

Abstract
Mre11 nuclease plays a central role in the repair of cytotoxic and mutagenic DNA double-strand breaks (DSBs). As x-ray structural information has only been available for the Pyrococcus furiosus enzyme (PfMre11), the conserved and variable features of this nuclease across the domains of life have not been experimentally defined. Our crystal structure and biochemical studies demonstrate that TM1635 from Thermotoga maritima, originally annotated as a putative nuclease, is the Mre11 endo/exonuclease from T. maritima (TmMre11) and the first such structure from eubacteria. TmMre11 and PfMre11 display similar overall structures, despite sequence identity in the twilight zone of only ∼20%. However, they differ substantially in their DNA specificity domains and in their dimeric organization. Residues in the nuclease domain are highly conserved, but those in the DNA specificity domain are not. The structural differences likely affect how Mre11s from different organisms recognize and interact with single-stranded DNA, double-stranded DNA and DNA hairpin structures during DNA repair. The TmMre11 nuclease active site has no bound metal ions, but is conserved in sequence and structure with exception of a histidine that is important in PfMre11 nuclease activity. Nevertheless, biochemical characterization confirms that TmMre11 possesses both endonuclease and exonuclease activities on ssDNA and dsDNA substrates, respectively.
Keywords: Mre11, SbcD, endonuclease, exonuclease, crystal structure, DNA repair, structural genomics, Thermotoga maritima
Introduction
Double-stranded DNA damage can occur in intermediate steps during normal cellular processes, such as meiosis, mating type switching and V(D)J recombination 1,2,3,4, as well as during environmental insults, such as exposure to ionizing radiation and genotoxic chemicals. Mre11 (Meiotic recombination 11) and SbcD proteins are orthologs involved in double-strand DNA break (DSB) detection and repair by Homologous Recombination and Non-Homologous End Joining (NHEJ) of damaged DNA. Mre11 is the central player in the eukaryotic triad of MRN proteins (Mre11-Rad50-Nbs1/Xsr2) and in the prokaryotic MR (Mre11-Rad50) assembly that are crucial to the repair process.5,6 Mre11 has broad substrate recognition and can act on single-stranded DNA (ssDNA), double-stranded DNA (dsDNA) and DNA hairpin structures. Its functionality is characterized by ssDNA endonuclease activity and ATP-dependent dsDNA 3′-5′ exonuclease activity. These enzymes can process and tether DNA ends during gene repair. 6,7,8,9,10,11,12
Mre11 enzymes are present in all kingdoms of life and share significant sequence similarities in their nuclease domain. 13,14,15,16,17,18 In vivo studies of Mre11 activity in Saccharomyces cerevisiae and Schizosaccharomyces pombe have demonstrated that Mre11 is critical for survival when DNA is damaged by ionizing radiation and gene toxins. In humans, mutations in Mre11 and Nsb1 cause ataxia telangiectasia-like disorder (ATLD) and Nijmegen breakage syndrome (NBS), 19,20,21,22 which result in a greater susceptibility to cancer. Mre11 is directly implicated in some cancers and disrupting the activity of Mre11 or other MRN members may provide effective therapeutic interventions in cancer treatment. 23,24 Yeast has been used as an in vivo platform for probing Mre11 function and for EM studies. 9,25 X-ray crystallographic studies of Mre11, which have so far been confined to the PfMre11, have provided a structural basis for understanding the complex activity of this important enzyme.
The crystal structure of PfMre11 with metal and dAMP in the active site (PDB accession id 1ii7) defined the secondary structures of the nuclease and DNA specificity domains, the relative spatial orientation of the two domains of the protein, the dimeric organization in the crystal structure, and helped to elucidate the mechanism of the nuclease activity. The structure of the PfMre11 nuclease-defective H85L mutant (PfMre11-3, PDB id 1s8e) aided in clarifying the role of His85 as a proton donor in cleavage of the DNA phosphodiester backbone. 12,26 PfMre11 crystal structures, as well as small angle x-ray scattering (SAXS) solution structures, revealed a consistent dimeric organization. Recently, PfMre11 structures in complex with synaptic and branched DNA complexes (PDB ids 3dsc and 3dsd) 27 also indicated a role for the dimer in assembly of the functional complex and how 17 residues located in six DNA recognition loops interact with DNA.
Despite these advances, questions have remained about structure-function relationships in Mre11s. For example, how does the variable size and sequence of Mre11s from other organisms translate into structural and functional differences? What oligomeric architectures are adopted by different Mre11s, and what structural features define the DNA specificity and the ability to discriminate between ssDNA, dsDNA and DNA hairpin substrates during DNA repair? We present here the crystal structure and biochemical characterization of TmMre11 (product of gene tm1635). Notably, TmMre11 provides a second Mre11 protein crystal structure to probe Mre11 structure-function relationships. This new structure reveals a unique dimeric assembly and structural differences in the nuclease and DNA specificity domains may be important for modulating Mre11 activity and aid in understanding the key features of Mre11 in higher eukaryotes and humans.
Results and Discussion
Sequence analysis
TmMre11 (TM1635, UniProt: Q9X1X0_THEMA) has two domains. The N-terminal nuclease domain is a member of PFAM family PF00149 (calcineurin-like phosphoesterase family). 28 PF00149 is currently comprised of 9072 unique proteins from 1153 species spanning all kingdoms of life (as of March 2009). The C-terminal specificity domain has been assigned to the protein family PF04152 (Mre11 DNA-binding domain). According to the Conserved Domain Database, 29 TmMre11 is a member of COG0420 which is comprised of 281 exonucleases in 230 species. The most conserved residues in this family are in the vicinity of the metal chelating site containing the sequence DXH(X)25GDXXD(X)25GNHD/E. 30,31 Seven proteins possess a PF00149 domain in T. maritima, but only one is a probable Mre11 nuclease based on sequence similarity in a PSI-BLAST 32 search. The tm1635 gene is located next to the tm1636 gene that encodes for a putative Rad50 ATPase, TM1636 (UniProt: Q9X1X1/Rad50_THEMA), which presumably forms the MR complex with TM1635.
Sequence clustering at the 50% sequence identity level (by UniProt) reveals that Mre11s from Thermotoga neapolitana, Thermotoga petrophila, Marinitoga peizophila and Thermotoga sp are closest to TmMre11. The equivalent 50% sequence identity cluster for PfMre11 (which has only ∼20 % overall sequence identity to TmMre11) includes Mre11s from Pyrococcus kodakaraensis, Thermococcus barophilus, Thermococcus onnurineus and Pyrococcus horikoshii. Human (HuMre11) and yeast Mre11 (ScMre11) are larger in size (∼700 aa) compared to TmMre11 (385 aa) and PfMre11 (426 aa). TmMre11 is one of the smallest Mre11 proteins and possibly closer to the minimal ancestral unit. The overall sequence identity of TmMre11 to HuMre11 and ScMre11 is < 20%.
