

Abstract
Rationale
Several studies have examined the role of different neurotransmitter systems in modulating risk-taking behavior.
Objective
This investigation was aimed to determine whether the benzodiazepine lorazepam dose-dependently alters risk-taking behavior and underlying neural substrates.
Methods
Fifteen healthy, non-smoking, individuals (6 females, 9 males), aged 18–39 years (mean 27.6 +/− 1.4 years) with 12–18 years of education (mean 15.6 +/− 0.3 years) underwent functional magnetic resonance imaging while performing a risk-taking decision-making task.
Results
Our results show that lorazepam did not affect risky behavior at 0.25 and 1mg, but dose-dependently attenuated activation in (a) the amygdala and medial prefrontal cortex during the response selection phase, and in (b) the bilateral insular cortex and amygdala during the outcome (i.e., rewarded or punished) phase. Furthermore, a lorazepam-induced increase in insular cortex activation was associated with less risky responses.
Conclusions
Taken together, our findings support the idea that GABAergic modulation in limbic and paralimbic structures is important during both the response selection and outcome phase of risk-taking decision-making.
Keywords: Risk-taking, decision-making, fMRI, insula, amygdala, medial prefrontal cortex, GABAergic, lorazepam
Introduction
Risk refers to a situation involving exposure to danger or the possibility of loss or injury. Risk-taking refers to the propensity to act in situations that contain risk, and is comprised of a set of cognitive, affective, and action processes, which aim to establish a balance between potential losses and benefits of an action. In particular, these processes involve constructs such as hazard, probability, consequence, or potential adversity or threat (Slovic, 1987;Slovic, 2000;Slovic, 2000). According to Kahneman and Tversky (1979;1984;2000), risk or loss aversion (i.e., losses exert greater control over decisions compared to gains) is a pervasive human norm, i.e., individuals, when making a decision, are more strongly influenced by the potential of losses associated with an action than by the potential of gains. In other words, from a normative perspective, individuals tend to be risk-averse. Thus, it is important to understand maladaptive (excessive or deficient) risk-taking behavior because it often characterizes groups of individuals with vulnerabilities for psychiatric problems. For example, altered risk-taking behavior has been observed in substance dependence, where data show that these individuals are more likely to select actions associated with large short-term gains (and large long-term losses) preferentially to those with small short-term gains (and small long-term losses) (Bechara and Damasio, 2002;Grant, Contoreggi et al., 2000). Furthermore, they have a tendency to select risky options (Lane and Cherek, 2000) and show an altered temporal horizon of risks and benefits (i.e., a steeper temporal discounting function) (Petry, Bickel et al., 1998;Madden, Bickel et al., 1999).
Functional neuroimaging studies of risk-taking have shown that several neural substrates activate in relation to the degree of risk. Among these structures are the orbital and dorsolateral prefrontal cortex, anterior cingulate, insula, inferior parietal cortex and thalamus, predominantly on the right side, and cerebellum predominantly on the left side (Ernst, Bolla et al., 2002;Ernst, Nelson et al., 2004;Ernst, Dickstein et al., 2004). The notion of risk is closely associated with the potential of an aversive outcome or punishment. Therefore, it is not surprising that some key components of this neural circuitry, e.g. the anterior cingulate / medial prefrontal cortex, anterior insula, and orbitofrontal cortex have also been implicated in higher order processing of pain (Craig, 2003;Craig, 2002). Thus, it appears that top down modulation of aversive stimuli (pain) may also be involved in risk-related processing. Consistent with this idea, other investigators have reported that the medial-frontal area may be important for risk-level adjustments (Gehring and Willoughby, 2002), findings supported by observations that risk anticipation is closely linked to activation in the medial frontal gyrus (Fukui, Murai et al., 2005). Similarly, choosing high-risk over low-risk decisions was associated with increased activity in both anterior cingulate and orbitofrontal cortices (Cohen, Heller et al., 2005;Ernst, Nelson, McClure, Monk, Munson, Eshel, Zarahn, Leibenluft, Zametkin, Towbin, Blair, Charney, and Pine, 2004), and the amygdala was activated during risk-taking following a choice that entailed risk or loss (Kahn, Yeshurun et al., 2002). Lastly, altered risk-taking behavior may relate to a balance in the engagement of reward-processing areas, such as the nucleus accumbens, and of anxiety-related processing areas, such as the amygdala (Ernst, Nelson et al., 2005).
Several studies have begun to examine the role of different neurotransmitter systems in modulating risk-taking behavior. For example, systemic administration of alprazolam, a GABA-ergic agonist, in healthy volunteers produced dose-related changes in subjective effects, response rates, and, most importantly, dose-dependently increased selection of the risky response option (Lane, Tcheremissine et al., 2005). Acute marijuana administration produced measurable changes in risky decision-making under laboratory conditions and a change in sensitivity to both reinforced and losing outcomes (Lane, Cherek et al., 2005). Others have examined the effects of alcohol on risk-taking behavior showing that alcohol administration impairs the ability to factor in the magnitude of gains and the likelihood of winning when the losses are large (George, Rogers et al., 2005). Furthermore, alcohol dose-dependently increased the selection of risky responses in an experimental paradigm (Lane, Cherek et al., 2004) as well as during simulated driving (Burian, Liguori et al., 2002). Adrenergic attenuation by systemic administration of a beta-adrenergic antagonist, propranolol, reduced the discrimination between large and small possible losses when the probability of winning was relatively low and the probability of losing was high (Rogers et al 2004), which may be due to reduction of the punishment signal. Attenuation of serotonergic modulation by tryptophan depletion did not change risk-related behaviors but resulted in a reduced ability to discriminate between magnitudes of expected gains (but not expected losses) and did not affect the ability to discriminate the probability of gains or losses (Harmer, Rogers et al., 2003). In combination, these studies show that risk-taking can be increased by GABAergic modulation, alcohol, adrenergic attenuation, and stimulation of cannabinoid receptors. These data are all consistent with the idea that the representation of future physiological aversive states characterized by anxiety, sympathetic arousal, and possibly pain, is crucial for the degree of risk-taking.
The aim of this investigation was to determine whether the benzodiazepine lorazepam (a GABA-ergic agonist) dose-dependently alters risk-taking behavior and neural substrates underlying risk-taking. We hypothesized that participants would show an increase in risky response selection associated with an attenuation of neural substrates signaling aversive outcomes. We employed primarily a region of interest approach since an effective method to prevent false-positive findings is to focus on a-priori hypothesized areas (Stein, Pankiewicz et al., 1998). The a priori areas included the insula, amygdala,medial prefrontal cortex, and nucleus accumbens. The latter area was selected due to its role in reward sensitivity and uncertainty as well as its interconnections with the amygdala and medial prefrontal cortex (Knutson and Cooper, 2005;Knutson, Fong et al., 2001). Similarly, the medial prefrontal cortex is known to be involved in cognitive control, conflict monitoring, outcome evaluation and uncertainty (Ridderinkhof, Ullsperger et al., 2004). Based on our previous work (Paulus, Feinstein et al., 2005), we hypothesized that lorazepam would attenuate insula and amygdala activation in a dose-dependent manner. Support for these hypotheses would enable us to better understand the influence of inhibitory neurotransmitters such as GABA on processing of risk-related behavior.
