enhanced airway eosinophilia 48 h after allergen challenge was paralleled by increased release of the eosinopoietic IL-5 and GM-CSF, as well as IgG1 and TARC in the airways of mice exposed to both stress and allergen challenge.
Previous studies suggest that in response to LPS-stimulation, the dendritic cells derived from mice exposed to social stress, released increased levels of TNFα and IL-6 as compared with control animals. Interestingly, CD11c+/CD11b+ dendritic cells42, 99 were also less sensitive to the inhibitory effects of glucocorticoids than cells from control mice33, 35, 52-54, 100 and expressed increased levels of NF-κB. Thus, myeloid dendritic cells may become primed during stress by activating NF-κB and by nuclear translocation of p65. Although the exact mechanism remains unclear, it is plausible that increased corticosterone induces GR downregulation in these cells73, 101, 102. Because activated GR constitutively inhibits NF-κB function via several different mechanisms (see above), a decrease in GR expression may promote NF-κB upregulation and activation. Dendritic cells “primed” by stress then become less responsive to corticosteroids. Our studies show that dendritic cell priming is amplified in the presence of allergic inflammation. Since enhancement of allergic airway inflammation in part requires interaction among dendritic cells and T cells of the submucosal airway tissue, dendritic cells and/or T cells may also be necessary for the glucocorticoid insensitivity seen after exposure to stress. It is also possible that stress and asthma-induced immune cell glucocorticoid insensitivity are due to aberrant glucocorticoid receptor-NF-κB interaction.
Role of the epithelial product SP-D in asthmatic inflammation
Stress and asthma may also affect glucocorticoid responsiveness of structural cells of the airways, such as epithelial and smooth muscle cells. These cells play an important orchestrating role and participate both in the initiation and resolution of the asthmatic airway response. An interesting potential link between the innate immune system and stress-induced modifications in corticosteroid responsive epithelial cell functions could be the hydrophilic epithelial product, surfactant protein D (SP-D)103-105.
SP-D is a member of the collectin family, composed of an N-terminal collagen and a C-terminal lectin domain. This protein is a constitutive mediator of antigen clearance and is capable of interacting with cellular components of both the innate and adaptive immune systems on mucosal surfaces105. SP-D is synthesized and secreted primarily by type II alveolar epithelial cells, Clara cells and cells of submucosal glands in the lung. We have recently shown that patients suffering from chronic obstructive pulmonary disease expressed significantly lower levels of SP-D in their BAL than healthy controls106. These studies also indicated that corticosteroid treatment significantly increased SP-D levels. It is possible that the beneficial effects of glucocorticoid therapy are partially mediated by enhanced SP-D expression in the lung. Indeed a cohort of evidence now supports the potent immunosuppressive activities of this lung collectin107. In vivo studies on SP-D deficient mice demonstrated enhanced susceptibility to inflammation in infectious and inflammatory models, including allergic airway sensitization105 and showed that one of the most prominent pathological characteristics in the SP-D deficient lung is the abnormal activation of alveolar macrophages and dendritic cells105, 108. That SP-D plays a protective role on immune cells was confirmed using treatment with recombinant protein that suppressed activation of lymphocytes98 and dendritic cells108 and inhibited airway inflammation in vivo.
In response to acute inflammatory stimuli there is a protective surge in SP-D release in the airways95, 109. Although the exact transcriptional activation pathways responsible for enhanced SP-D synthesis are not known, proinflammatory cytokines, second messengers as well as corticosteroids have been implicated106, 110. In a recent study from our laboratory mice receiving allergic sensitization as well as repeated social stress produced significantly less SP-D in response to allergen inhalation than control (non-stressed) animals111. We propose that stress-induced altered corticosteroid responsiveness of epithelial cells of the airways may inhibit SP-D production (Figure 3A). Stress also abrogates corticosteroid responsiveness of proinflammatory dendritic cells by increasing NF-kB expression and function (Figure 3B). Diminished SP-D levels will then result in a failure of protection against dendritic cell and Th2-cell activation and the consequent allergic airway response (Figure 3C). The clinical relevance of these studies may foster the development of novel immune modulatory agents that will effectively treat airways inflammation in a glucocorticoid-insensitive state.
Figure 3.