Structure determination
The crystal structure of TmMre11 was determined from a truncated construct (residues 1-324 of the 385 residues of the full-length protein) by MAD phasing to a resolution of 2.2 Å (Fig 1). This construct is similar in size to that used for the PfMre11 crystal structure, where it was shown that this deletion did not affect nuclease activity. 12 Data collection, model, and refinement statistics are summarized in Table 1. 33,34 The final model consists of two TmMre11 subunits (chains A and B) and 159 water molecules in the asymmetric unit (ASU). The two subunits are very similar in structure and superimpose with an RMSD of 0.8 Å over 292 Cα residues. Residues 93-98, 141-147, 188-195 and 311-312 in chain A and 1-6, 95-98, 141-149, 187-195 and 311-312 in chain B were disordered and were not modeled. The Matthews' coefficient 35 is 2.75 Å3/Da, with an estimated solvent content of 55%. The Ramachandran plot produced by Molprobity 36 showed that 95.7% and 0.5% of amino acids are in the favored and outlier regions, respectively.
Fig. 1. Crystal structure of TmMre11.

(A) Stereo ribbon diagram of the TmMre11 monomer color-coded from N-terminus (yellow) to C-terminus (green). TmMre11 is a ladle-shaped molecule with two domains. Helices H1–H8 and β-strands β1-β15 that comprise the Nuclease Domain (nuc_domain) (approximately residues 1-270) and Specificity Domain (spec_domain) (approximately residues 271-324) are indicated. The nuc_domain has a β-sandwich architecture formed by two 6-stranded β-sheets surrounded by α-helices. The spec_domain is formed by a 3-stranded β-sheet and two α-helices. (B) Diagram showing the secondary structure elements of TmMre11 superimposed on its primary sequence, adapted from PDBsum (http://www.ebi.ac.uk/pdbsum), where α-helices are sequentially labeled (H1, H2, H3 etc), β-strands labeled (β1, β2, β3, etc) and β-hairpins by red loops.
Table 1.
Crystallographic data and refinement statistics for TmMre11 (PDB 2q8u)
| Space group | P21 | P21 | ||
|---|---|---|---|---|
| Unit cell parameters | a = 47.60 Å, | a = 47.73 Å, | ||
| b = 113.41 Å, | b = 114.13 Å, | |||
| c = 80.95 Å, | c = 81.71 Å, | |||
| β = 101.77° | β = 101.86° | |||
| Data collection | λ1 Se | λ2 MAD-Se | λ3 MAD-Se | λ4 MAD-Se |
| Wavelength (Å) | 0.9809 | 0.9184 | 0.9792 | 0.9789 |
| Resolution range (Å) | 37.5-2.20 | 29.5-2.40 | 29.5-2.40 | 29.5-2.40 |
| Number of observations | 109,289 | 126,788 | 126,646 | 126,320 |
| Number of unique reflections | 42,158 | 33,290 | 33,269 | 33,270 |
| Completeness (%) | 98.8 (99.5)a | 99.3 (96.8)a | 99.2 (95.8)a | 99.2 (96.5)a |
| Mean I/σ(I) | 12.5 (2.2)a | 15.5 (2.1)a | 14.3 (2.0)a | 14.1 (1.9)a |
| Rsym on I (%) | 4.3 (53.3)a | 5.8 (59.1)a | 6.1 (60.9)a | 6.9 (66.0)a |
| Highest resolution shell(Å) | 2.26-2.20 | 2.46-2.40 | 2.46-2.40 | 2.46-2.40 |
| Model and refinement statistics | ||||
| Resolution range (Å) | 37.5-2.20 | Data set used in refinement | λ1 Se | |
| No. of reflections (total) | 42,148b | Cutoff criteria | |F|>0 | |
| No. of reflections (test) | 2,120 | Rcryst | 0.198 | |
| Completeness (% total) | 98.7 | Rfree | 0.241 | |
|
Stereochemical parameters | ||||
| Restraints (RMS observed) | ||||
| Bond angle (°) | 1.43 | |||
| Bond length (Å) | 0.016 | |||
| Average isotropic B-value (Å2) | 56.4 | |||
| ESU based on Rfree (Å) | 0.191 | |||
| Protein residues / atoms | 600 / 4858 | |||
| Water molecules | 159 | |||
Highest resolution shell
Rsym =Σ|Ii-<Ii>|/Σ|Ii|, where Ii is the scaled intensity of the ith measurement and <Ii> is the mean intensity for that reflection.
Rcryst = Σ‖Fobs|-|Fcalc‖/Σ|Fobs|, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively.
Rfree = as for Rcryst, but for 5.0% of the total reflections chosen at random and omitted from refinement.
Typically, the number of unique reflections used in refinement is slightly less that the total number that were integrated and scaled. Reflections are excluded due to systematic absences, negative intensities, and rounding errors in the resolution limits and cell parameters.
Overall structure
TmMre11 is a ladle-shaped molecule with two domains that meet at the nuclease active site. The N-terminal nuclease domain (nuc_domain, residues 1-270) is comprised of two 6-stranded β-sheets (β1- β12), which form a β-sandwich surrounded by 5 prominent α-helices (H1- H5). Several conserved phosphodiesterase motifs are located in the loops that link the core β-strands to the surrounding α-helices. The overall fold and the active site location resemble the calcineurin-like Ser/Thr phosphoesterase family (SCOP superfamily 56300: metal-dependent phosphatases) and APE1 (DNA base excision repair apurinic endonuclease 1). Although APE1 and Mre11 are both DNA repair enzymes, they possess different mechanisms of action. The APE1 mechanism involves binding of a conserved glutamic acid to Mg2+, whereas Mre11 uses seven residues 37 to coordinate a di-Mn2+ that is similar to the di-metal (Fe or Zn) mediated activity in Ser/Thr phosphatases. 38 The C-terminal DNA specificity domain (spec_domain, residues 271-324) is comprised of three β-strands (β13- β15) flanked by two α-helices (H7 and H8). This domain is unique to Mre11 and not present in other phosphatases or APE1.
The nuc_domain from both molecules in the ASU superimpose extremely well, with an RMSD of 0.4 Å over the 240 Cα residues of the domain, with the only significant deviations around Asn93 and His94 (disordered in molecule A) and Lys148 and Asn149 (disordered in molecule B). The spec_domain from both subunits in the ASU superimpose slightly less well with an RMSD of 0.7 Å over 52 Cα residues, with the only significant deviation at Pro311 and Asp312 in a loop just prior to helix H8.
Comparison with the PfMre11 structure and human and yeast Mre11 proteins
Sequence similarities between the TmMre11, PfMre11, HuMre11 and ScMre11 were analyzed by performing a step-wise, multiple sequence alignment39 (CLUSTALPROF option with the BLOSUM matrix and a gap penalty of 1.0, The Biology Workbench 40) of the human and yeast protein sequences to the structure-based sequence alignment 41 of TmMre11 and PfMre11 (Fig 2A). This combined alignment was then mapped to the TmMre11 structure. 42
Fig. 2. Comparisons of different Mre11 proteins.