Methods
Subjects
The UCSD Institutional Review Board approved study procedures. All participants provided written informed consent and were paid for their participation. Fifteen healthy, non-smoking, individuals (6 females, 9 males), aged 18–39 years (mean 27.6 +/− 1.4 years) with 12–18 years of education (mean 15.6 +/− 0.3 years), were studied. Participants did not have medical or psychiatric disorders as determined by medical history and a structured clinical interview for DSM-IV diagnoses. Each subject was required to consume less than 150 mg/day caffeine and less than 14 alcohol-containing drinks per week. Subject had no history of drug or alcohol abuse and did not report use of benzodiazepines in the prior month. All participants passed a urine drug screen prior to study drug administration (to assure that individuals were not using other psychoactive substances at time of testing that could interfere with the study drug) and presented to the MRI facility between 8a and 4p with at least one week between scans.
Study Design
The study was performed in a double-blinded manner. Participants underwent each of three conditions in randomized order between 1–3 weeks apart. We selected doses that minimally disrupt performance on behavioral tasks and memory (≤ 1.0 mg) but are used to treat anxiety. Subjects arrived at the MRI facility 60–90 minutes prior to the MRI scan. Upon arrival, subjects' vital signs were assessed and subjects received orally placebo, 0.25 mg or 1.0 mg lorazepam suspension mixed in diet, decaffeinated cola. The risky gains decision-making task was conducted approximately 80 minutes after the oral administration of placebo or lorazepam to coincide with peak blood concentrations of lorazepam (Kyriakopoulos, Greenblatt et al., 1978).
Assessments and fMRI paradigm
Task
The Risky Gains task has been previously described elsewhere (Paulus, Rogalsky et al., 2003). Briefly, subjects are presented with a sequence of three numbers in ascending order (20, 40, and 80). Each number is presented on the screen for one second and, if the subject presses a button while that number is shown on the screen, he/she receives the number of points shown on the screen. The subjects are informed that when a 40 or 80 appears, there is a chance that it will be in red color, which indicates that that the trial ends and that the subject loses 40 or 80 points, respectively. Thus, although the subject may gain more points per trial by waiting until a 40 or 80 appears on the screen, there is also a risk of losing 40 or 80 points. The probabilities of presenting a negative 40 or 80 are such that a subject's final score would be identical if they were to consistently select 20, 40, or 80. Thus, there is no inherent advantage to selecting the risky response (40 or 80) over the safe response (20). Each trial lasts 3.5 seconds irrespective of the subject's choice and the subject receives rewarding feedback (stimulus on the screen and auditory sound) immediately after selecting a response.
Behavioral and Psychological Measures
The 96 trials of the Risky-Gains task consist of three trial types, which were presented in subject-independent, randomized order: (1) a 20, 40 or 80 non-punished trial type (n=54), (2) a 40 punished trial type (n=24), and (3) an 80 punished trial type (n=18). The primary dependent measure to assess the degree of risk-taking and the response to punishment is the probability of selecting a safe (20) or a risky (40 or 80) response as a function of the previous trial outcome (punished versus non-punished).
Subjects completed the state form of the Spielberger State-Trait Anxiety Inventory (Spielberger, 1983) (STAIS), and visual analog scales (VAS) for anxiety, tension, alertness, and trembling before and after the MRI scan.
All subjects completed a practice run immediately prior to the fMRI session to familiarize themselves with the task. Participants were observed by the experimenter during this initial run to assure adequate understanding and completion of the task.
Image Acquisition
During the task, one BOLD-fMRI run was collected for each subject using a 1.5-Tesla Siemens (Erlangen, Germany) scanner (T2*-weighted echo planar imaging, TR = 2000 ms, TE = 40 ms, 64 × 64 matrix, 20 4-mm axial slices, 256 repetitions). During the same experimental session, a T1-weighted image (MPRAGE, TR = 11.4 ms, TE = 4.4 ms, flip angle = 10°, FOV = 256 × 256, 1 mm3 voxels) was obtained for anatomical reference. For preprocessing, voxel time series were interpolated to correct for non-simultaneous slice acquisition within each volume and corrected for three-dimensional motion.
FMRI analysis pathway
The data were preprocessed and analyzed with the software AFNI (Cox, 1996). The echo-planar images were realigned to the 128th acquired scan and time corrected for slice acquisition order. To exclude the voxels showing an artifact related to signal drop, a combined threshold/cluster-growing algorithm was applied to the mean of the functional images to compute a whole brain mask. This screened out non-brain voxels and voxels falling within the artifact region. A randomized fast-event related design was used with 6 resting trials interspersed between the 96 risky-gains trials. The preprocessed time series data for each individual were analyzed using two multiple regression models which we refer to as “risk-related” and “outcome-related” activation patterns. For the risk-related multiple regression model the five regressors of interest were constructed from the behavioral data obtained from each subject during the task. Specifically, response regressors were defined from the onset of the trial until the individual selected an option and, for punished trials, until the appearance of negative 40 or 80. These five regressors are referred to as (1) selecting 20 (safe response) (2) selecting 40 (risky response); (3) selecting 80 (risky response); (4) punished with −40; and (5) punished with −80. The subsequent time period, which included outcome and inter-trial interval as well as the null-trials served as the baseline condition for this analysis. For the outcome related multiple regression model, two regressors, which quantify risk-taking behavior with gains and losses respectively, were used to evaluate the neural systems response to the experienced outcome: (1) selecting a risky response (40 or 80) which resulted in reward and (2) selecting a risky response which resulted in punishment (−40 or −80). These regressors were set to 1 from the onset of the outcome until the end of the trial. The time interval that included both selection of the response and the inter-trial interval as well as the null trials served as the baseline condition for this analysis. Both sets of regressors of interest were convolved with a modified gamma variate function modeling a prototypical hemodynamic response (Boynton, Engel et al., 1996) prior to inclusion in the regression model. In addition three regressors were used to account for residual motion (in the roll, pitch, and yaw direction). Regressors for baseline and linear trends were used to eliminate slow signal drifts. The AFNI program 3dDeconvolve was used to calculate the estimated voxel-wise response amplitude. Finally, a subject-specific linear contrast was used to identify brain activation associated with selecting a risky response (40 or 80) versus a safe response (20). A Gaussian filter with FWHM 6 mm was applied to the voxel-wise percent signal change data to account for individual variations of the anatomical landmarks. Data of each subject were normalized to Talairach coordinates.
