

Abstract
Saliva is a body fluid with important functions in oral and general health. A consortium of three research groups catalogued the proteins in human saliva collected as the ductal secretions: 1166 identifications—914 in parotid and 917 in submandibular/sublingual saliva—were made. The results showed that a high proportion of proteins that are found in plasma and/or tears are also present in saliva along with unique components. The proteins identified are involved in numerous molecular processes ranging from structural functions to enzymatic/catalytic activities. As expected, the majority mapped to the extracellular and secretory compartments. An immunoblot approach was used to validate the presence in saliva of a subset of the proteins identified by mass spectrometric approaches. These experiments focused on novel constituents and proteins for which the peptide evidence was relatively weak. Ultimately, information derived from the work reported here and related published studies can be used to translate blood-based clinical laboratory tests into a format that utilizes saliva. Additionally, a catalogue of the salivary proteome of healthy individuals allows future analyses of salivary samples from individuals with oral and systemic diseases, with the goal of identifying biomarkers with diagnostic and/or prognostic value for these conditions; another possibility is the discovery of therapeutic targets.
Keywords: Human saliva, parotid, submandibular, sublingual, ductal secretion, proteomics, mass spectrometry
Introduction
The components of human saliva have evolved to carry out many important and well-recognized functions.1–8 Amylase and lipases initiate digestion. Mucins play numerous roles, such as aiding in lubrication, protection and healing of the oral mucosa, formation of the food bolus, and phonation. As major components of the salivary pellicle that coats the tooth, these molecules, primarily MUC5b and MUC7, in conjunction with the acidic proline-rich proteins, also help regulate bacterial adhesion.9 In this regard, other salivary components, including immunoglobulins, lysozyme, lactoferrin, lactoperoxidase, cystatins, histatins, and proline-rich glycoproteins, have significant antimicrobial activity. Statherins and histatins, which inhibit calcium phosphate precipitation, help maintain tooth integrity and contribute to the buffering capacity of saliva, which is also regulated by salivary electrolytes. Other proteins have more specialized functions. For example, gustin, also known as carbonic anhydrase VI, has important roles in tastebud development and function and, thus, in regulating taste sensation.10 The proline-rich proteins and histatins neutralize dietary tannins.11 Secretory leukocyte proteinase inhibitor blocks HIV infection.12 Thus, the composition and flow rate of human saliva govern oral health. This can be seen in the ill effects of hyposalivation, a condition that affects several populations, including individuals diagnosed with Sjögren’s syndrome, patients who have been treated with ionizing radiation for head and neck cancers, and the elderly.5
Whole saliva is a complex mixture of microbial products, mucosal exudate, gingival crevicular fluid, desquamated epithelial cells, and exocrine secretions of the major and minor (so-called based on their size) salivary glands. The major salivary glands include the paired parotid and submandibular (SM) glands as well as the paired sublingual (SL) glands located in the floor of the mouth. The minor salivary glands are found throughout the oral cavity, including the lower lip, tongue, palate, and cheeks, as well as the upper pharynx.4,13 The relative contribution of individual glands to whole saliva varies greatly, depending on several factors. For example, during unstimulated flow, the SM, parotid, minor salivary, and SL glands secrete, on a volumetric basis, approximately 65, 20, 10, and 5% of whole saliva, respectively. However, during stimulated flow, the parotid secretions account for 50% of salivary volume.4 This observation is in accord with the cellular composition of the glands. The parotid consists of primarily serous acini, whereas the SM and SL glands produce both mucous and serous secretions. Finally, it is interesting to note that the ducts that deliver the glandular secretions to the oral cavity are not passive conduits but also contain secretory vesicles whose contents enrich saliva.14 The vesicles contribute important components such as kallikrein,15 a proteinase that participates in the processing of a wide variety of substrates, as well as molecules that play important antimicrobial roles.16,17
Given the numerous variables with regard to the sources and composition of human saliva, many different approaches will be required to compile a comprehensive catalogue of the proteins that make up this body fluid. In general, mass spectrometry (MS)-based methods are of greatest utility because they are unbiased, requiring no prior knowledge of protein composition. Tandem MS-based approaches (MS/MS) in combination with liquid chromatography (LC) and electro-spray ionization (ESI) have been used with database search algorithms to identify proteins.18,19 This process involves digestion of proteins either before or after fractionation for analysis by LC or multidimensional LC-MS/MS. These so-called bottom-up methods have been used for a wide variety of studies of protein complexes, organelles, cells, tissues, and body fluids.20–25 In addition, one- and two-dimensional gel electrophoresis in combination with both LC-MS/MS and matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) MS have been employed to identify proteins.26
Here we describe the initial results of a National Institute of Dental and Craniofacial Research (NIDCR)-funded effort in which these methods were used to compile a catalogue of human salivary proteins. Three groups of investigators were involved—The Scripps Research Institute/University of Rochester; University of California—Los Angeles/University of Southern California; and University of California—San Francisco. We envision that this catalogue will be a valuable resource for investigators who are interested in the biology of the oral cavity and upper gastrointestinal tract or in developing diagnostic tests that utilize this body fluid.