(A) Combination of structure- and sequence-based sequence alignment to analyze similarities and differences between Mre11 proteins from T. maritima, P. furiosus, human and S. cerevisiae. The TmMre11 and PfMre11 proteins have been aligned by a structure-based sequence alignment using their 3D coordinates (Protein Data Bank files) as input. This alignment was then used as an anchor for the sequence-based sequence alignment of the HuMre11 and ScMre11. The TmMre11 chain B, which has residues 1-6 disordered, was used since it has all of the catalytic site residues modeled in the structure. Chain A has residues 1-6, but does not have the active site His94. Therefore, in the figure, the alignment for TmMre11 starts after the dots at residue 7. All the proteins show regions of conserved residues in the nuc_domain, which are involved in endo- and exonucleolytic cleavage of DNA. No significant sequence conservation is found in the spec_domain, which suggests that this may lead to differences in recognition of single-stranded DNA, double-stranded DNA and DNA hairpin structures during the DNA repair process in different species. (B) Left panel: The structure of TmMre11 (blue) superimposed on PfMre11 (gray, red and green) reveals their overall similarity. However, the spec_dom of TmMre11 is smaller with 3 β-strands as opposed to 5 β-strands in PfMre11 (gray and red). The nuc_domain of PfMre11 also has an extra insertion (green) that is not found in TmMre11. Right panel: The spec_domain of TmMre11 (blue) and PfMre11 (gray) based on superimposing only by the nuc_domain of the two proteins reveals that the relative orientation of the spec_domain with respect to the nuc_domain is different in the two proteins. Thus, the combination of the smaller spec_domain and slightly different juxtaposition of the two domains in TmMre11 is likely to impact substrate recognition. (C) Comparison of the nuclease active site of TmMre11 (blue) with PfMre11 (gray). The PfMre11 His85 residue (magenta) that is critical in nuclease activity is above the plane of the other residues His206, His208, His10, His173 and Asp49 (all green) that form the rest of the active site and coordinate the metal (orange spheres). In TmMre11, His94 (red) corresponds to the PfMre11 His85 and is positioned differently that could be due to the lack of a metal in the active site. The other active site residues in the TmMre11 are His216, His218, His16, His180 and Asp58 (all cyan). The inset depicts a larger, focused view of the active site.
The TmMre11 structure superimposes reasonably well (Fig 2B-left) with PfMre11 (PDB id 1ii7, using molecule A from both protein structures) with an RMSD of 2.8 Å over 269 Cα atoms and a structure-based sequence alignment of ∼26% sequence identity 43 in contrast to the sequence-based alignment of ∼20% sequence identity The nuc_domains from TmMre11 and PfMre11 superimpose with an RMSD of 2.6 Å over 217 Cα atoms (27% sequence identity), whereas the spec_domains superimpose with an RMSD of 2.3 Å over 53 Cα atoms (19% sequence identity). If TmMre11 is superimposed on PfMre11 using only the nuc_domain, a small rotation of the spec_domain is evident (Fig 2B-right). Similar results are observed when different combinations of molecules are used for comparison, e.g. molecule B from the TmMre11 with molecule A from the PfMre11. This observation suggests some flexibility in the connection between the nuc_domain and the spec_domain, which could act as a hinge, allowing conformational changes in the presence of substrate.
TmMre11 is smaller and more compact than PfMre11 and does not have an extra β-strand insertion in the nuc_domain between β4 and β5 (residues 116-119, TmMre11 nomenclature). More interestingly, TmMre11 only has a 3-stranded β-sheet in the spec_domain. It lacks two additional β-strands in PfMre11 that are formed from an insertion in the nuc_domain between residues 234-249 (Fig 2B). Similar insertions are also present in HuMre11 and ScMre11, although smaller in length, and are only partially conserved in sequence with PfMre11 (Fig 2A). Since the spec_domain is involved in substrate specificity, these prominent structural differences may be associated with functional differences between the two proteins. Future biochemical experiments can be designed to test this hypothesis.
The most sequence conserved region in TmMre11 compared to other Mre11s is the nuclease active site in the nuc_domain that includes the phosphoesterase motifs. Experiments in S. cerevisiae have demonstrated that substitutions in these conserved residues result in heightened sensitivity to infrared radiation with defects in the repair of dsDNA breaks. 44 PfMre11 residues involved in nuclease activity (His85, Asn84, His10, His173, His206, His208, Asp8 and Asp49) are identical in TmMre11 (His94, Asn93, His16, His180, His216, His218, Asp14 and Asp58 respectively) and are also conserved in HuMre11 and ScMre11 (Fig 2A). Most of these residues are also well conserved amongst the 332 seed members of this protein family (based on PFAM alignment) and are, therefore, not unique to Mre11.
Biochemical studies with PfMre11 have revealed that the enzyme performs ssDNA endonuclease and dsDNA exonuclease activities at this active site. 12 Unlike the PfMre11 structures, the TmMre11 crystal structure lacks a di-metal at the active site and thus represents the apo form of the enzyme. The spatial locations of the five basic residues (His206, His208, His10, His173 and Asp49), that are involved in direct Mn2+ coordination and comprise the five phosphodiesterase motifs in the PfMre11, are equivalent to TmMre11 His216, His218, His16, His180 and Asp58 (Fig 2C). This metal coordination in PfMre11 is approximately planar. Asn84 and His85 are positioned just above the plane where they are involved in binding to a dAMP molecule in the presence of Mn2+. It is believed that PfMre11 His85 acts in completing the cleavage of the DNA phosphate backbone by donating a proton to the departing 3′-OH group. 12 In biochemical and crystallographic studies of the His85->Leu85 mutation in PfMre11 (PfMre11-3, corresponding to a disease mutation in human Mre11), the protein retained DNA binding ability, but its exonuclease activity was abolished. 26 However, the electron density around Leu85 was diffuse in the PfMre11-H85L structure, which suggested the mutation disrupted metal binding and led to loss of nuclease activity. Interestingly, in TmMre11, the loop that contains Asn93 and His94 (corresponding to PfMre11 Asn84 and His85) is disordered in chain A of the crystal structure, but is observed in chain B. Its location results in His94 being farther above the central plane of the active site compared to His85 of PfMre11. This structural difference is most likely due to the absence of a coordinated metal and/or dAMP in the TmMre11 crystal structure, though it could also possibly represent a difference in active site geometry that may have some functional implications. However, all except one of the PfMre11 structures are with metal bound and all show the same active site geometry.