Statistical Analyses
Two sets of analyses were carried out after data of each subject were normalized to Talairach coordinates. First, a priori regions of interest (defined by the Talairach Daemon atlas; Lancaster, Woldorff et al., 2000) in the bilateral amygdala, medial prefrontal cortex (Brodmann Areas 24 and 32), insula, and nucleus accumbens were used as masks for both the decision- and outcome-related analysis. Based on these areas of interest, it was determined via simulations that a voxel-wise a-priori probability of 0.05 would result in a corrected cluster-wise activation probability of 0.05 if a minimum volume of 128 μl and 2 connected voxels (in the amygdala) or 512 μl and 8 connected voxels (in all other ROIs) was considered. Using the thresholding and clustering techniques described above, the corrected voxel-wise probabilities are as follows: amygdala p < 0.012, insular cortex p < 0.00006859, medial prefrontal cortex p < 0.00014493 and nucleus accumbens p < 0.0084. The areas of interest were superimposed on each individual's voxel-wise percent signal change brain image. Only activations within the areas of interest, which also satisfied the volume and voxel connection criteria were extracted and used for further analysis. The voxel-wise percent signal change data were entered into a mixed model ANOVA with dose as a fixed factor and subjects as a random factor.
Second, a whole-brain analysis was conducted with the signal contrast of risky (selecting a 40 or 80 and receiving a gain of 40 or 80 points) versus safe (selecting 20 and receiving a sure gain of 20 points) responses as the main dependent measure. Similarly, another whole-brain analysis explored the outcome-related activation of a punished risky response (waiting to select 40 or 80 and being punished) versus a reward (or non-punished; selecting a 40 or 80 and obtaining the points) risky response. A threshold adjustment method based on Monte-Carlo simulations was used to guard against identifying false positive areas of activation (Forman, Cohen et al., 1995). Based on these simulations, it was determined that a voxel-wise a-priori probability of 0.05 would result in a corrected cluster-wise activation probability of 0.05 if a minimum volume of 1000 μl and a connectivity radius of 4.0 mm was considered. Finally, the average percent signal difference (i.e., regressor of interest coefficient divided by the baseline regressor multiplied by 100) was extracted from regions of activation that were found to survive this threshold/cluster method. These voxel-wise percent signal change data were also entered into two mixed model ANOVA with dose as a fixed factor and subjects as a random factor and risky vs safe responses, and punished vs rewarded responses as dependent measures, respectively.
All behavioral analyses were carried out with SPSS 10.0 (Norusis, 1990). A repeated measures multivariate ANOVA, with dose (placebo, 0.25, 1.0 mg) as the within-subjects factor, was used to analyze the behavioral measures. Behavioral measures are reported as main effect of dose. A difference score between the 1.0 mg and the placebo condition was created to determine whether the dose-related effect of lorazepam on individual's brain activation was associated with individual differences in behavioral variables.
Results
Behavioral and Psychological Ratings Results
Subjects selected a risky response (40 or 80) 55% (+/−4%) of the time. One subject never selected a risky response during the 1.0 mg lorazepam condition and was, therefore, excluded from the decision-related analyses. There was no significant effect of lorazepam on the frequency of selecting a risky response (see Table 1). Although there was a trend (F(1,13)= 3.15, p=0.1, eta2 = 0.19) for the frequency of risky responses to be decreased after punishment (Figure 1), there was no interaction between the drug condition and the effect of punishment on selecting a risky response (F(2,12) = 0.24, p = 0.79, eta2 = 0.04). Lorazepam had no significant effect on performance (number of total points gained during the Risky Gains task) or response latency (F(1,13)= 0.03, p=0.96, eta2 = 0.006). Therefore, contrary to our expectation, lorazepam had no significant effect on the frequency of selecting a risky response and did not affect post-punishment induced reduction of risky responses. There was no significant main effect of lorazepam (F=1.6, p=0.22, eta2 =0.11) or lorazepam by time administration (i.e., before or after the scanner) interaction (F=.03, p=.97, eta2 =0.003) on the STAIS. In comparison there was a main effect of dose on VAS ratings of sleep (F=4.4 p=.02, eta2 =0.238) and tension (F=4.2, p=.03, eta2 =0.276). Specifically, there was greater sleepiness with higher dose and greater tension with .25mg (inverted u-shape), respectively. Furthermore, a dose by time interaction on tiredness (F=7.2, p< 01, eta2 =.396) was detected, suggesting greater tiredness after 1mg of lorazepam.
Table 1.
Effect of lorazepam on risky responses, punished responses and total score.
| Placebo | 0.25 mg Lorazepam | 1.0 mg Lorazepam | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | F(dose) | p | |
| Fraction Risky Responses | 0.58 | 0.14 | 0.60 | 0.15 | 0.59 | 0.13 | 0.26 | 0.77 |
| Fraction Punished Trials | 0.19 | 0.07 | 0.21 | 0.09 | 0.20 | 0.09 | 0.77 | 0.46 |
| Total Score | 1913 | 392 | 1695 | 455 | 1731 | 421 | 1.06 | 0.35 |
Figure 1.
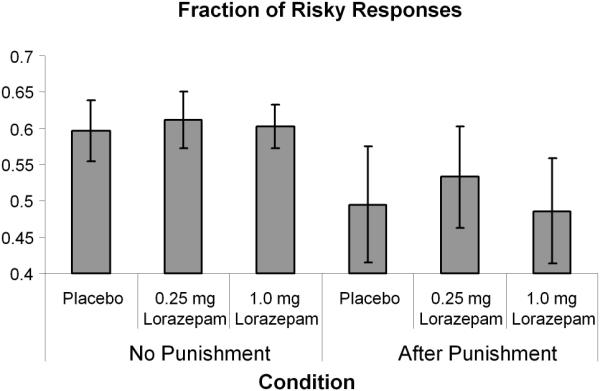
Punished and non-punished risky responses for 0mg, 0.25mg, and 1mg of lorazepam.
Neuroimaging Results
Dose-dependent alteration of decision-related neural activation patterns
A-priori region of interest analyses examining risk-related responses (i.e., selecting a risky versus safe response) revealed significant dose-dependent decreases in left amygdala (F(2,28) = 4.05, p = 0.028, eta2 = 0.225) and medial prefrontal cortex (F(2,28) = 4.51, p = 0.02, eta2 = 0.244), which includes the rostral part of the anterior cingulate (Figure 2). In contrast, lorazepam induced a dose-dependent increase in the left insular cortex. This dose-dependent increase in insular cortex (F(2,28) = 6.59, p = 0.004, eta2 = 0.32) was associated with a decrease in selection of risky responses (comparison of 1.0 mg dose of lorazepam relative to the placebo condition; r2 = 0.395 [Figure 3]). Thus, a stronger increase in insular activation was associated with a more risk-averse behavior, which is consistent with our previous findings in this task (Paulus, Rogalsky, Simmons, Feinstein, and Stein, 2003). No voxels survived thresholding in the nucleus accumbens.
Figure 2.

Lorazepam effect regions of interest analysis: Areas of significant BOLD activation in limbic areas during Risky versus Safe responses.
Figure 3.