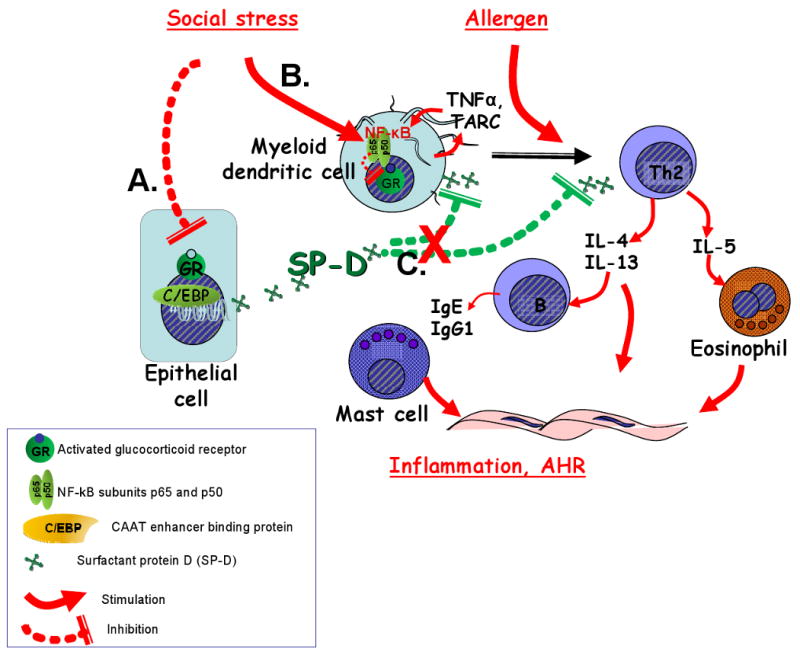
Proposed mechanism of how stress-induced glucocorticoid insensitivity could affect the innate immune system during the allergic airway response. (A): Altered corticosteroid responsiveness of airway epithelial cells may inhibit SP-D production. (B): Stress abrogates corticosteroid responsiveness of proinflammatory dendritic cells. (C): Diminished SP-D levels will result in a failure to protect against dendritic cell and Th2-cell activation and the consequent allergic airway response.
Therapeutic relevance
When inhaled corticosteroids appear ineffective in the treatment of asthma, health care providers often impugn that the patients are nonadherent112. Other explanations, however, may be operative. Potentially, the patients are adherent to therapy, yet given environmental stress, there may exist a transient glucocorticoid-insensitive state that renders such therapy ineffective113. Although asthma exacerbation has long been thought to be affected by emotional states, evidence has only recently emerged to identify cellular and molecular mechanisms responsible for stress-induced glucocorticoid insensitivity.
For example it is conceivable that impaired SP-D production by epithelial cells in the lung contributes to the enhancement of allergic airway inflammation during stress and that therapeutic replacement or induction of SP-D will be a potentially effective adjunct or alternative approach. It is unlikely however that a reduced SP-D synthesis is solely responsible for the stress-induced changes in the pulmonary innate immune system and the significance of altered corticosteroid responsiveness of airway epithelial cells during social stress needs further clarification.
As discussed, NF-κB activation, a pivotal signaling mechanism, mediates airway inflammation. Accordingly, IKK2 inhibitors or potentially p38 MAP kinase inhibitors could serve as nonsteroidal anti-inflammatory agents that could be effective in stress-enhanced airway inflammation. In parallel studies, we have shown that after exposure to IFNγ and TNFα, human airway smooth muscle cells and human precision cut lung slices become glucocorticoid-insensitive114, yet IKK2 inhibitors and p38 MAP kinase inhibitors remain effective in decreasing cytokine-induced chemokine/cytokine secretion 114-116. Undoubtedly, more work is necessary in this area; however, the identification of novel therapeutic targets to overcome stress-mediated glucocorticoid insensitivity may offer substantial value in the management of asthma.
Conclusions
Stress profoundly affects the course of airway inflammation. In asthma, evidence now suggests that social stress prolongs allergen-induced inflammatory changes, increases asthma exacerbations and contributes to asthma morbidity and mortality. Recent studies highlighted the role of altered corticosteroid responsiveness of T cells, dendritic cells and airway epithelial cells in stress-induced enhancement of inflammation. Central to this process is the shift in the balance of the mutual transrepression of the GR and NF-κB that results in steroid insensitivity and an increase in immune cell activation. Further studies are necessary to identify the therapeutic targets that can ameliorate stress effects on the immune system so that the “allostatic function” maybe restored.
Acknowledgments
Declaration of all sources of funding: Dr Haczku was receiving grant support from NIH R01AI055593, R01HL076646 and ALA CI. Dr Panettieri was supported by the Mind, Body, Brain, and Health Initiative.