Oligomeric form and DNA binding residues
PfMre11 was determined to be a dimer in solution by size-exclusion chromatography and by small-angle x-ray scattering. 12 All crystal structures of PfMre11 with and without DNA (PDB ids 1ii7, 1s8e, 3dsc and 3dsd) displayed a consistent dimeric organization with a total buried interface of ∼1450 Å2 (PISA45) formed from 21 residues involved in salt-bridges, hydrogen bonds and hydrophobic interactions (Fig 3). In contrast, 5 of the 7 constructs (residues 1-324, 1-327, 1-333, 1-336 and 1-345), engineered to obtain diffraction quality crystals of the TmMre11, were found by size-exclusion chromatography coupled with static light scattering to be monomers in solution (n.b. the remaining 2 constructs were not tested). However, crystal packing analysis (PISA 45) of TmMre11 predicts that a dimer formed from the two molecules in the asymmetric unit, with a buried surface area of ∼1580Å2 and a Complexation Significance Score of 1.0 (ranges from 0 to 1 as complexation relevance increases), is the significant oligomeric form. This interface involves ∼17 residues (Val68, Leu71, His72, Leu75, Leu78, Lys79, Met82, Leu99, Leu101, Phe102, Asn104, Phe105, Val106, Ser108, Ile109, Ser110 and Ile113, that are involved in hydrophobic interactions and hydrogen bonds. This dimeric arrangement (mediated by α-helices H2 and H3) is significantly different from PfMre11 (also mediated by H2 and H3, but with different relative orientations). This TmMre11 dimer interface is dependent on Phe102 and Phe105 in H3 (Fig 3). Phe102 is present as a Leu or Ile residue in PfMre11, HuMre11 and ScMre11, His72 is conserved in HuMre11, and Leu75, Leu78, Lys79 and Ile113 are conserved in TmMre11, PfMre11, HuMre11 and ScMre11. The conservation of these residues across different species suggests that they may be functionally important and the TmMre11 dimer observed in the crystal structure may represent a biologically relevant oligomer.
Fig. 3. Oligomerization in TmMre11 and PfMre11.

(A) The TmMre11 dimer is viewed along the non-crystallographic 2-fold axis of the dimer (blue and green). (B) The PfMre11 dimer (gray and red) is depicted in approximately the same orientation as the TmMre11 dimer, revealing the difference in dimeric assembly as observed in the crystal structures. (C) Details of the dimer interface of TmMre11 are shown. Phe102 (orange) and Phe105 (cyan) in helices H3 and H3′ play a key role in assembly of the dimer interface. His72 (yellow) is also present in the HuMre11. Leu75 (grey), Leu78 (pink), Lys79 (blue) and Ile113 (magenta) are found in TmMre11, PfMre11, HuMre11 and ScMre11. Conservation of these residues at the dimer interface suggests that they may be needed for dimer formation.
Recent crystal structures of PfMre11 bound to DNA in synaptic and branched DNA complexes 27 have revealed the protein-DNA interactions. Of the 17 residues (Tyr13, Glu14, Phe16, His17, Lys18, Pro19, His52, Ser57, Arg55, Gln89, Lys111, Trp150, Tyr325, Lys327, Lys318, Arg90 and Tyr301) in the six DNA recognition loops, the following residues are conserved in the same location in TmMre11: Trp22, His61, Ser67, Lys322 and Tyr298 (corresponding to Phe16, His52, Ser57, Lys327 and Tyr301 in PfMre11) (Fig 2A). In addition, Arg26 and Pro27 in TmMre11 are in the vicinity of Lys18 and Pro19 in PfMre11. All seven of these residues are also conserved in the other four Mre11s that are within the TmMre11 sequence cluster. However, the 17 PfMre11 DNA binding residues are only partially conserved in HuMre11 and ScMre11. For example, PfMre11 Arg90 is involved in a key interaction with minor groove DNA, but is not conserved in HuMre11 or ScMre11. Similarly, PfMre11 His17, Lys18, Gln89, Lys111 (present in the extra β-strands corresponding to the region between β4 and β5 in the nuc_domain of TmMre11), Trp150, Tyr301, Lys318 and Tyr325 are also not conserved in the HuMre11 or ScMre11. Thus, TmMre11, PfMre11 HuMre11 and ScMre11 are likely to possess interesting differences in activity and in their DNA substrate specificity. Additionally, it is likely that Mre11s from different organisms have DNA recognition and specificity modes that are organism specific and vary with the type of repair substrates that they encounter, but nevertheless still retain a similar nuclease mechanism. This notion is supported by an analysis of the sequence-structure properties of the least conserved region in TmMre11, the spec_domain. A PSI-BLAST search was performed (3 iterations of the PSSM-Position Specific Scoring Matrix with standard search parameters) using residues 271-385 of the full-length sequence of the spec_domain, which revealed that the sequence of this region is very specific (53%-96% sequence identity, e-value=2e-39 to 8e-35) to TmMre11 and the corresponding proteins from T. petrophila RKU-1, M. piezophila KA3, T. sp. RQ2 and T. neapolitana DSM 4359. Sequence alignment in the spec_domain of these five TmMre11-like Mre11s indicates that 11 residues (Lys273, Tyr276, Tyr277, Lys278, Lys279, Lys286, Arg289, Phe291, Arg293, Phe295 and Tyr303) are conserved. Of these, Tyr276, Lys278, Lys286 and Phe295 correspond in PfMre11 to Lys277, Lys279, Lys290 and Lys298 (same location, but two are different in sequence), and could be involved in DNA binding (Fig 4A). The rest of the 11 residues do not have any charged (Lys/Arg) or aromatic residue equivalents in PfMre11. The next most significant matches (sequence identity falls from 53% to 26%) are with proteins from T. lettingae TMO (24% sequence identity) and Thermus thermophilus HB8 (26% sequence identity). Mapping of the evolutionarily conserved residues using 374 unique Mre11/SbcD proteins for alignment (using the full protein sequence in the TmMre11 structure, e-value=1e-164 to 0.007 and sequence identities of 91%-21% compared to TmMre11), 46 indicates a similar lack of conservation in the spec_domain and the presence of highly conserved regions in the nuc_domain (Fig 4B).
Fig. 4. DNA binding residues in TmMre11.

(A) Residues at the nuclease active site (cyan) are conserved between the TmMre11 and PfMre11. Several other TmMre11 residues (Trp22, His61, Ser67, Lys322, Tyr298, Arg26 and Pro27) that are the counterparts of DNA binding residues in PfMre11 are shown. Additionally, other residues in the spec_domain that are conserved in TmMre11 and orthologs from T. petrophila RKU-1, M. piezophila KA3, T. sp. RQ2 and T. neapolitana DSM 4359 and that may be involved in substrate recognition and discrimination are shown. (B) Surface representation of the TmMre11 monomer showing color-coded according to amino acid conservation based on comparison to 374 unique Mre11/SbcD proteins. The highest conservation is seen in the nuc_domain (red spheres) and least conservation in the spec_domain (cyan spheres).
Biochemical characterization of TmMre11
As TM1635 was originally annotated as a putative nuclease, we performed Mre11 nuclease assays on the protein when the crystal structure revealed it to be a possible Mre11. For endonuclease activity, two assays were performed on the full-length TmMre11 (residues 1-385) on ssDNA substrate. In one set of experiments, the activity of TmMre11 was tested on bacteriophage φX174 at four different temperatures 4 °C, 25 °C, 37 °C and 50 °C and at different time points (Fig 5A). TmMre11 is active on this substrate and its activity is higher as the temperature increases, as expected for a thermophile. In another set of assays, the endonuclease activity of TmMre11 was compared to that of PfMre11 at 50 °C and 37 °C (Fig 5B). Both enzymes showed higher activity at 50 °C vs. 37 °C. At 37 °C, PfMre11 did not appear to have significant activity in the first 30 minutes and TmMre11 had higher activity at 37°C compared to PfMre11. At 50 °C, the activity of TmMre11 was slightly higher than PfMre11.