Materials and Methods
The protocols for collecting human saliva were approved by each institution’s Committee on Human Research, and written informed consent was obtained from all donors. Additional details of the methods used by the three research groups are given as Supporting Information.
Summary: The Scripps Research Institute and the University of Rochester
Noncannulated ductal saliva was obtained from three donors (2 males, 1 female), aged 45–49 at the time of collection. Salivary secretion was stimulated with 0.4% citrate and collected on ice between 7 and 10 a.m. for 30–120 min using a cup-like device for parotid secretions27 and a Block and Brotman collector for SM and SL secretions.28 The samples were immediately dialyzed (3.5-kDa molecular mass cutoff) and lyophilized. For identification of medium- and high-copy-number proteins, small volumes (<10 mL) were processed separately and directly analyzed by MS. To enhance the identification of low-copy-number salivary components, large volumes (10–100 mL) of saliva were collected as described above and pooled for large-scale protein fractionations as described below. For isolation of low-molecular-mass proteins ≤20 kDa, an ethanol-soluble fraction of saliva was prepared. In this case, saliva was collected from a single male donor and prepared as described in the Supplemental Methods (Supporting Information).
The detection of low-copy-number salivary proteins was facilitated by fractionating a large quantity of saliva over multiple columns. Parotid saliva was separated under native conditions. SM/SL saliva was first denatured in 4 M guanidine-HCl and 50 mM Tris, pH 8.0, to dissociate the high-molecular-mass mucin complexes (MUC7 and MUC5b). Both parotid and SM/SL secretions were size-fractionated using either tangential flow ultrafiltration (MiniTan II, Millipore) or size exclusion chromatography on Sephadex G100/G200 or Sepharose CL-6B. Further separations were achieved using anion-exchange chromatography (DE52, DEAE Sepharose CL-6B, or DEAE Ceramic HyperD F), reversed-phase high-pressure LC (C4 resin), immobilized metal chelate affinity chromatography (Talon, BD Clontech), and hydrophobic charge interaction chromatography (MEP Hypercel), as described in the Supplemental Methods (Supporting Information). Purifications were performed either by gravity flow or by using a Biologic DuoFlow Maximizer purification system (Bio-Rad). MS analyses were performed after a single chromatographic fractionation and after multidimensional separations. LC-MS/MS was used to characterize these fractions.
Specialized protein separation techniques were also used. These included metal chelate affinity chromatography on Talon resin to remove the histatins, followed by chromatography of the eluate on a resin coupled with single-chain antibodies that specifically recognized α-amylase. A subset of the samples was also separated by two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (2-D SDS-PAGE), chromato-focusing, ZOOM isoelectric focusing (IEF), and one-dimensional SDS-PAGE with subsequent pixelation of the bands. Samples were reduced and alkylated before digestion with LysC and redigestion with trypsin. Depending on sample amount, 10–100 μg of a digest prepared from each sample was analyzed by multidimensional protein identification technology.25 As peptides were eluted from the microcapillary column, they were electrosprayed directly into an LCQ Deca, LCQ DecaXP, or LTQ 2-D ion trap mass spectrometer (ThermoFinnigan) or analyzed on an LTQ-Orbitrap instrument.29
Tandem mass spectra were analyzed as follows. Poor-quality spectra were removed from the data set using an automated spectral quality assessment algorithm.30 The remaining spectra were searched with the SEQUEST algorithm, version 27,18 against the European Bioinformatics Institute (EBI) human International Protein Index (IPI) database (version 3.01; release date November 1, 2004) (ftp://ftp.ebi.ac.uk/pub/databases/IPI/current) and concatenated to a decoy database in which the sequence for each entry in the original database was reversed.31 SEQUEST results were assembled and filtered using the DTA-Select program at threshold settings that generated a peptide false-positive rate of 0.35%.32 Only proteins identified with at least two peptide hits, with each peptide containing a tryptic terminus, were accepted. Under these conditions, the estimated false-positive rate at the protein level was <3%.