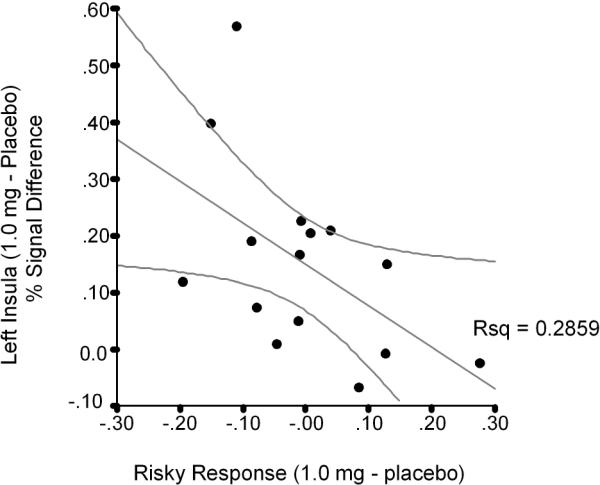
Correlation scatter plot between lorazepam's effect on insula activation during Risky versus Safe responses and percentage of risky responses.
Whole brain analysis (Table 2) revealed a distributed neural substrate with significantly more activation during the selection of a risky response relative to the selection of a safe response. These areas included those previously reported (Paulus, Rogalsky, Simmons, Feinstein, and Stein, 2003) such as the bilateral anterior insula and inferior frontal gyrus (see Table 2 for Brodmann areas). Moreover, anterior cingulate and posterior parietal cortical areas were significantly more activated during risky responses. Lorazepam had two types of effects on these neural substrates. First, a nonlinear dose response (with the 0.25 mg dose showing the largest activation) was found in the middle and inferior frontal gyri, part of the anterior cingulate, and the caudate. Second, a dose-dependent linear decrease in activation was observed in anterior and rostral parts of the cingulate and middle frontal gyri.
Table 2.
Whole brain analyses of lorazepam during decision phase (effect on Risky versus Safe responses).
| X | y | z | L/R | Area | BA | F-value |
|---|---|---|---|---|---|---|
| Main Effect of Task: Risky-Safe Option | ||||||
| −43 | 8 | 26 | L | Inferior Frontal Gyrus / Anterior Insula |
9 13 |
39.98 |
| 1 | −10 | 4 | R | Thalamus | 47.52 | |
| 41 | 11 | 24 | R | Inferior Frontal Gyrus Anterior Insula |
9 13 |
40.25 |
| 1 | 12 | 38 | R | Cingulate Gyrus | 32 | 31.12 |
| 16 | −63 | 39 | R | Precuneus | 7 | 31.39 |
| −55 | −31 | 23 | L | Inferior Parietal Lobule | 40 | 35.08 |
| 50 | −32 | 6 | R | Superior Temporal Gyrus | 22 | 36.55 |
| −9 | −61 | 49 | L | Precuneus | 7 | 19.06 |
| 0 | −27 | 24 | R | Cingulate Gyrus | 23 | 31.30 |
| −27 | −71 | 27 | L | Precuneus | 31 | 17.99 |
| Nonlinear Dose Effect of Lorazepam | ||||||
| 57 | 24 | −14 | R | Inferior Frontal Gyrus | 47 | 8.34 |
| 18 | 3 | 44 | R | Cingulate Gyrus | 24 | 12.95 |
| 14 | 27 | 6 | R | Caudate | 7.30 | |
| −39 | 14 | 44 | L | Middle Frontal Gyrus | 6 | 6.61 |
| Linear Dose Effect of Lorazepam | ||||||
| 27 | −38 | −15 | R | Fusiform Gyrus | 37 | 5.65 |
| 9 | −4 | 40 | R | Cingulate Gyrus | 24 | 8.21 |
| 50 | −11 | 28 | R | Precentral Gyrus | 6 | 11.92 |
| −18 | 26 | 47 | L | Superior Frontal Gyrus | 8 | 3.46 |
| −8 | −2 | 5 | L | Thalamus | 4.72 | |
| 32 | 41 | 16 | R | Middle Frontal Gyrus | 10 | 4.51 |
| −21 | 1 | −11 | L | Parahippocampal Gyrus | 34 | 4.05 |
| −39 | 7 | 1 | L | Insula | 13 | 6.59 |
| 9 | 7 | 11 | R | Caudate | 4.19 | |
Dose-dependent alteration of outcome-related activation patterns
A-priori anatomically constrained region of interest analyses showed that lorazepam dose-dependently decreased punished versus non-punished-related activation in bilateral insula (left insula: F(2,12)= 11.72, p=0.002, eta2 = 0.66; right insula: F(2,12)= 4.08, p=0.04, eta2 = 0.40) and right amygdala (F(2,12)= 14.24, p=0.001, eta2 = 0.70 Figure 5) but there was no significant effect of lorazepam on the nucleus accumbens.
Figure 5.

Lorazepam effect regions of interest analysis: Areas of significant BOLD activation in limbic areas during Punished vs non-Punished.
When examining the outcome-related activation on risky responses, i.e., the difference between punished versus non-punished risky choices, we observed strong bilateral caudate activation (see Figure 4). These activations included the entire extension of the caudate rostrally from the head to the tail. Lorazepam had no significant effect of changing the win versus lose related activation differences in these structures (left caudate: F(2,12)= 2.78, p=0.11, eta2 = 0.31; right caudate: F(2,12)= 1.36, p=0.39, eta2 = 0.18). Whole brain analyses revealed that lorazepam dose-dependently affected win versus lose related activation in bilateral insula and medial prefrontal cortex (among other areas), with the 1.0 mg lorazepam dose significantly attenuating the win-related activation in these structures.
Figure 4.

Lorazepam effect whole brain analysis: Areas of significant BOLD activation during Punished vs non-Punished.
Relationship between brain activation and changes in subjective ratings
Functional regions of interest derived from the whole brain analyses and the a-priori constrained region of interest analyses were subjected to a stepwise linear regression to predict lorazepam induced changes in state anxiety or changes in visual analog ratings scales. None of the dose-dependent lorazepam activation differences predicted changes in subjective rating scales (data not shown).
Discussion
This investigation yielded three main results. First, lorazepam did not affect risky behavior at the doses tested. Second, lorazepam dose-dependently attenuated activation in (a) the amygdala and medial prefrontal cortex during the decision phase and (b) in the bilateral insular cortex and amygdala during the outcome (i.e., rewarded or punished) phase. Third, a lorazepam-induced activation increase in insular cortex was associated with less risky responses. Taken together, this investigation supports the idea that GABAergic modulation in limbic and paralimbic structures, which include bilateral amygdala, insula and medial prefrontal cortex, is important during both the response selection (i.e., decision) and outcome phase of risk-taking decision-making.