Fig. 5. Biochemical characterization of TmMre11.
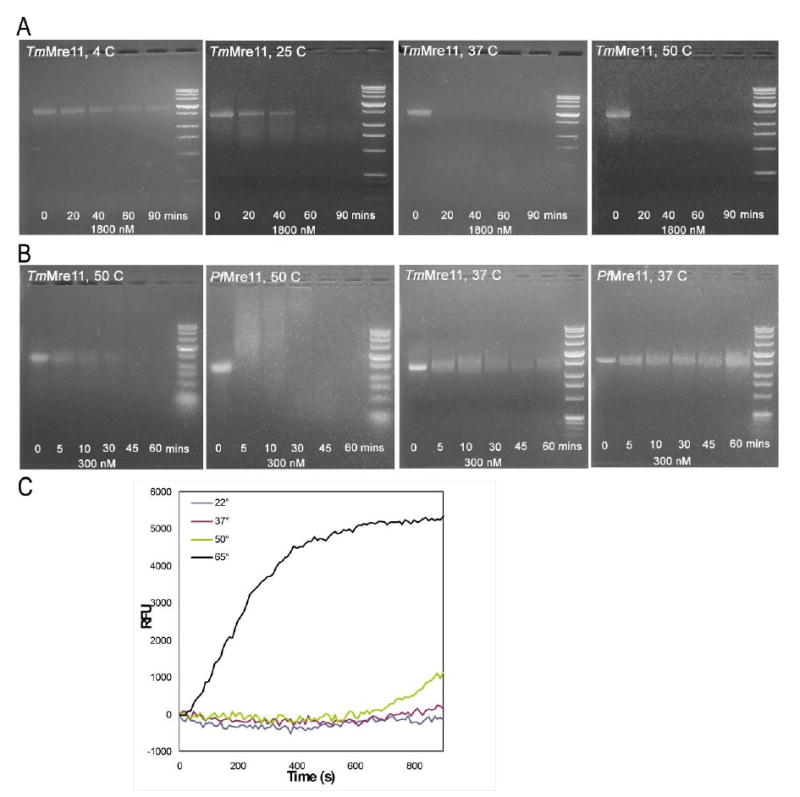
TmMre11 was originally annotated in UniProt as a putative nuclease. Assays were performed to verify Mre11 activity. (A) Endonuclease assay using TmMre11 with single-stranded bacteriophage φX174 DNA as substrate. From left to right, the gels are for reactions at 4 °C, 25 °C, 37 °C and 50 °C, respectively. The lanes on each gel, from left to right, are for incubation times of 0 (no enzyme control), 20, 40, 60 and 90 minutes, respectively. The right lane on each gel is the DNA standard. At lower temperatures, the DNA substrate is not substantially cleaved even over a long time period and the corresponding bands remain visible in the gel. At the higher temperatures, the disappearance of the substrate band indicates that endonucleolytic activity has occurred. Thus, TmMre11 is active on ssDNA substrate and the activity is directly proportional to the temperature of the reaction, which is expected for a thermophile. (B) Comparison of the endonuclease activities of TmMre11 and PfMre11 on bacteriophage φX174 at 37 °C and 50 °C. From left to right, the gels are for TmMre11 and PfMre11 at 50 °C followed by the reaction at 37 °C. On each gel, from left to right, the lanes are the control reaction (no enzyme), products at time 5, 10, 30, 45 and 60 minutes and the DNA marker standard. Both enzymes, being thermophilic, display higher activity at 50 °C, with TmMre11 showing slightly higher activity at this temperature. At 37 °C, PfMre11 does not appear to have significant activity in the first 30 minutes of the reaction and TmMre11 has higher activity. (C) Exonuclease assay on double-stranded DNA. The reaction graph depicts, in the y-axis, the RFU (Relative Fluorescence Unit) monitored in the release of 2-aminopurine bases from the 2-aminopurine containing DNA duplex due to exonuclease activity. The x-axis shows the time (in seconds) of the period (15 minutes) over which the reaction was allowed to proceed. TmMre11 shows efficient exonuclease activity at 65 °C and weaker activity at 50 °C. The 50 °C reaction has ∼10% increase in the last 5 minutes and the 65 °C reaction has ∼60% linear increase during that time.
For exonuclease activity, a fluorescence assay was performed to test the exonucleolytic release of 2-aminopurine from blunt dsDNA at 65 °C, 50 °C, 37 °C and 22 °C, following the methods used previously for characterization of PfMre11. Fluorescence emission data were collected for 15 minutes for each reaction. The two reactions at 37 °C and 22 °C did not show any fluorescence, indicating no exonuclease activity at the lower temperatures. At 50 °C, a low level of activity was observed after 10 minutes. At 65 °C, a constant increase in fluorescence was observed over the period of data collection, indicating efficient exonuclease activity (Fig 5C). In contrast, previous studies on PfMre11 revealed significant exonuclease activity at both 65°C and 50°C, with the optimum activity observed beyond 65°C. Difference in the size and structure of the spec_domain and the lack of conservation of residues implicated in DNA binding could account for some of the differences in the enzyme activity on the same substrates.
Endonuclease assays were also performed on TmMre11 mutants. To confirm that the observed nuclease activity was from TmMre11 and not due to the presence of any contaminating nuclease in the purified protein, we performed the endonuclease assay at 50 °C on the His94Ser active site mutant (Fig 6A). This residue is equivalent to the His85 of PfMre11 as discussed earlier. As expected, this substitution abolished the endonuclease activity and, thereby, confirmed that the results obtained in Fig 5A, 5B and 5C are from TmMre11 and not a contaminating nuclease. This result also confirmed that His94 is essential in TmMre11 function (and is probably involved in dAMP interaction, as observed for the His85 in PfMre11). Assays on mutants corresponding to two residues that are most likely involved in metal coordination, His180Ser (Fig 6B) and His216Ser (Fig 6C), show a partial loss of endonuclease activity. In PfMre11, mutation of His52, which is primarily involved in exonuclease activity, reveals a partial loss of endonuclease activity. 27 In TmMre11, the endonuclease assay on the His61Ser mutant (His61 is the counterpart of PfMre11 His52, Fig2A and Fig4A) also reveals a partial loss of endonuclease function (Fig 6D).
Fig. 6. Endonuclease assays on TmMre11 mutants.

Endonuclease assays were performed at 50 °C with single-stranded bacteriophage φX174 DNA as substrate. The lanes, from left to right, are for incubation times of 0 (no enzyme control), 5, 10, 20, 30 and 40 minutes, respectively. The left lane is the DNA standard. (A) The TmMre11-His94Ser mutant (which most likely interacts with dAMP and corresponds to His85 of PfMre11) results in total loss of function and no cleavage of substrate is observed, demonstrating that this residue is critical for biochemical activity. (B) and (C) Mutation of two of the five putative metal binding residues, His180Ser and His216Ser result in partial loss of function; substrate cleavage appears to occur ∼20 minutes after start of the reaction. (D) The His61Ser mutant (corresponds to His52 in PfMre11, whose primary role is in exonuclease activity, and mutation leads to partial loss of endonuclease activity) shows a partial loss of endonuclease function.