Summary: University of California—Los Angeles and University of Southern California
Ten adult subjects (6 females and 4 males), of various ethnic and racial backgrounds, ranging in age from 22 to 30 years, were recruited. Saliva collection took place on a monthly basis and was performed between the hours of 9 and 11 a.m. Parotid saliva was obtained as the ductal secretion using a cup-like device.27 Stimulated parotid, SM, and SL salivas were collected by the repeated application of an aqueous citric acid solution (2%). Separate SM and SL secretions were acquired using a saliva collector, described by Wolff and co-workers,33 that was fitted with a sterile 100-μL pipet tip. Collection volumes over a 10-min period ranged from 500 to 2000 μL for parotid saliva, 50–100 μL for SL saliva, and 100–500 μL for SM saliva.
The saliva samples were separated by a number of methods, including 2-D SDS-PAGE, reversed-phase LC on a C4 column, and ZOOM IEF. For some experiments, individual isoelectric point (pI) fractions from ZOOM IEF fractionation were trypsin-digested, and the tryptic peptides were subjected to either online (LC-MS/MS) or off-line strong cation-exchange chromatography (with ammonium formate). LC-MS/MS of peptide mixtures was performed on an Applied Biosystems (Foster City, CA) QSTAR Pulsar XL (QqTOF) mass spectrometer equipped with a nanoelectrospray interface (Protana, Odense, Denmark) and an LC Packings (Sunnyvale, CA) nano-LC system.
Proteins were identified using the Mascot database search engine (Matrix Science, London, U.K.). All searches were performed against the EBI human IPI database (version 3.03; release date February 5, 2005). For the protein sequence searches, the following variable modifications were set: carbamidomethylation of cysteines, cyclization of N-terminal carbamoylmethyl cysteines, and oxidization of methionines. For saliva samples prefractionated by in-solution IEF, DMA modification of cysteines was added to the variable modification list. In all searches, one missed tryptic cleavage was allowed, and a mass tolerance of 0.3 Da was set for the precursor and product ions. A Mascot score of >25 with a p-value <0.05 was considered a significant match. False-positive rates were determined using the method of Elias and co-workers34 and described on the Matrix Science Web site (www.matrixscience.com). Briefly, false-positive rates (FPR) were calculated by multiplying the number of false-positive identifications (hits to the decoy database constructed from randomized sequences) and dividing by the number of total identifications. Thus, FPR = FP/(FP + TP), where FP is the number of false-positive hits, and TP is the number of true-positive hits. The estimated FPR was determined to be ≤2%. The MS/MS spectra for each peptide were manually examined to verify the identification.
Summary: University of California—San Francisco
Saliva samples were obtained from 10 adult subjects (5 males and 5 females). SM/SL and parotid gland secretions were collected simultaneously between 2 and 4 p.m. Parotid saliva was obtained using a Lashley cup.27,35 A combination of SM/SL salivas was collected using a custom-fitted Block and Brotman collector that fit snugly on the floor of the mouth.28 Gland secretion was enhanced by gustatory stimulation with a solution of 2% citric acid. All samples were collected on ice for a maximum duration of 30 min.