Abbreviations
- ACTH
Adrenocorticotropic hormone
- Af
Aspergillus fumigatus
- AHR
Airway hyperreactivity
- AP-1
Activator protein 1
- BAL
Bronchoalveolar lavage
- CD4
Cluster of differentiation 4
- C/EBP
CCAAT enhancer binding protein
- CRH
Corticotrophin-releasing hormone
- Foxp3
forkhead box P3
- GILZ
Glucocorticoid-inducible leucine zipper
- GM-CSF
Granulocyte macrophage colony stimulating factor
- GR
Glucocorticoid receptor
- GRE
Glucocorticoid response elements
- HPA
Hypothalamic-pituitary-adrenal axis
- HSP90
Heat shock protein 90
- IFN
Interferon
- IκBα
NF-κB inhibitor α
- IL
Interleukin
- LPS
Lipopolysaccharide
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Rorγt
Retinoid-related orphan receptor γt
- RU486
Mifepristone (11β-[p-(Dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one)
- SDR
Social disruption stressor
- SP
Surfactant protein
- STAT
Signal transducers and activators of transcription
- TARC
Thymus and activation regulated chemokine
- Tbet
T box expressed in T cells
- TGF
Transforming growth factor
- Th
T helper
- TNF
Tumor necrosis factor
Footnotes
Clinical implications: Social stress prolongs allergen-induced airway inflammation, increases asthma exacerbations and contributes to asthma morbidity and mortality.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kandel ER. A new intellectual framework for psychiatry. Am J Psychiatry. 1998;155:457–69. doi: 10.1176/ajp.155.4.457. [DOI] [PubMed] [Google Scholar]
- 2.Adler NE, Boyce T, Chesney MA, Cohen S, Folkman S, Kahn RL, et al. Socioeconomic status and health. The challenge of the gradient. Am Psychol. 1994;49:15–24. doi: 10.1037//0003-066x.49.1.15. [DOI] [PubMed] [Google Scholar]
- 3.Wright RJ, Subramanian SV. Advancing a multilevel framework for epidemiologic research on asthma disparities. Chest. 2007;132:757S–69S. doi: 10.1378/chest.07-1904. [DOI] [PubMed] [Google Scholar]
- 4.Wright RJ. Health effects of socially toxic neighborhoods: the violence and urban asthma paradigm. Clin Chest Med. 2006;27:413–21. doi: 10.1016/j.ccm.2006.04.003. v. [DOI] [PubMed] [Google Scholar]
- 5.Subramanian SV, Ackerson LK, Subramanyam MA, Wright RJ. Domestic violence is associated with adult and childhood asthma prevalence in India. Int J Epidemiol. 2007;36:569–79. doi: 10.1093/ije/dym007. [DOI] [PubMed] [Google Scholar]
- 6.Marshall GD. Neuroendocrine mechanisms of immune dysregulation: applications to allergy and asthma. Ann Allergy Asthma Immunol. 2004;93:S11–7. doi: 10.1016/s1081-1206(10)61482-2. [DOI] [PubMed] [Google Scholar]
- 7.Miller BD, Wood BL. Emotions and family factors in childhood asthma: psychobiologic mechanisms and pathways of effect. Adv Psychosom Med. 2003;24:131–60. doi: 10.1159/000073785. [DOI] [PubMed] [Google Scholar]
- 8.Marshall GD, Jr, Agarwal SK. Stress, immune regulation, and immunity: applications for asthma. Allergy Asthma Proc. 2000;21:241–6. doi: 10.2500/108854100778248917. [DOI] [PubMed] [Google Scholar]
- 9.Badoux A, Levy DA. Psychologic symptoms in asthma and chronic urticaria. Ann Allergy. 1994;72:229–34. [PubMed] [Google Scholar]
- 10.Lehrer PM, Isenberg S, Hochron SM. Asthma and emotion: a review. J Asthma. 1993;30:5–21. doi: 10.3109/02770909309066375. [DOI] [PubMed] [Google Scholar]
- 11.Isenberg SA, Lehrer PM, Hochron S. The effects of suggestion and emotional arousal on pulmonary function in asthma: a review and a hypothesis regarding vagal mediation. Psychosom Med. 1992;54:192–216. doi: 10.1097/00006842-199203000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Miller BD. Depression and asthma: a potentially lethal mixture. J Allergy Clin Immunol. 1987;80:481–6. doi: 10.1016/0091-6749(87)90080-7. [DOI] [PubMed] [Google Scholar]
- 13.Beggs PJ, Curson PH. An integrated environmental asthma model. Arch Environ Health. 1995;50:87–94. doi: 10.1080/00039896.1995.9940884. [DOI] [PubMed] [Google Scholar]
- 14.Kolbe J, Garrett J, Vamos M, Rea HH. Influences on trends in asthma morbidity and mortality: the New Zealand experience. Chest. 1994;106:211S–5S. doi: 10.1378/chest.106.4_supplement.211s. [DOI] [PubMed] [Google Scholar]
- 15.Rosenkranz MA, Busse WW, Johnstone T, Swenson CA, Crisafi GM, Jackson MM, et al. From The Cover: Neural circuitry underlying the interaction between emotion and asthma symptom exacerbation. Proc Natl Acad Sci U S A. 2005;102:13319–24. doi: 10.1073/pnas.0504365102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu LY, Coe CL, Swenson CA, Kelly EA, Kita H, Busse WW. School examinations enhance airway inflammation to antigen challenge. Am J Respir Crit Care Med. 2002;165:1062–7. doi: 10.1164/ajrccm.165.8.2109065. [DOI] [PubMed] [Google Scholar]