Conclusions
T. maritima proteins have been often used as pertinent examples for x-ray crystallographic studies of proteins involved in DNA damage responses including recombination repair by RuvB, 47 nucleotide excision repair by UvrC, 48 and deaminated base excision repair by endonuclease V. 49 Here, we employed TmMre11 to further examine the structure-function relationships for Mre11 in the initiation of double-strand break repair. TmMre11 provides a critical second Mre11 system to investigate Mre11 function as part of the complex with Rad50 ABC ATPase, which also has general implications for understanding ABC ATPase partnerships and functions. 50
Mre11 enzymatic and structural activities include end detection, end processing and alignment of DNA ends. 5 Notably, crystal structure comparisons of TmMre11 and PfMre11 reveal different dimeric assemblies that result in quaternary structure plasticity in the dimer interface region, which is implicated in differential recognition of DNA double-strand breaks and collapsed replication forks. This dimer interface plasticity identified here suggests that variations in dimer assembly may explain the proposed requirement for Mre11 to act symmetrically on DNA double-strand breaks with two ends formed by radiation and oxidative damage, but asymmetrically on DNA double-strand breaks with a single end formed during replication. 27 However, this hypothesis can only convincingly be tested by experimentally obtaining structural information of different dimeric arrangements of each enzyme with different DNA substrates. Furthermore, our results from size-exclusion chromatography coupled with static light scattering support the existence of a stable monomeric form of TmMre11 in solution, suggesting that the Mre11 dimer may dissociate into monomers during its role in recombination. Such dissociation of the Mre11 dimer would provide a possible mechanism to release Mre11 from DNA, allowing transfer for repair pathway progression, including possible loading of Rad51 and RPA in preparation for homologous recombination. The structural and sequence differences in the DNA specificity domains are likely to affect the recognition of different substrates (ssDNA, dsDNA and DNA hairpins) for repair. Thus, this new Mre11 structure reveals novel structural features and provides a new foundation for examination of TmMre11 interactions with DNA and with its Rad50 partner.
Materials and Methods
Protein production and crystallization
The full-length TM1635 gene (GenBank: AAD36702.1, GI: 4982208; Swiss-Prot: Q9X1X0) was amplified by polymerase chain reaction (PCR) from T. maritima genomic DNA using a phosphorylated forward primer (5′- GTGATTAATTTGAAGGAGCTCAAAATAC-3′) and a phosphorylated reverse primer (5′-GGCCTCACTCTTTTTCACCTCATCGAG-3′). The blunt ends of the PCR product were ligated to vector pMH1 (an arabinose-inducible derivative of Invitrogen's pBAD/Thio vector which encodes an amino-terminal MGSDKIHHHHHH tag) predigested with Pml I and Psi I. 51 As the full-length protein did not crystallize, six C-terminally truncated gene constructs encompassing residues 1-324, 1-327, 1-330, 1-333, 1-336, and 1-345 were made by PIPE mutagenesis, 52 using one universal forward primer (5′-taagtttaaacggtctccagcttggctgttttgg-3′) and one truncation-specific reverse primer for each construct (1-324 [5′-gaccgtttaaacttaCTCTATCTTCACCAGATTATCTATCTC-3′], 1-327 [5′-gaccgtttaaacttaTGACTTTCTCTCTATCTTCACCAGA-3′], 1-330 [5′-gaccgtttaaacttaCTCTCTTCTTGACTTTCTCTCTATCTTC-3′], 1-333 [5′-gaccgtttaaacttaCTCCTCTATCTCTCTTCTTGACTTTCTC-3′], 1-336 [5′-gaccgtttaaacttaTCGAAGCACCTCCTCTATCTCTCTTC-3′], and 1-345 [5′-gaccgtttaaacttaTTCTTCTTTGAACTCTTCCGGGCTTTC-3′]; upper case sequence is gene specific). From previous biochemical studies of PfMre11, deletions of the C-terminal residues are not expected to affect the nuclease activity of the enzyme. The construct encoding residues 1-324 was used for structure solution and was produced and crystallized as follows. Protein expression was performed in a selenomethionine-containing medium, with suppression of normal methionine synthesis, using the Escherichia coli strain GeneHogs (Invitrogen). At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/ml and the cells were harvested. After one freeze/thaw cycle, the cells were homogenized in Lysis Buffer [50 mM HEPES pH 8.0, 50 mM NaCl, 10 mM imidazole, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)] and passed through a Microfluidizer (Microfluidics). The lysate was clarified by centrifugation at 32,500g for 30 min and loaded onto nickel-chelating resin (GE Healthcare) pre-equilibrated with Lysis Buffer. The resin was washed with Wash Buffer [50 mM HEPES pH 8.0, 300 mM NaCl, 40 mM imidazole, 10% (v/v) glycerol, 1 mM TCEP] and the protein was eluted with Elution Buffer [20 mM HEPES pH 8.0, 300 mM imidazole, 10% (v/v) glycerol, 1 mM TCEP]. The eluate was buffer exchanged with HEPES Crystallization Buffer [20 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP] and concentrated for crystallization assays to 18 mg/ml by centrifugal ultrafiltration (Millipore).
Protein used for the biochemical assays (full-length construct, residues 1-385) was produced as follows. Expression was performed in LB media in 6l shaker flasks. The cells were grown at 37 °C with shaking at 250 rpm. At OD 0.5-0.6, L-(+)-arabinose was added to a final concentration of 0.02% (w/v) and the cells were grown for an additional 3-5 hours. In addition to nickel-chelating purification, the protein was further purified by ion-exhange and size-exclusion chromatography. For the ion-exchange step, the eluate from the nickel-chelating column was diluted 10× in 20 mM Tris pH 8.0, 50 mM NaCl, 5% (v/v) glycerol and 1 mM TCEP and loaded on to a 10 ml Resource Q column (GE Healthcare). Following a wash in 24 ml of the same buffer, the protein was eluted by a linear salt gradient from 50-500 mM NaCl. For the size-exclusion step, protein was loaded on a 120 ml Superdex 200 column (GE Healthcare) and purified in 20 mM HEPES pH 8.0, 200 mM NaCl and 1 mM TCEP. The His61Ser, His94Ser, His180Ser, and His216Ser substituted TmMre11 were produced by site-directed mutagenesis using standard protocols (QuikChange kit, Stratagene) and verified by sequencing (Eton Bioscience).