Both lorazepam doses tested in this study did not affect risk-taking behavior. These results contrasts with a previous study, which showed that alprazolam dose-dependently decreased risky responses (Lane, Tcheremissine, Lieving, Nouvion, and Cherek, 2005). These apparently contradictory findings may be reconciled by highlighting several differences between that study and the current investigation. First, in the Lane et al. (2005b) study, alprazolam did not robustly affect risk-taking unless a dose of 2 mg was used, which would correspond to approximately 4 mg of lorazepam. Thus, the doses used in the current study may not have been sufficiently high to affect behavior. It should be noted that higher doses of lorazepam affect alertness (Greenblatt, Harmatz et al., 1988) and other cognitive functions such as memory (Sperling, Greve et al., 2002). Therefore, one may not be able to differentiate the behavioral effects on risk-taking from those on sedation. Second, subjects in this study had no history of any DSM-IV Axis I diagnosis. In contrast, several subjects in the above mentioned study (Lane, Tcheremissine, Lieving, Nouvion, and Cherek, 2005) were reported to have a history of drug abuse/dependence. We have previously shown that individuals who are at higher risk for stimulant use show increased risk-taking behavior (Leland and Paulus, 2005). This suggests that individuals in Lane and colleagues' study may have had a higher baserate of selecting risky responses, and/or that their risk-taking propensity may interact with the effects of a benzodiazepine. Third, there are significant task-related differences between the paradigm used by Lane and colleagues and the risky-gains task. Although it is important to probe risk-taking using different types of decision-making tasks due to the variability of such behavior in different contexts (Monterosso, Ehrman et al., 2001), these behaviors may be differentially sensitive to GABAergic modulation. Finally, we did not find a selective dose-dependent difference to punishment, which may be due to the fact that the overall effect of punishment on decreasing subsequent risky responses was modest. Lastly, there may have been the possibility that some risk-taking preferences may have been in transition across the three testing sessions. Thus, if risk levels were unstable during the primary dosing and testing, it could have obscured the acute drug effects on risk-taking behavior.
Lorazepam had the expected effect on decision (risk)-related processing in the amygdala and the medial prefrontal cortex / anterior cingulate. However, in contrast to our hypothesis, lorazepam dose-dependently increased risk-related activation in the insular cortex which, in the left hemisphere, was inversely associated with the rate of risky responses. Consistent with our previous findings (Paulus, Rogalsky, Simmons, Feinstein, and Stein, 2003), heightened insular cortex activation may be related to increased anticipation of aversive outcomes when processing the consideration of selecting a risky response. In contrast, the dose-dependent attenuation of amygdala activation during the response selection phase may reflect an altered appraisal of inherent value of the risky choice (Kahn, Yeshurun, Rotshtein, Fried, Ben Bashat, and Hendler, 2002). Other investigators have emphasized the importance of the amygdala for the subjective assessment of incentive value of the choice (Arana, Parkinson et al., 2003), as opposed to the role of the insular cortex, which is thought to instantiate somatic markers (Bechara, 2001). According to the somatic marker hypothesis (Damasio, 1996), certain stimuli initiate a state that is associated with pleasurable or aversive somatic markers which serve as a guide to bias the selection towards certain (pleasurable) actions. Moreover, the amygdala, in the context of choice selection, has also been implicated in processing emotional consequences of the possibility of alternative outcomes, which is often referred to as regret (Coricelli, Critchley et al., 2005), and is related to choice-related processing of missing or unavailable information (Hsu, Bhatt et al., 2005). We speculate that lorazepam-related dose-dependent attenuation of the amygdala during response-related processing may have counterbalanced the increase in insular cortex activation and result in no observable behavioral changes of risky response selection. Future studies may need to examine the effect of lorazepam on the functional connectivity between the amygdala and insular as well as the amygdala and medial prefrontal cortex to better delineate the role of GABA in risk-related behavior.
The dose-dependent attenuation of risk-related activation in the medial prefrontal cortex / anterior cingulate is consistent with the idea that lorazepam decreases conflict processing. Various types of conflict tests have been used in animal studies of anxiety-related behaviors (Millan, 2003). In particular, a recent study using a new conflict procedure with rhesus monkeys showed a high correlation between therapeutic potencies and the ability of benzodiazepines to increase suppressed responding. The cingulate cortex has been hypothesized to contribute to the internal monitoring of planning and controlling functions of the prefrontal cortex as well as the regulation of affective signals coming from limbic areas (Frith and Done, 1988). Others have attributed a conflict-monitoring function to this structure (Botvinick, Nystrom et al., 1999) due to its role implementing strategic processes to reduce cognitive conflicts (Carter, Macdonald et al., 2000). The anterior cingulate cortex is also important for assessing the value of an action for obtaining a predicted outcome (Richmond, Liu et al., 2003). Moreover, this medial prefrontal structure may have an important role in maintaining action-outcome associations, when the action is only probabilistically associated with an outcome (Rushworth, Walton et al., 2004). Taking into account the prominent role of the anterior cingulate during cognitive and action monitoring, GABAergic modulation of this structure during risk-related response processing may reflect the degree to which a response alternative signals conflict and initiates the utilization of strategic processes to reduce such state (i.e., conflict). In contrast to animal conflict procedures, processing of risk-related responding during the risky gains decision-making task incorporates both conflict (the possibility of a punished outcome) and uncertainty. Recent studies indicate that risk (probabilistic aversive outcomes) and ambiguity (incomplete information) may depend on partially distinct neural substrates (Hsu, Bhatt, Adolphs, Tranel, and Camerer, 2005;Huettel, Stowe et al., 2006). Therefore, future investigations of GABAergic modulation may need to utilize paradigms that disambiguate uncertainty from punished responding to better delineate the effects of benzodiazepines on these processes in humans.
Fear is an adaptive response to potentially dangerous, i.e., risky, stimuli that threaten to perturb homeostasis (Millan, 2003). GABAergic neurons constitute the major mode of inhibitory transmission throughout the CNS, acting as a “brake” under conditions of stress (Millan, 2003). Interestingly, the highest densities of benzodiazepine receptors in human brain have been reported in cortical, limbic and paralimbic areas (Zezula, Cortes et al., 1988;Marowsky, Fritschy et al., 2004;Quirk and Gehlert, 2003). Acute administration of lorazepam has strong dose-related effects on sleep, mood, drug liking, and abuse liability (Roache and Griffiths, 1987) as well as cognitive functions such as learning (Rush, Higgins et al., 1993) and attention (Rush, Higgins et al., 1994), which is associated with attenuation of fronto-temporal activation (Coull, Frith et al., 1999). However, the effects of benzodiazepines on fear-related experimental paradigms have been mixed (Riba, Rodriguez-Fornells et al., 2001;Baas, Grillon et al., 2002).We have previously shown that lorazepam dose-dependently attenuates amygdala and insular cortex during emotional face processing (Paulus, Feinstein, Castillo, Simmons, and Stein, 2005) and the current study extends this finding to include attenuation of risk-related processing.
Of interest, lorazepam did not affect striatal activation (either dorsal or ventral) related to gain versus loss when contrasting the outcome-related activation for selecting a risky response and receiving 40 or 80 points versus being punished by losing 40 or 80 points. This observation supports the general idea that lorazepam did not indiscriminately attenuate brain activation. This finding is consistent with our previous results, which suggested that lorazepam did not affect primary visual cortex activation (Paulus, Feinstein, Castillo, Simmons, and Stein, 2005).