- 17.Miller BD, Wood BL. Psychophysiologic reactivity in asthmatic children: a cholinergically mediated confluence of pathways. J Am Acad Child Adolesc Psychiatry. 1994;33:1236–45. doi: 10.1097/00004583-199411000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Miller GE, Gaudin A, Zysk E, Chen E. Parental support and cytokine activity in childhood asthma: the role of glucocorticoid sensitivity. J Allergy Clin Immunol. 2009;123:824–30. doi: 10.1016/j.jaci.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 19.Wright RJ, Cohen S, Carey V, Weiss ST, Gold DR. Parental stress as a predictor of wheezing in infancy: a prospective birth-cohort study. Am J Respir Crit Care Med. 2002;165:358–65. doi: 10.1164/ajrccm.165.3.2102016. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg S, Paton JY, Ahola S, McCann DC, McGuinness D, Hillary CR, et al. The role of acute and chronic stress in asthma attacks in children. Lancet. 2000;356:982–7. doi: 10.1016/S0140-6736(00)02715-X. [DOI] [PubMed] [Google Scholar]
- 21.Wright RJ, Finn P, Contreras JP, Cohen S, Wright RO, Staudenmayer J, et al. Chronic caregiver stress and IgE expression, allergen-induced proliferation, and cytokine profiles in a birth cohort predisposed to atopy. J Allergy Clin Immunol. 2004;113:1051–7. doi: 10.1016/j.jaci.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 22.Chen E, Miller GE. Stress and inflammation in exacerbations of asthma. Brain Behav Immun. 2007;21:993–9. doi: 10.1016/j.bbi.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vig RS, Forsythe P, Vliagoftis H. The role of stress in asthma: insight from studies on the effect of acute and chronic stressors in models of airway inflammation. Ann N Y Acad Sci. 2006;1088:65–77. doi: 10.1196/annals.1366.023. [DOI] [PubMed] [Google Scholar]
- 24.Selye H. A syndrome produced by diverse nocuous agents. Nature. 1936;138:32. doi: 10.1176/jnp.10.2.230a. [DOI] [PubMed] [Google Scholar]
- 25.Selye H. A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci. 1998;10:230–1. doi: 10.1176/jnp.10.2.230a. [DOI] [PubMed] [Google Scholar]
- 26.McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- 27.Miller G, Chen E, Cole SW. Health psychology: developing biologically plausible models linking the social world and physical health. Annu Rev Psychol. 2009;60:501–24. doi: 10.1146/annurev.psych.60.110707.163551. [DOI] [PubMed] [Google Scholar]
- 28.Datti F, Datti M, Antunes E, Teixeira NA. Influence of chronic unpredictable stress on the allergic responses in rats. Physiol Behav. 2002;77:79–83. doi: 10.1016/s0031-9384(02)00811-9. [DOI] [PubMed] [Google Scholar]
- 29.Joachim RA, Quarcoo D, Arck PC, Herz U, Renz H, Klapp BF. Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 2003;65:811–5. doi: 10.1097/01.psy.0000088582.50468.a3. [DOI] [PubMed] [Google Scholar]
- 30.Joachim RA, Sagach V, Quarcoo D, Dinh T, Arck PC, Klapp BF. Upregulation of tumor necrosis factor-alpha by stress and substance p in a murine model of allergic airway inflammation. Neuroimmunomodulation. 2006;13:43–50. doi: 10.1159/000094394. [DOI] [PubMed] [Google Scholar]
- 31.Joachim RA, Sagach V, Quarcoo D, Dinh QT, Arck PC, Klapp BF. Effect of stress on eotaxin and expression of adhesion molecules in a murine model of allergic airway inflammation. J Neuroimmunol. 2007;182:55–62. doi: 10.1016/j.jneuroim.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Pincus-Knackstedt MK, Joachim RA, Blois SM, Douglas AJ, Orsal AS, Klapp BF, et al. Prenatal stress enhances susceptibility of murine adult offspring toward airway inflammation. J Immunol. 2006;177:8484–92. doi: 10.4049/jimmunol.177.12.8484. [DOI] [PubMed] [Google Scholar]
- 33.Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H, et al. Expression of glucocorticoid resistance following social stress requires a second signal. J Leukoc Biol. 2003;74:507–13. doi: 10.1189/jlb.0303090. [DOI] [PubMed] [Google Scholar]
- 34.Avitsur R, Stark JL, Dhabhar FS, Padgett DA, Sheridan JF. Social disruption-induced glucocorticoid resistance: kinetics and site specificity. J Neuroimmunol. 2002;124:54–61. doi: 10.1016/s0165-5728(02)00010-3. [DOI] [PubMed] [Google Scholar]
- 35.Avitsur R, Stark JL, Dhabhar FS, Sheridan JF. Social stress alters splenocyte phenotype and function. J Neuroimmunol. 2002;132:66–71. doi: 10.1016/s0165-5728(02)00310-7. [DOI] [PubMed] [Google Scholar]