The purified protein was crystallized using the nanodroplet vapor diffusion method 53 with standard Joint Center for Structural Genomics (JCSG) crystallization protocols. 51 Sitting drops composed of 200 nl protein mixed with 200 nl crystallization solution (see below) were equilibrated against a 50-μl reservoir at 277K. Screening for diffraction was carried out using the Stanford Automated Mounting system (SAM) 54 at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA). Two crystals were used for structure determination. The first crystal, with dimensions of ∼ 0.1 × 0.1 × 0.1 mm3 and harvested after 27 days, grew from 2.5% (w/v) polyethylene glycol (PEG) 6000 and 0.1 M MES pH 5.14 and diffracted to 2.4 Å [1,2-ethanediol was used as cryoprotectant at a final concentration of 10% (v/v)], and was used for the initial phasing. The second crystal, with dimensions of ∼ 0.2 × 0.2 × 0.2 mm3 and harvested after 43 days, grew from 10.3% (w/v) PEG 6000 and 0.1 M MES pH 5.57 and diffracted to 2.2 Å [1,2-ethanediol was used as cryoprotectant at a final concentration of 25% (v/v)], and was used for the final crystallographic refinement. Both sets of diffraction data were indexed in monoclinic space group P21 (Table 1). For determination of the oligomeric state in solution, the various constructs of TM1635 were analyzed using a 1 × 30 cm2 Superdex 200 column (GE Healthcare) coupled with miniDAWN static light scattering and Optilab differential refractive index detectors (Wyatt Technology). The mobile phase consisted of 20 mM Tris pH 8.0, 150 mM NaCl, and 0.02% (w/v) sodium azide. The molar mass was calculated using ASTRA 5.1.5 software (Wyatt Technology).
Data collection, structure solution, and refinement
Multi-wavelength anomalous diffraction (MAD) data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL, SLAC National Laboratory, Menlo Park, CA) on beamline 11-1 at wavelengths corresponding to the high energy remote (λ2), inflection point (λ3) and peak (λ4) of a selenium MAD experiment to 2.4 Å resolution and for a higher resolution data set for refinement (λ1) to 2.2 Å resolution using the BLU-ICE 55 data collection environment. The data sets were collected at 100K using a MarMosaic 325 CCD detector (Mar USA). The MAD data were integrated and reduced using XDS 56 and scaled with the program XSCALE. Phasing was performed with SHELXD 57and autoSHARP 58. Density modification was performed with RESOLVE 59 and iterative RESOLVE 60 was used for automatic model building to 2.4 Å resolution. The higher resolution data set from the second crystal was integrated with MOSFLM 61 and scaled with SCALA 33. Model completion and crystallographic refinement were performed with this data set (λ1) using COOT 62 and REFMAC5. 63 The refinement protocol included the 2.4 Å experimental phase restraints in the form of Hendrickson-Lattman coefficients from SHARP, NCS restraints (positional weight 0.5, thermal weight 2) and TLS refinement with one TLS group per molecule. Data and refinement statistics are summarized in Table 1.
Validation and deposition
The quality of the crystal structure was analyzed using the JCSG Quality Control server. This server verifies: the stereochemical quality of the model using AutoDepInputTool, 64 MolProbity, 36 and WHATIF 5.0; 65 agreement between the atomic model and the data using SFcheck 4.0 66 and RESOLVE, 59 the protein sequence using CLUSTALW, 39 atom occupancies using MOLEMAN2, 67 consistency of NCS pairs, and evaluates difference in Rcryst/Rfree, expected Rfree/Rcryst and maximum/minimum B-factors by parsing the refinement log-file and PDB header. Protein quaternary structure analysis was performed using the PISA server. 45 Fig 1B was adapted from an analysis using PDBsum, 68 and all others were prepared with PyMOL. 69 Atomic coordinates and experimental structure factors for TmMre11 have been deposited in the PDB under the accession code 2q8u.
Endonuclease assays
For the first set of endonuclease assays (Fig 5A), endonucleolytic cleavage of circular ssDNA φX174 DNA (Virion DNA New England Biolabs) was tested with TmMre11 at 4 °C, 25 °C, 37 °C and 50 °C and time points of 0 (control), 20, 40, 60 and 90 minutes. 12 μL reactions contained 2 μg of purified TmMre11 (full-length, residues 1-385, ∼1800 nM assuming a dimer to be the functional form) and 1.5 μg of circular φX174 DNA in 25 mM HEPES pH7.0, 25 mM NaCl and 5 mM MnCl2. For the second set of assays (Fig 5B) for direct comparison of endonucleolytic cleavage of φX174 DNA by TmMre11 and PfMre11 (residues 1-342) at 50 °C and 37 °C, the reactions (10ul) contained 300 nM of purified TmMre11 or PfMre11, and 1.5 ug of circular φX174 DNA in 25 mM HEPES pH 7.0, 25 mM NaCl and 5mM MnCl2. Time points were taken at 0 (control), 5, 10, 30, 45 and 60 minutes. The first time point was taken at 5 min because the activity of both enzymes is high at 50 °C and stopping the reaction quickly enables differences to be detected early in the reaction. For endonuclease assays on the TmMre11 mutants (Fig 6), the experiments were performed as in Fig 5A except that time points were taken at 0 (control), 5, 10, 20, 30 and 40 minutes. A tube containing 12 ul of buffer and 1.5 ug of circular DNA was used as a control for all the experiments.
At the indicated time points, reactions were stopped by addition of EDTA 20 mM and 2 μL of 6X loading buffer containing 40% sucrose, 2.5 % SDS and 2.5 mg/mL Proteinase K. Reaction products were separated on 1.0 % agarose in 1X TAE and visualized with ethidium bromide staining.
Exonuclease assays
Steady state 2-aminopurine (2-AP) fluorescence 3′-5′ exonuclease assays were performed on a Fluoromax-3 (Horiba/Yobin-Yvon) fluorimeter with constant wavelength excitation at 310 nM and emission monitored at 375 nM. An increase in fluorescence at 375 nM is observed with exonucleolytic release of 2-AP from duplex DNA, and is monitored over a time period of 15 minutes. Reactions (400 uL) contained 250 nM protein, 1 uM 2-AP blunt substrate, 50 mM Tris 7.5, 150 mM NaCl, 0.1 % polyethylene glycol 6000, 2.5 % glycerol and 1nM MnCl2. Reaction was tested at four different temperatures, 22 °C, 37 °C, 50 °C and 65 °C. Blunt dsDNA substrate was formed by annealing equimolar amounts of exo5′1 (5′-GGCGTGCCTTGGGCGCGCTGCGGGCGG(2-AP)G-3′) with exo3′1 (5′-CTCCGCCCGCAGCGCGCCCAAGGCACGCC-3′). Reaction graphs were plotted by normalizing the fluorescence data for every reaction to the first data point.
Acknowledgments
The project was sponsored by the National Institute of General Medical Sciences Protein Structure Initiative (P50 GM62411, U54 GM074898). Portions of this research were performed at the Stanford Synchrotron Radiation Lightsource (SSRL). The SSRL is a national user facility at the SLAC National Accelerator Laboratory operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health (National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences). Mre11 research in the Tainer group is funded by CA117638 and CA092584. We are grateful to Chiharu Hitomi and Dr. Julie Tubbs in the Tainer group for assistance with site-directed mutagenesis and biochemical assays. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. Genomic DNA from Thermotoga maritima MSB8 (DSM3109) (ATCC #43589D-5) was obtained from the American Type Culture Collection (ATCC).