The samples were analyzed using a variety of workflows, including 2-D SDS-PAGE followed by MS analyses using published methods.36 Additional fractionation procedures included ZOOM IEF, strong cation-exchange chromatography on a polysulfethyl A column, and ultrafiltration. For ultrafiltration, saliva samples were transferred to 10-kDa-cutoff Vivaspin ultrafiltration devices and centrifuged at 10 000g at 4 °C for 60 min. The filtrate (native peptides, fraction I) was stored at −20 °C until further analysis. The retentate was washed twice with 25 mM ammonium bicarbonate containing 150 mM NaCl to release trapped peptides. Both filtrates were combined and stored as fraction II (released peptides). The remaining proteins were recovered by adding more of the same buffer, after which the filter device was inverted and the centrifugation step was repeated. Proteins were reduced with dithiothreitol, and the sulfhydryl groups of the cysteine residues were alkylated by adding iodoacetamide as described above. Subsequently, 0.1% (w/w) sequencing-grade trypsin was added, and the sample was incubated overnight at 37 °C. The reaction was stopped by the addition of 5% trifluoroacetic acid. Tryptic peptides (fraction III) were recovered as the ultrafiltrate of the reaction mixture. Tryptic and native peptides in these fractions were analyzed by either LC-MALDI TOF/TOF MS (4800, Applied Biosystems, Foster City, CA) or LC-QqTOF MS on an Applied Biosystems QSTAR Pulsar XL instrument equipped with a nanoelectrospray interface.37
MS data were analyzed using a laboratory information system, created in-house, that uses Mascot Distiller for spectral processing and peak detection. Peptide identifications were accomplished using the Mascot algorithm (version 2.1) to search against human proteins in the Swiss-Prot (version 50; release date May 2, 2006) and IPI (version 3.15; release date February 22, 2006) databases. For the protein sequence searches, carbamidomethylation of cysteines was set as a fixed modification, and the following variable modifications were used: deamidation of asparagines and glutamine residues, and oxidization of methionine sand cyclization of N-terminal glutamines. For saliva samples prefractionated by in-solution IEF, DMA modification of cysteine was used as a fixed modification. In all searches, up to three missed tryptic cleavages were allowed, and a mass tolerance of 150 ppm and 0.1 Da was set for the precursor and product ions, respectively. Peptide-spectral matches with expectation values <0.05 were considered significant. To estimate the rate of false-positive identifications, we searched all spectra against a decoy database created by randomizing each protein in IPI with the Mascot decoy tool (http://www.matrixscience.com/help/decoy_help.html).31 In all cases, the peptide false-positive identification rate was <3%. The MS/MS spectra for each peptide were manually examined to verify the identification.
Data Submission and Centralization
Data collected by the participating groups were submitted to the proteomics database hosted by the University of California—Los Angeles (http://www.hspp.ucla.edu). The central repository (proteomics database) stores the information according to proposed guidelines for reporting MS data. Recorded information includes saliva sample source (parotid or SM/SL), peptide and protein identifications, post-translational modifications, MS instrumentation used in the analysis, peak list file records, and database search programs.38,39 Quality of the peptide and protein identifications was determined by the individual research groups; no further validation was performed in the central database. The data were submitted in an XML format using a template that was specially created for this purpose. The relational database schema, XML template, and Web interface to the central repository can be viewed through the http://www.hspp.ucla.edu Web site that is dedicated to this project. In addition, The Scripps Research Institute (http://fields.scripps.edu/public/project/saliva) and the University of California—San Francisco (http://www.salivarium.ucsf.edu) host Web sites that include information specific to their research groups.
Data Standardization and Integration
The three research groups used the IPI database to derive peptide and protein identifications. IPI, however, is updated frequently, and each group used a different version. For purposes of integration, protein identifications submitted by the three groups were standardized to version 3.24 of IPI (release date December 1, 2006). Specifically, the submitted protein identifications were mapped to this database by an approach previously utilized by the Human Proteome Organization (HUPO) Plasma Proteome Project.40 Each protein identification was submitted with the protein accession number and a list of experimentally observed peptide sequences. These peptide lists were searched against IPI v.3.24. An IPI v.3.24 protein was selected whenever its sequence contained subsequences of residues identical to all the experimentally determined peptides associated with the original submitted protein identification. Peptide lists that could not be matched exactly with a protein in IPI v.3.24 were excluded from further analysis. In total, 2.9% of the submitted peptides were excluded during the standardization process, which was attributable to the 5% allowed mismatch between database entries.41 This value is similar to the reported 2% of peptide sequences lost in the plasma proteome integration process.40
Clustering of Equivalent Protein Identifications
Frequently, a peptide sequence list for a protein matched more than one entry in the reference database. Peptide-based evidence cannot distinguish between these redundant identifications, which likely represent protein homologues and isoforms. Therefore, protein identifications were grouped into clusters whenever identifications were made with identical peptide lists. Following a strategy established by the HUPO Plasma Proteome Project, these clusters of equivalent protein identifications were further grouped together whenever the peptide list for a cluster was a subset of the peptide list of another cluster.40 Finally, the resulting groups of clusters were used to select a single representative for the entire group. The choice of the representative protein was based on the idea that the protein most likely present in the sample is the one that is reported by the maximum number of research groups with the maximum amount of peptide evidence. Specifically, the representative protein for a grouped set of clusters was chosen by applying the following procedures sequentially: (A) Determine the number of research groups (one, two, or three) reporting each protein in the cluster. The proteins with the maximum number of reports are chosen from the cluster. (B) Measure the number of distinct peptide sequences reported for each protein identification in the cluster. The protein identifications with the highest number of distinct peptide sequences are selected. (C) The protein that has a well-defined description in the IPI v.3.24 database or that is cross-referenced to the UniProt/Swiss-Prot databases is selected in preference to one described as “hypothetical,” “putative,” “fragment,” “similar to,” “kDa” or “CDNA.” (D) Select the protein with the lowest IPI accession number.