- 36.Eells JB, Misler JA, Nikodem VM. Early postnatal isolation reduces dopamine levels, elevates dopamine turnover and specifically disrupts prepulse inhibition in Nurr1-null heterozygous mice. Neuroscience. 2006;140:1117–26. doi: 10.1016/j.neuroscience.2005.12.065. [DOI] [PubMed] [Google Scholar]
- 37.Sonoda J, Chida Y, Sudo N, Kubo C. Social disruption stress exacerbates alpha-galactosylceramide-induced hepatitis in mice. Neuroimmunomodulation. 2005;12:375–9. doi: 10.1159/000091131. [DOI] [PubMed] [Google Scholar]
- 38.Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21:458–66. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeVries AC, Glasper ER, Detillion CE. Social modulation of stress responses. Physiol Behav. 2003;79:399–407. doi: 10.1016/s0031-9384(03)00152-5. [DOI] [PubMed] [Google Scholar]
- 40.Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39:247–57. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- 41.Yeh WY, Cheng Y, Chen CJ. Social patterns of pay systems and their associations with psychosocial job characteristics and burnout among paid employees in Taiwan. Soc Sci Med. 2009;68:1407–15. doi: 10.1016/j.socscimed.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 42.Bailey MT, Kierstein S, Sharma S, Spaits M, Kinsey SG, Tliba O, et al. Social stress enhances allergen-induced airway inflammation in mice and inhibits corticosteroid responsiveness of cytokine production. J Immunol. 2009;182:7888–96. doi: 10.4049/jimmunol.0800891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey MT, Kierstein S, Spaits M, Sheridan JF, Panettieri RAJ, Haczku A. Social Stress Enhances Allergen-Induced Airway Inflammation in Mice. Am J Respir Crit Care Med. 2006:A237. doi: 10.4049/jimmunol.0800891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okuyama K, Ohwada K, Sakurada S, Sato N, Sora I, Tamura G, et al. The distinctive effects of acute and chronic psychological stress on airway inflammation in a murine model of allergic asthma. Allergol Int. 2007;56:29–35. doi: 10.2332/allergolint.O-06-435. [DOI] [PubMed] [Google Scholar]
- 45.Bowers SL, Bilbo SD, Dhabhar FS, Nelson RJ. Stressor-specific alterations in corticosterone and immune responses in mice. Brain Behav Immun. 2008;22:105–13. doi: 10.1016/j.bbi.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forsythe P, Ebeling C, Gordon JR, Befus AD, Vliagoftis H. Opposing effects of short- and long-term stress on airway inflammation. Am J Respir Crit Care Med. 2004;169:220–6. doi: 10.1164/rccm.200307-979OC. [DOI] [PubMed] [Google Scholar]
- 47.Georen S, Ahnblad P, Stjarne P, Wikstrom AC, Stierna P. Significance of endogenous glucocorticoid sensitivity for airway eosinophilia in a murine model of allergy. Acta Otolaryngol. 2005;125:378–85. doi: 10.1080/00016480410025261. [DOI] [PubMed] [Google Scholar]
- 48.Silverman ES, Breault DT, Vallone J, Subramanian S, Yilmaz AD, Mathew S, et al. Corticotropin-releasing hormone deficiency increases allergen-induced airway inflammation in a mouse model of asthma. J Allergy Clin Immunol. 2004;114:747–54. doi: 10.1016/j.jaci.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 49.Hermann G, Tovar CA, Beck FM, Sheridan JF. Kinetics of glucocorticoid response to restraint stress and/or experimental influenza viral infection in two inbred strains of mice. J Neuroimmunol. 1994;49:25–33. doi: 10.1016/0165-5728(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 50.Dobbs CM, Feng N, Beck FM, Sheridan JF. Neuroendocrine regulation of cytokine production during experimental influenza viral infection: effects of restraint stress-induced elevation in endogenous corticosterone. J Immunol. 1996;157:1870–7. [PubMed] [Google Scholar]
- 51.Hermann G, Beck FM, Sheridan JF. Stress-induced glucocorticoid response modulates mononuclear cell trafficking during an experimental influenza viral infection. J Neuroimmunol. 1995;56:179–86. doi: 10.1016/0165-5728(94)00145-e. [DOI] [PubMed] [Google Scholar]
- 52.Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol. 2004;148:106–15. doi: 10.1016/j.jneuroim.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 53.Quan N, Avitsur R, Stark JL, He L, Lai W, Dhabhar F, et al. Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol. 2003;137:51–8. doi: 10.1016/s0165-5728(03)00042-0. [DOI] [PubMed] [Google Scholar]