Abbreviations used
- Tm
Thermotoga maritima
- Pf
Pyrococcus furiosus
- Hu
human
- Sc
Saccharomyces cerevisiae
- nuc_domain
nuclease domain
- spec_domain
specificity domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 2.Bierne H, Ehrlich SD, Michel B. Deletions at stalled replication forks occur by two different pathways. EMBO J. 1997;16:3332–3340. doi: 10.1093/emboj/16.11.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gellert M. Recent advances in understanding V(D)J recombination. Adv Immunol. 1997;64:39–64. doi: 10.1016/s0065-2776(08)60886-x. [DOI] [PubMed] [Google Scholar]
- 4.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85:509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 6.Hopfner KP, Putnam CD, Tainer JA. DNA double-strand break repair from head to tail. Curr Opin Struct Biol. 2002;12:115–122. doi: 10.1016/s0959-440x(02)00297-x. [DOI] [PubMed] [Google Scholar]
- 7.Connelly JC, Leach DR. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells. 1996;1:285–291. doi: 10.1046/j.1365-2443.1996.23024.x. [DOI] [PubMed] [Google Scholar]
- 8.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- 10.Connelly JC, Leach DR. Tethering on the brink: the evolutionarily conserved Mre11-Rad50 complex. Trends Biochem Sci. 2002;27:410–418. doi: 10.1016/s0968-0004(02)02144-8. [DOI] [PubMed] [Google Scholar]
- 11.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 12.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 13.Cromie GA, Connelly JC, Leach DR. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell. 2001;8:1163–1174. doi: 10.1016/s1097-2765(01)00419-1. [DOI] [PubMed] [Google Scholar]
- 14.D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 15.Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair. 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bentchikou E, Servant P, Coste G, Sommer S. Additive effects of SbcCD and PolX deficiencies in the in vivo repair of DNA double-strand breaks in Deinococcus radiodurans. J Bacteriol. 2007;189:4784–4790. doi: 10.1128/JB.00452-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharples GJ, Leach DR. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- 19.Nijmegen breakage syndrome. The International Nijmegen Breakage Syndrome Study Group. Arch Dis Child. 2000;82:400–406. doi: 10.1136/adc.82.5.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci U S A. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 22.Chahwan C, Nakamura TM, Sivakumar S, Russell P, Rhind N. The fission yeast Rad32 (Mre11)-Rad50-Nbs1 complex is required for the S-phase DNA damage checkpoint. Mol Cell Biol. 2003;23:6564–6573. doi: 10.1128/MCB.23.18.6564-6573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomaki K, Blomqvist C, Heikkila P, Lukas J, Nevanlinna H, Bartek J. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhee JG, Li D, Suntharalingam M, Guo C, O'Malley BW, Jr, Carney JP. Radiosensitization of head/neck squamous cell carcinoma by adenovirus-mediated expression of the Nbs1 protein. Int J Radiat Oncol Biol Phys. 2007;67:273–278. doi: 10.1016/j.ijrobp.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Moreau S, Morgan EA, Symington LS. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics. 2001;159:1423–1433. doi: 10.1093/genetics/159.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arthur LM, Gustausson K, Hopfner KP, Carson CT, Stracker TH, Karcher A, Felton D, Weitzman MD, Tainer J, Carney JP. Structural and functional analysis of Mre11-3. Nucleic Acids Res. 2004;32:1886–1893. doi: 10.1093/nar/gkh343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35:D237–240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aravind L, Koonin EV. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998;26:3746–3752. doi: 10.1093/nar/26.16.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connelly JC, Kirkham LA, Leach DR. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc Natl Acad Sci U S A. 1998;95:7969–7974. doi: 10.1073/pnas.95.14.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Tickle IJ, Laskowski RA, Moss DS. Error estimates of protein structure coordinates and deviations from standard geometry by full-matrix refinement of gammaB- and betaB2-crystallin. Acta Crystallogr D Biol Crystallogr. 1998;54:243–252. doi: 10.1107/s090744499701041x. [DOI] [PubMed] [Google Scholar]
- 35.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 36.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 38.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramaniam S. The Biology Workbench--a seamless database and analysis environment for the biologist. Proteins. 1998;32:1–2. [PubMed] [Google Scholar]
- 41.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 42.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 43.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 44.Bressan DA, Olivares HA, Nelms BE, Petrini JH. Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics. 1998;150:591–600. doi: 10.1093/genetics/150.2.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 46.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Putnam CD, Clancy SB, Tsuruta H, Gonzalez S, Wetmur JG, Tainer JA. Structure and mechanism of the RuvB Holliday junction branch migration motor. J Mol Biol. 2001;311:297–310. doi: 10.1006/jmbi.2001.4852. [DOI] [PubMed] [Google Scholar]
- 48.Truglio JJ, Rhau B, Croteau DL, Wang L, Skorvaga M, Karakas E, DellaVecchia MJ, Wang H, Van Houten B, Kisker C. Structural insights into the first incision reaction during nucleotide excision repair. EMBO J. 2005;24:885–894. doi: 10.1038/sj.emboj.7600568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalhus B, Arvai AS, Rosnes I, Olsen OE, Backe PH, Alseth I, Gao H, Cao W, Tainer JA, Bjoras M. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat Struct Mol Biol. 2009;16:138–143. doi: 10.1038/nsmb.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hopfner KP, Tainer JA. Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr Opin Struct Biol. 2003;13:249–255. doi: 10.1016/s0959-440x(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 51.Lesley SA, Kuhn P, Godzik A, Deacon AM, Mathews I, Kreusch A, Spraggon G, Klock HE, McMullan D, Shin T, Vincent J, Robb A, Brinen LS, Miller MD, McPhillips TM, Miller MA, Scheibe D, Canaves JM, Guda C, Jaroszewski L, Selby TL, Elsliger MA, Wooley J, Taylor SS, Hodgson KO, Wilson IA, Schultz PG, Stevens RC. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc Natl Acad Sci U S A. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 53.Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. An approach to rapid protein crystallization using nanodroplets. J Appl Crystallogr. 2002;35:278–281. [Google Scholar]
- 54.Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. An automated system to mount cryo-cooled protein crystals on a synchrotron beamline, using compact sample cassettes and a small-scale robot. J Appl Crystallogr. 2002;2002:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallog. 1993;26:795–800. [Google Scholar]
- 57.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 58.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 59.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terwilliger TC. Improving macromolecular atomic models at moderate resolution by automated iterative model building, statistical density modification and refinement. Acta Crystallogr D Biol Crystallogr. 2003;59:1174–1182. doi: 10.1107/S0907444903009922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography. 1992;(26) [Google Scholar]
- 62.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 63.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 65.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 66.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 67.Kleywegt GJ. Validation of protein crystal structures. Acta Crystallogr D Biol Crystallogr. 2000;56:249–265. doi: 10.1107/s0907444999016364. [DOI] [PubMed] [Google Scholar]
- 68.Laskowski RA, Chistyakov VV, Thornton JM. PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 2005;33:D266–268. doi: 10.1093/nar/gki001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA, USA: 2002. [Google Scholar]