This investigation had several limitations. First, the lack of a behavioral effect of lorazepam on risk-related response selection makes it more difficult to interpret dose-dependent activation differences because we were unable to relate a lorazepam-induced behavioral change to a neural activation change. However, activation differences that are not accompanied by behavioral differences can be very informative because they speak to the neural processes that may underlie the overtly observed behavior. Although we acknowledge that the observed neural activation differences may be due to a number of different factors (which cannot be disambiguated with the current experimental design and with BOLD-fMRI technology) they are, nevertheless, important. In particular, some investigators have proposed a “risk-as-feelings” hypothesis, which highlights the role of affect experienced at the moment of decision making (Loewenstein, Weber et al., 2001). The differential activity in limbic and paralimbic structures is consistent with the notion that lorazepam altered the affective state associated with risk-related behavior. Therefore, lorazepam may not change risky behaviors (on this task) but may affect the feelings associated with selecting a risky alternative. Future studies will need to better delineate these effects. Moreover, the lack of behavioral differences eliminates possible performance confounds on the effects of lorazepam. The results presented in this manuscript are limited to explain risk-related brain processes and future investigations may apply other risk-taking decision-making paradigms better suited to produce significant interactions between lorazepam and behavior. Second, our experimental design lacked of a baseline risk-taking session prior to dosing, which raises the possibility that some risk-taking preferences may have been temporally unstable. These instabilities could obscure the acute drug effects on risk-taking behavior. Third, the risky gains decision-making task does not allow for differentiation between uncertainty-related processing and reward or punishment-related processing. A recently published article reported several regions (including the anterior insula) that showed a selective increase in activation uncertainty when compared to risky decision making (Huettel, Stowe, Gordon, Warner, and Platt, 2006). Future studies should develop adequate paradigms to clarify the commonalities and discrepancies between the uncertainty and risk-taking neural networks. Fourth, the AFNI 3dDeconvolve program used to calculate the response amplitude assumes that the overlapping BOLD responses accumulate with linear additivity and, potentially, nonlinear effects may contribute to the altered hemodynamic responses. However, in computational simulation, we found little accumulation of the hemodynamic response. This is primarily due to the fact that our regressors are based on the subjects' response latency and the individual variations from trial to trial. Moreover, if nonlinearities (due to the task set up) are contributing to the effect, this would be similar across the different treatment conditions. Fifth, the Monte-Carlo threshold adjustment method (Forman, Cohen, Fitzgerald, Eddy, Mintun, and Noll, 1995) used to extract the activation clusters has been reported to possibly underestimate the spatial extent of autocorrelation or the variability of the autocorrelation function, thus, possibly interfering with the critical levels for a significant cluster size (Petersson, Nichols et al., 1999). Finally, the lack of jittering in our paradigm may cause a certain degree of overlap between the decision and outcome periods. Therefore, other paradigms will need to be used in the future to better delineate whether lorazepam differentially affects uncertainty or reward/punishment processing.
In summary, this investigation used three techniques - human psychopharmacology, functional neuroimaging, and an experimental paradigm targeted to probe risk-related responding - to elucidate the role of GABAergic modulation on brain structures important for processing risk. The results are consistent with the hypothesis that GABAergic modulation attenuates activation of neural substrates signaling aversive outcomes, which may help to explain why these drugs are anxiolytic. Future investigations will need to determine whether these effects are specific to benzodiazepines, are characteristic of other anxiolytic drugs, and can be used to predict potential anxiolytic efficacy of novel compounds.
Acknowledgements
We would like to acknowledge the invaluable help of Thuy Le. This work was supported by grants from NIH (DA13186, MPP; MH65413, MBS).
Reference List
- 1.Arana FS, Parkinson JA, Hinton E, Holland AJ, Owen AM, Roberts AC. Dissociable contributions of the human amygdala and orbitofrontal cortex to incentive motivation and goal selection. J Neurosci. 2003 Oct 22;23:9632–9638. doi: 10.1523/JNEUROSCI.23-29-09632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baas JM, Grillon C, Bocker KB, Brack AA, Morgan CA, III, Kenemans JL, Verbaten MN. Benzodiazepines have no effect on fear-potentiated startle in humans. Psychopharmacology (Berl) 2002;161:233–247. doi: 10.1007/s00213-002-1011-8. [DOI] [PubMed] [Google Scholar]
- 3.Bechara A. Neurobiology of decision-making: risk and reward. Semin.Clin.Neuropsychiatry. 2001;6:205–216. doi: 10.1053/scnp.2001.22927. [DOI] [PubMed] [Google Scholar]
- 4.Bechara A, Damasio H. Decision-making and addiction (part I): impaired activation of somatic states in substance dependent individuals when pondering decisions with negative future consequences. Neuropsychologia. 2002;40:1675–1689. doi: 10.1016/s0028-3932(02)00015-5. [DOI] [PubMed] [Google Scholar]
- 5.Botvinick M, Nystrom LE, Fissell K, Carter CS, Cohen JD. Conflict monitoring versus selection-for-action in anterior cingulate cortex. Nature. 1999 Nov 11;402:179–181. doi: 10.1038/46035. [DOI] [PubMed] [Google Scholar]
- 6.Boynton GM, Engel SA, Glover GH, Heeger DJ. Linear systems analysis of functional magnetic resonance imaging in human V1. J Neurosci. 1996 Jul 1;16:4207–4221. doi: 10.1523/JNEUROSCI.16-13-04207.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burian SE, Liguori A, Robinson JH. Effects of alcohol on risk-taking during simulated driving. Hum.Psychopharmacol. 2002;17:141–150. doi: 10.1002/hup.384. [DOI] [PubMed] [Google Scholar]