- 54.Avitsur R, Kavelaars A, Heijnen C, Sheridan JF. Social stress and the regulation of tumor necrosis factor-alpha secretion. Brain Behav Immun. 2005;19:311–7. doi: 10.1016/j.bbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 55.Chida Y, Sudo N, Sonoda J, Hiramoto T, Kubo C. Early-life psychological stress exacerbates adult mouse asthma via the hypothalamus-pituitary-adrenal axis. Am J Respir Crit Care Med. 2007;175:316–22. doi: 10.1164/rccm.200607-898OC. [DOI] [PubMed] [Google Scholar]
- 56.Ashwell JD, King LB, Vacchio MS. Cross-talk between the T cell antigen receptor and the glucocorticoid receptor regulates thymocyte development. Stem Cells. 1996;14:490–500. doi: 10.1002/stem.140490. [DOI] [PubMed] [Google Scholar]
- 57.McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- 58.Wilckens T, De Rijk R. Glucocorticoids and immune function: unknown dimensions and new frontiers. Immunol Today. 1997;18:418–24. doi: 10.1016/s0167-5699(97)01111-0. [DOI] [PubMed] [Google Scholar]
- 59.Quan N, Avitsur R, Stark JL, He L, Shah M, Caligiuri M, et al. Social stress increases the susceptibility to endotoxic shock. J Neuroimmunol. 2001;115:36–45. doi: 10.1016/s0165-5728(01)00273-9. [DOI] [PubMed] [Google Scholar]
- 60.Joachim RA, Sagach V, Quarcoo D, Dinh QT, Arck PC, Klapp BF. Neurokinin-1 receptor mediates stress-exacerbated allergic airway inflammation and airway hyperresponsiveness in mice. Psychosom Med. 2004;66:564–71. doi: 10.1097/01.psy.0000132878.08780.93. [DOI] [PubMed] [Google Scholar]
- 61.Pujols L, Mullol J, Picado C. Alpha and beta glucocorticoid receptors: relevance in airway diseases. Curr Allergy Asthma Rep. 2007;7:93–9. doi: 10.1007/s11882-007-0005-3. [DOI] [PubMed] [Google Scholar]
- 62.Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]
- 63.Adcock IM, Ford PA, Bhavsar P, Ahmad T, Chung KF. Steroid resistance in asthma: mechanisms and treatment options. Curr Allergy Asthma Rep. 2008;8:171–8. doi: 10.1007/s11882-008-0028-4. [DOI] [PubMed] [Google Scholar]
- 64.Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 65.Geng CD, Pedersen KB, Nunez BS, Vedeckis WV. Human glucocorticoid receptor alpha transcript splice variants with exon 2 deletions: evidence for tissue- and cell type-specific functions. Biochemistry. 2005;44:7395–405. doi: 10.1021/bi047485e. [DOI] [PubMed] [Google Scholar]
- 66.Lin CW, Nakane M, Stashko M, Falls D, Kuk J, Miller L, et al. trans-Activation and repression properties of the novel nonsteroid glucocorticoid receptor ligand 2,5-dihydro-9-hydroxy-10-methoxy-2,2,4-trimethyl-5-(1-methylcyclohexen-3-y 1)-1H-[1]benzopyrano[3,4-f]quinoline (A276575) and its four stereoisomers. Mol Pharmacol. 2002;62:297–303. doi: 10.1124/mol.62.2.297. [DOI] [PubMed] [Google Scholar]
- 67.Lucibello FC, Slater EP, Jooss KU, Beato M, Muller R. Mutual transrepression of Fos and the glucocorticoid receptor: involvement of a functional domain in Fos which is absent in FosB. Embo J. 1990;9:2827–34. doi: 10.1002/j.1460-2075.1990.tb07471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. Embo J. 2001;20:7168–73. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol. 2009;300:7–16. doi: 10.1016/j.mce.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117:522–43. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 71.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006;26:7913–28. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–63. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 73.Kalinyak JE, Dorin RI, Hoffman AR, Perlman AJ. Tissue-specific regulation of glucocorticoid receptor mRNA by dexamethasone. J Biol Chem. 1987;262:10441–4. [PubMed] [Google Scholar]
- 74.Breslin MB, Geng CD, Vedeckis WV. Multiple promoters exist in the human GR gene, one of which is activated by glucocorticoids. Mol Endocrinol. 2001;15:1381–95. doi: 10.1210/mend.15.8.0696. [DOI] [PubMed] [Google Scholar]
- 75.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 76.Durham SR, Till SJ, Corrigan CJ. T lymphocytes in asthma: bronchial versus peripheral responses. J Allergy Clin Immunol. 2000;106:S221–6. doi: 10.1067/mai.2000.110154. [DOI] [PubMed] [Google Scholar]
- 77.Wills-Karp M. IL-12/IL-13 axis in allergic asthma. J Allergy Clin Immunol. 2001;107:9–18. doi: 10.1067/mai.2001.112265. [DOI] [PubMed] [Google Scholar]
- 78.Haczku A. T cells and eosinophils in asthma. Acta Microbiol Immunol Hung. 1998;45:19–29. [PubMed] [Google Scholar]
- 79.Gould CL, Lyte M, Williams J, Mandel AD, Sonnenfeld G. Inhibited interferon-gamma but normal interleukin-3 production from rats flown on the space shuttle. Aviat Space Environ Med. 1987;58:983–6. [PubMed] [Google Scholar]
- 80.Corrigan CJ, Haczku A, Gemou-Engesaeth V, Doi S, Kikuchi Y, Takatsu K, et al. CD4 T-lymphocyte activation in asthma is accompanied by increased serum concentrations of interleukin-5. Effect of glucocorticoid therapy. Am Rev Respir Dis. 1993;147:540–7. doi: 10.1164/ajrccm/147.3.540. [DOI] [PubMed] [Google Scholar]
- 81.Tsicopoulos A, Hamid Q, Haczku A, Jacobson MR, Durham SR, North J, et al. Kinetics of cell infiltration and cytokine messenger RNA expression after intradermal challenge with allergen and tuberculin in the same atopic individuals. J Allergy Clin Immunol. 1994;94:764–72. doi: 10.1016/0091-6749(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 82.Haczku A, Chung KF, Sun J, Barnes PJ, Kay AB, Moqbel R. Airway hyperresponsiveness, elevation of serum-specific IgE and activation of T cells following allergen exposure in sensitized Brown-Norway rats. Immunology. 1995;85:598–603. [PMC free article] [PubMed] [Google Scholar]
- 83.Haczku A, Moqbel R, Jacobson M, Kay AB, Barnes PJ, Chung KF. T-cells subsets and activation in bronchial mucosa of sensitized Brown-Norway rats after single allergen exposure. Immunology. 1995;85:591–7. [PMC free article] [PubMed] [Google Scholar]
- 84.Haczku A, Macary P, Haddad EB, Huang TJ, Kemeny DM, Moqbel R, et al. Expression of Th-2 cytokines interleukin-4 and -5 and of Th-1 cytokine interferon-gamma in ovalbumin-exposed sensitized Brown-Norway rats. Immunology. 1996;88:247–51. doi: 10.1111/j.1365-2567.1996.tb00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haczku A, Macary P, Huang TJ, Tsukagoshi H, Barnes PJ, Kay AB, et al. Adoptive transfer of allergen-specific CD4+ T cells induces airway inflammation and hyperresponsiveness in brown-Norway rats. Immunology. 1997;91:176–85. doi: 10.1046/j.1365-2567.1997.d01-2221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hamelmann E, Takeda K, Haczku A, Cieslewicz G, Shultz L, Hamid Q, et al. Interleukin (IL)-5 but not immunoglobulin E reconstitutes airway inflammation and airway hyperresponsiveness in IL-4-deficient mice. Am J Respir Cell Mol Biol. 2000;23:327–34. doi: 10.1165/ajrcmb.23.3.3796. [DOI] [PubMed] [Google Scholar]
- 87.Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med. 1997;185:2143–56. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Louahed J, Zhou Y, Maloy WL, Rani PU, Weiss C, Tomer Y, et al. Interleukin 9 promotes influx and local maturation of eosinophils. Blood. 2001;97:1035–42. doi: 10.1182/blood.v97.4.1035. [DOI] [PubMed] [Google Scholar]
- 89.McLane MP, Haczku A, van de Rijn M, Weiss C, Ferrante V, MacDonald D, et al. Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am J Respir Cell Mol Biol. 1998;19:713–20. doi: 10.1165/ajrcmb.19.5.3457. [DOI] [PubMed] [Google Scholar]
- 90.Eisenbarth SC, Piggott DA, Bottomly K. The master regulators of allergic inflammation: dendritic cells in Th2 sensitization. Curr Opin Immunol. 2003;15:620–6. doi: 10.1016/j.coi.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 91.Agarwal SK, Marshall GD., Jr Stress effects on immunity and its application to clinical immunology. Clin Exp Allergy. 2001;31:25–31. [PubMed] [Google Scholar]
- 92.Berin MC, Eckmann L, Broide DH, Kagnoff MF. Regulated production of the T helper 2-type T-cell chemoattractant TARC by human bronchial epithelial cells in vitro and in human lung xenografts. Am J Respir Cell Mol Biol. 2001;24:382–9. doi: 10.1165/ajrcmb.24.4.4360. [DOI] [PubMed] [Google Scholar]
- 93.Stevens TL, Bossie A, Sanders VM, Fernandez-Botran R, Coffman RL, Mosmann TR, et al. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature. 1988;334:255–8. doi: 10.1038/334255a0. [DOI] [PubMed] [Google Scholar]
- 94.Penna G, Vulcano M, Sozzani S, Adorini L. Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum Immunol. 2002;63:1164–71. doi: 10.1016/s0198-8859(02)00755-3. [DOI] [PubMed] [Google Scholar]
- 95.Haczku A, Atochina EN, Tomer Y, Chen H, Scanlon ST, Russo S, et al. Aspergillus fumigatus-induced allergic airway inflammation alters surfactant homeostasis and lung function in BALB/c mice. Am J Respir Cell Mol Biol. 2001;25:45–50. doi: 10.1165/ajrcmb.25.1.4391. [DOI] [PubMed] [Google Scholar]
- 96.Haczku A, Atochina EN, Tomer Y, Cao Y, Campbell C, Scanlon ST, et al. The late asthmatic response is linked with increased surface tension and reduced surfactant protein B in mice. Am J Physiol Lung Cell Mol Physiol. 2002;283:L755–65. doi: 10.1152/ajplung.00062.2002. [DOI] [PubMed] [Google Scholar]