- 8.Carter CS, Macdonald AM, Botvinick M, Ross LL, Stenger VA, Noll D, Cohen JD. Parsing executive processes: strategic vs. evaluative functions of the anterior cingulate cortex. Proc.Natl.Acad.Sci.U.S.A. 2000 Feb 15;97:1944–1948. doi: 10.1073/pnas.97.4.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen MX, Heller A, Ranganath C. Functional Connectivity with Anterior Cingulate and Orbitofrontal Cortices During Decision-Making. Cognitive Brain Research. 2005 doi: 10.1016/j.cogbrainres.2005.01.010. in press. [DOI] [PubMed] [Google Scholar]
- 10.Coricelli G, Critchley HD, Joffily M, O'Doherty JP, Sirigu A, Dolan RJ. Regret and its avoidance: a neuroimaging study of choice behavior. Nat.Neurosci. 2005;8:1255–1262. doi: 10.1038/nn1514. [DOI] [PubMed] [Google Scholar]
- 11.Coull JT, Frith CD, Dolan RJ. Dissociating neuromodulatory effects of diazepam on episodic memory encoding and executive function. Psychopharmacology (Berl.) 1999;145:213–222. doi: 10.1007/s002130051051. [DOI] [PubMed] [Google Scholar]
- 12.Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Computers and Biomedical Research. 1996;29:162–173. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- 13.Craig AD. How do you feel? Interoception: the sense of the physiological condition of the body. Nat.Rev.Neurosci. 2002;3:655–666. doi: 10.1038/nrn894. [DOI] [PubMed] [Google Scholar]
- 14.Craig AD. A new view of pain as a homeostatic emotion. Trends Neurosci. 2003;26:303–307. doi: 10.1016/s0166-2236(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 15.Damasio AR. The somatic marker hypothesis and the possible functions of the prefrontal cortex. Philos.Trans.R.Soc.Lond.B.Biol.Sci. 1996 Oct 29;351:1413–1420. doi: 10.1098/rstb.1996.0125. [DOI] [PubMed] [Google Scholar]
- 16.Ernst M, Bolla K, Mouratidis M, Contoreggi C, Matochik JA, Kurian V, Cadet JL, Kimes AS, London ED. Decision-making in a Risk-taking Task. A PET Study. Neuropsychopharmacology. 2002;26:682–691. doi: 10.1016/S0893-133X(01)00414-6. [DOI] [PubMed] [Google Scholar]
- 17.Ernst M, Dickstein DP, Munson S, Eshel N, Pradella A, Jazbec S, Pine DS, Leibenluft E. Reward-related processes in pediatric bipolar disorder: a pilot study. J.Affect.Disord. 2004;82(Suppl 1):S89–S101. doi: 10.1016/j.jad.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 18.Ernst M, Nelson EE, Jazbec S, McClure EB, Monk CS, Leibenluft E, Blair J, Pine DS. Amygdala and nucleus accumbens in responses to receipt and omission of gains in adults and adolescents. Neuroimage. 2005 May 1;25:1279–1291. doi: 10.1016/j.neuroimage.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 19.Ernst M, Nelson EE, McClure EB, Monk CS, Munson S, Eshel N, Zarahn E, Leibenluft E, Zametkin A, Towbin K, Blair J, Charney D, Pine DS. Choice selection and reward anticipation: an fMRI study. Neuropsychologia. 2004;42:1585–1597. doi: 10.1016/j.neuropsychologia.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Forman SD, Cohen JD, Fitzgerald M, Eddy WF, Mintun MA, Noll DC. Improved assessment of significant activation in functional magnetic resonance imaging (fMRI): use of a cluster-size threshold. Magnetic Resonance in Medicine. 1995;33:636–647. doi: 10.1002/mrm.1910330508. [DOI] [PubMed] [Google Scholar]
- 21.Frith CD, Done DJ. Towards a neuropsychology of schizophrenia. Br.J.Psychiatry. 1988;153:437–443. doi: 10.1192/bjp.153.4.437. [DOI] [PubMed] [Google Scholar]
- 22.Fukui H, Murai T, Fukuyama H, Hayashi T, Hanakawa T. Functional activity related to risk anticipation during performance of the Iowa Gambling Task. Neuroimage. 2005 Jan 1;24:253–259. doi: 10.1016/j.neuroimage.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Gehring WJ, Willoughby AR. The medial frontal cortex and the rapid processing of monetary gains and losses. Science. 2002 Mar 22;295:2279–2282. doi: 10.1126/science.1066893. [DOI] [PubMed] [Google Scholar]
- 24.George S, Rogers RD, Duka T. The acute effect of alcohol on decision making in social drinkers. Psychopharmacology (Berl) 2005 Jul 20;:1–10. doi: 10.1007/s00213-005-0057-9. [DOI] [PubMed] [Google Scholar]
- 25.Grant S, Contoreggi C, London ED. Drug abusers show impaired performance in a laboratory test of decision making. Neuropsychologia. 2000;38:1180–1187. doi: 10.1016/s0028-3932(99)00158-x. [DOI] [PubMed] [Google Scholar]
- 26.Greenblatt DJ, Harmatz JS, Dorsey C, Shader RI. Comparative single-dose kinetics and dynamics of lorazepam, alprazolam, prazepam, and placebo. Clin.Pharmacol.Ther. 1988;44:326–334. doi: 10.1038/clpt.1988.158. [DOI] [PubMed] [Google Scholar]
- 27.Harmer CJ, Rogers RD, Tunbridge E, Cowen PJ, Goodwin GM. Tryptophan depletion decreases the recognition of fear in female volunteers. Psychopharmacology (Berl) 2003;167:411–417. doi: 10.1007/s00213-003-1401-6. [DOI] [PubMed] [Google Scholar]
- 28.Hsu M, Bhatt M, Adolphs R, Tranel D, Camerer CF. Neural systems responding to degrees of uncertainty in human decision-making. Science. 2005 Dec 9;310:1680–1683. doi: 10.1126/science.1115327. [DOI] [PubMed] [Google Scholar]
- 29.Huettel SA, Stowe CJ, Gordon EM, Warner BT, Platt ML. Neural signatures of economic preferences for risk and ambiguity. Neuron. 2006 Mar 2;49:765–775. doi: 10.1016/j.neuron.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 30.Kahn I, Yeshurun Y, Rotshtein P, Fried I, Ben Bashat D, Hendler T. The role of the amygdala in signaling prospective outcome of choice. Neuron. 2002 Mar 14;33:983–994. doi: 10.1016/s0896-6273(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 31.Kahneman D, Tversky A. Choices, Values and Frames. Cambridge University Press; Cambridge, UK: 2000. [Google Scholar]
- 32.Kahneman D, Tversky A. Prospect theory: An analysis of decision under risk. Econometrica. 1979;47:263–291. [Google Scholar]
- 33.Kahneman D, Tversky A. Choices, values, and frames. American Psychologist. 1984;39:341–350. [Google Scholar]
- 34.Knutson B, Cooper JC. Functional magnetic resonance imaging of reward prediction. Curr.Opin.Neurol. 2005;18:411–417. doi: 10.1097/01.wco.0000173463.24758.f6. [DOI] [PubMed] [Google Scholar]
- 35.Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Neuroreport. 2001 Dec 4;12:3683–3687. doi: 10.1097/00001756-200112040-00016. [DOI] [PubMed] [Google Scholar]
- 36.Kyriakopoulos AA, Greenblatt DJ, Shader RI. Clinical Pharmacokinetics of Lorazepam - Review. Journal of Clinical Psychiatry. 1978;39:16–23. [PubMed] [Google Scholar]