- 97.Atochina EN, Beers MF, Tomer Y, Scanlon ST, Russo SJ, Panettieri RA, Jr, et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir Res. 2003;4:15. doi: 10.1186/1465-9921-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haczku A, Cao Y, Vass G, Kierstein S, Nath P, Atochina-Vasserman EN, et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol. 2006;176:3557–65. doi: 10.4049/jimmunol.176.6.3557. [DOI] [PubMed] [Google Scholar]
- 99.Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, et al. Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun. 2009;23:225–31. doi: 10.1016/j.bbi.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Engler H, Engler A, Bailey MT, Sheridan JF. Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. J Neuroimmunol. 2005;163:110–9. doi: 10.1016/j.jneuroim.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 101.Burnstein KL, Bellingham DL, Jewell CM, Powell-Oliver FE, Cidlowski JA. Autoregulation of glucocorticoid receptor gene expression. Steroids. 1991;56:52–8. doi: 10.1016/0039-128x(91)90124-e. [DOI] [PubMed] [Google Scholar]
- 102.Geng CD, Vedeckis WV. Steroid-responsive sequences in the human glucocorticoid receptor gene 1A promoter. Mol Endocrinol. 2004;18:912–24. doi: 10.1210/me.2003-0157. [DOI] [PubMed] [Google Scholar]
- 103.Crouch E, Rust K, Veile R, Donis-Keller H, Grosso L. Genomic organization of human surfactant protein D (SP-D). SP-D is encoded on chromosome 10q22.2-23. 1 J Biol Chem. 1993;268:2976–83. [PubMed] [Google Scholar]
- 104.Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol. 2001;63:521–54. doi: 10.1146/annurev.physiol.63.1.521. [DOI] [PubMed] [Google Scholar]
- 105.Haczku A. Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J Allergy Clin Immunol. 2008;122:861–79. doi: 10.1016/j.jaci.2008.10.014. quiz 80-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sims MW, Tal-Singer RM, Kierstein S, Musani AI, Beers MF, Panettieri RA, et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir Res. 2008;9:13. doi: 10.1186/1465-9921-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 108.Hortobagyi L, Kierstein S, Krytska K, Zhu X, Das AM, Poulain F, et al. Surfactant protein D inhibits TNF-alpha production by macrophages and dendritic cells in mice. J Allergy Clin Immunol. 2008;122:521–8. doi: 10.1016/j.jaci.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kierstein S, Poulain FR, Cao Y, Grous M, Mathias R, Kierstein G, et al. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res. 2006;7:85. doi: 10.1186/1465-9921-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cao Y, Tao JQ, Bates SR, Beers MF, Haczku A. IL-4 induces production of the lung collectin surfactant protein-D. J Allergy Clin Immunol. 2004;113:439–44. doi: 10.1016/j.jaci.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 111.Haczku A, Tong E, Krytska K, Kierstein S, Panettieri RA. Combination of stress and asthma inhibits the glucocorticoid receptor (GR) and CAAT enhancer binding protein (C/EBP)-mediated production of the immunoprotective surfactant protein D (SP-D) in the lung. Am J Respir Crit Care Med. 2008:A64. [Google Scholar]
- 112.Panettieri RA, Jr, Spector SL, Tringale M, Mintz ML. Patients' and primary care physicians' beliefs about asthma control and risk. Allergy Asthma Proc. 2009;30:519–28. doi: 10.2500/aap.2009.30.3281. [DOI] [PubMed] [Google Scholar]
- 113.Panettieri RA, Jr, Covar R, Grant E, Hillyer EV, Bacharier L. Natural history of asthma: persistence versus progression-does the beginning predict the end? J Allergy Clin Immunol. 2008;121:607–13. doi: 10.1016/j.jaci.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 114.Bhandare R, Damera G, Banerjee A, Flammer JR, Keslacy S, Rogatsky I, et al. Glucocorticoid Receptor Interacting Protein-1 Restores glucocorticoid Responsiveness in Steroid-resistant Airway Structural Cells. Am J Respir Cell Mol Biol. 2009 doi: 10.1165/rcmb.2009-0239RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Damera G, Tliba O, Panettieri RA., Jr Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22:353–9. doi: 10.1016/j.pupt.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Panettieri RA., Jr Asthma persistence versus progression: does airway smooth muscle function predict irreversible airflow obstruction? Allergy Asthma Proc. 2009;30:103–8. doi: 10.2500/aap.2009.30.3202. [DOI] [PubMed] [Google Scholar]