- 37.Lancaster JL, Woldorff MG, Parsons LM, Liotti M, Freitas CS, Rainey L, Kochunov PV, Nickerson D, Mikiten SA, Fox PT. Automated Talairach atlas labels for functional brain mapping. Hum Brain Mapp. 2000;10:120–131. doi: 10.1002/1097-0193(200007)10:3<120::AID-HBM30>3.0.CO;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lane SD, Cherek DR, Pietras CJ, Tcheremissine OV. Alcohol effects on human risk taking. Psychopharmacology (Berl) 2004;172:68–77. doi: 10.1007/s00213-003-1628-2. [DOI] [PubMed] [Google Scholar]
- 39.Lane SD, Cherek DR, Tcheremissine OV, Lieving LM, Pietras CJ. Acute marijuana effects on human risk taking. Neuropsychopharmacology. 2005;30:800–809. doi: 10.1038/sj.npp.1300620. [DOI] [PubMed] [Google Scholar]
- 40.Lane SD, Tcheremissine OV, Lieving LM, Nouvion S, Cherek DR. Acute effects of alprazolam on risky decision making in humans. Psychopharmacology (Berl) 2005 Apr 14; doi: 10.1007/s00213-005-2265-8. [DOI] [PubMed] [Google Scholar]
- 41.Lane SD, Cherek DR. Analysis of risk taking in adults with a history of high risk behavior. Drug & Alcohol Dependence. 2000;60:179–187. doi: 10.1016/s0376-8716(99)00155-6. [DOI] [PubMed] [Google Scholar]
- 42.Leland DS, Paulus MP. Increased risk-taking decision-making but not altered response to punishment in stimulant-using young adults. Drug Alcohol Depend. 2005 Apr 4;78:83–90. doi: 10.1016/j.drugalcdep.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 43.Loewenstein GF, Weber EU, Hsee CK, Welch N. Risk as feelings. Psychological Bulletin. 2001;127:267–286. doi: 10.1037/0033-2909.127.2.267. [DOI] [PubMed] [Google Scholar]
- 44.Madden GJ, Bickel WK, Jacobs EA. Discounting of delayed rewards in opioid-dependent outpatients: exponential or hyperbolic discounting functions? Exp.Clin.Psychopharmacol. 1999;7:284–293. doi: 10.1037//1064-1297.7.3.284. [DOI] [PubMed] [Google Scholar]
- 45.Marowsky A, Fritschy JM, Vogt KE. Functional mapping of GABA A receptor subtypes in the amygdala. Eur.J Neurosci. 2004;20:1281–1289. doi: 10.1111/j.1460-9568.2004.03574.x. [DOI] [PubMed] [Google Scholar]
- 46.Millan MJ. The neurobiology and control of anxious states. Prog.Neurobiol. 2003;70:83–244. doi: 10.1016/s0301-0082(03)00087-x. [DOI] [PubMed] [Google Scholar]
- 47.Monterosso J, Ehrman R, Napier KL, O'Brien CP, Childress AR. Three decision-making tasks in cocaine-dependent patients: do they measure the same construct? Addiction. 2001;96:1825–1837. doi: 10.1046/j.1360-0443.2001.9612182512.x. [DOI] [PubMed] [Google Scholar]
- 48.Norusis MJ. SPSS base system user's guide. SPSS Inc.; Chicago: 1990. [Google Scholar]
- 49.Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch.Gen.Psychiatry. 2005;62:282–288. doi: 10.1001/archpsyc.62.3.282. [DOI] [PubMed] [Google Scholar]
- 50.Paulus MP, Rogalsky C, Simmons A, Feinstein JS, Stein MB. Increased activation in the right insula during risk-taking decision making is related to harm avoidance and neuroticism. Neuroimage. 2003;19:1439–1448. doi: 10.1016/s1053-8119(03)00251-9. [DOI] [PubMed] [Google Scholar]
- 51.Petersson KM, Nichols TE, Poline JB, Holmes AP. Statistical limitations in functional neuroimaging. II. Signal detection and statistical inference. Philos.Trans.R.Soc.Lond B Biol.Sci. 1999 Jul 29;354:1261–1281. doi: 10.1098/rstb.1999.0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petry NM, Bickel WK, Arnett M. Shortened time horizons and insensitivity to future consequences in heroin addicts. Addiction. 1998;93:729–738. doi: 10.1046/j.1360-0443.1998.9357298.x. [DOI] [PubMed] [Google Scholar]
- 53.Quirk GJ, Gehlert DR. Inhibition of the amygdala: key to pathological states? Ann.N.Y.Acad.Sci. 2003;985:263–272. doi: 10.1111/j.1749-6632.2003.tb07087.x. [DOI] [PubMed] [Google Scholar]
- 54.Riba J, Rodriguez-Fornells A, Urbano G, Morte A, Antonijoan R, Barbanoj MJ. Differential effects of alprazolam on the baseline and fear-potentiated startle reflex in humans: a dose-response study. Psychopharmacology (Berl) 2001;157:358–367. doi: 10.1007/s002130100816. [DOI] [PubMed] [Google Scholar]
- 55.Richmond BJ, Liu Z, Shidara M. Neuroscience. Predicting future rewards. Science. 2003 Jul 11;301:179–180. doi: 10.1126/science.1087383. [DOI] [PubMed] [Google Scholar]
- 56.Ridderinkhof KR, Ullsperger M, Crone EA, Nieuwenhuis S. The role of the medial frontal cortex in cognitive control. Science. 2004 Oct 15;306:443–447. doi: 10.1126/science.1100301. [DOI] [PubMed] [Google Scholar]
- 57.Roache JD, Griffiths RR. Lorazepam and meprobamate dose effects in humans: behavioral effects and abuse liability. J Pharmacol.Exp.Ther. 1987;243:978–988. [PubMed] [Google Scholar]
- 58.Rush CR, Higgins ST, Bickel WK, Hughes JR. Acute effects of triazolam and lorazepam on human learning, performance and subject ratings. J Pharmacol.Exp.Ther. 1993;264:1218–1226. [PubMed] [Google Scholar]
- 59.Rush CR, Higgins ST, Bickel WK, Hughes JR. Acute behavioral effects of lorazepam and caffeine, alone and in combination, in humans. Behav.Pharmacol. 1994;5:245–254. doi: 10.1097/00008877-199406000-00003. [DOI] [PubMed] [Google Scholar]
- 60.Rushworth MF, Walton ME, Kennerley SW, Bannerman DM. Action sets and decisions in the medial frontal cortex. Trends Cogn Sci. 2004;8:410–417. doi: 10.1016/j.tics.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 61.Slovic P. Perception of risk. Science. 1987;236:280–285. doi: 10.1126/science.3563507. [DOI] [PubMed] [Google Scholar]
- 62.Slovic PE. Perception of Risk. Earthscan Publications Ltd; London: 2000. [Google Scholar]
- 63.Sperling R, Greve D, Dale A, Killiany R, Holmes J, Rosas HD, Cocchiarella A, Firth P, Rosen B, Lake S, Lange N, Routledge C, Albert M. Functional MRI detection of pharmacologically induced memory impairment. Proc.Natl.Acad.Sci.U.S.A. 2002 Jan 8;99:455–460. doi: 10.1073/pnas.012467899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spielberger CD. Manual for the State-Trait Anxiety Inventory (Form Y) Consulting Psychologists Press; Palo Alto, CA: 1983. [Google Scholar]
- 65.Stein EA, Pankiewicz J, Harsch HH, Cho JK, Fuller SA, Hoffmann RG, Hawkins M, Rao SM, Bandettini PA, Bloom AS. Nicotine-induced limbic cortical activation in the human brain: a functional MRI study. Am.J Psychiatry. 1998;155:1009–1015. doi: 10.1176/ajp.155.8.1009. [DOI] [PubMed] [Google Scholar]
- 66.Zezula J, Cortes R, Probst A, Palacios JM. Benzodiazepine receptor sites in the human brain: autoradiographic mapping. Neuroscience. 1988;25:771–795. doi: 10.1016/0306-4522(88)90036-x. [DOI] [PubMed] [Google Scholar]