The standardization, clustering, and representative protein selections were performed based on the protein and peptide identifications from the two salivary sources, parotid and SM/SL. The relationships between the salivary source and the respective peptide and protein identifications were constructed under the relational database. During the integration process, consideration was given to how to combine the identifications from the two salivary sources. In principle, the integration could be performed independently for parotid and for SM/SL. In this case, representative proteins would be selected as a result of two separate processes. Two concerns emerged: (1) Since the amount of peptide evidence is one criterion that is applied during the selection process, the independent procedures could choose two different representatives for proteins that are present in both sources at different abundances. (2) A protein that is identified in both sources (parotid, SM/SL) with different peptide evidence could cluster with different proteins. The representative proteins chosen for the two clusters would not necessarily be identical, especially in cases where ties existed during the selection procedures. For these reasons, the combined information from both sources was used for clustering and selection of representative proteins. Calculation of the number of distinct peptide sequences was based on the information from both sources. To create source-specific identifications after the representative protein selection, protein and peptide identifications derived from the specific salivary sources were extracted from the relational database.
Comparison of the Human Salivary Proteome with the Plasma and Tear Proteomes
As a first step, we downloaded a set of 889 high-confidence plasma protein identifications (http://www.bioinformatics.med.umich.edu/hupo/ppp) made by searching IPI v.2.01.42 The tear proteome data set with 491 protein identifications made from an unknown version of IPI was downloaded from http://genomebiology.com/2006/7/8/R72#IDAKPTNE.43 Then the peptide lists were mapped to IPI v.3.24 and clustered as described above. Because of changes in IPI database versions, the numbers of plasma and tear proteins were reduced to 657 and 467 proteins, respectively. Representative proteins for the plasma and tear protein clusters were selected as described above and compared with the parotid and SM/SL proteomes.
Immunoblotting
Immunoblotting was carried out as described previously.44 Briefly, samples of parotid and SM/SL salivas collected as the ductal secretions from 5 female and 4 male donors were electrophoretically separated on NuPAGE 4–12% BIS-TRIS polyacrylamide gradient gels (Invitrogen). In one experiment, the apolipoprotein E (apoE) analysis, an additional two samples that were collected from male donors were analyzed. Whole saliva samples were collected from 10 females and 2 males. To visualize the protein bands, some gels were stained with Coomassie brilliant blue. For immunoblotting, duplicate gels were transferred to nitrocellulose membranes, and nonspecific binding was blocked by incubating the transfers for 1 h in PBS-T-blotto containing 0.05% Tween-20 and 5% nonfat dried milk. Thereafter, the membranes were incubated at 4 °C in T-blotto containing the following primary antibodies (2 μg/mL unless otherwise indicated): goat polyclonal anti-human apoE (Chemicon International, Temecula, CA); murine monoclonal antibody (mAb) versus recombinant human tissue inhibitor of metalloproteinases 1 (TIMP-1) (Lab Vision Corp., Fremont, CA); rabbit polyclonal anti-human KLK-1 (Abcam, Inc., Cambridge, MA); murine mAb versus human carcinoembryogenic antigen (CEA) clone II-7 (Dako North America, Inc., Carpinteria, CA); murine mAb versus human prostatic acid phosphatase (1:5, v/v; AbD Serotec, Raleigh, NC); rabbit polyclonal anti-human calpain-1 (large subunit; EMD Chemicals, Inc., San Diego, CA); and murine mAb versus human HLA-G (produced in the Fisher laboratory).45 After overnight incubation, the blots were washed three times for 5 min each in PBS containing 0.05% Tween-20. Binding of primary antibodies was detected by incubating the membranes for 1 h with a horseradish peroxidase-conjugated secondary antibody (Jackson Immuno Research Laboratories, Inc.) to the relevant isotype diluted 1:1000 (v/v) in T-blotto. Immunoreactive bands were visualized using chemiluminescence.
Results
To allow comparison and analysis, the data sets from the three research groups were merged into a central repository hosted by the University of California—Los Angeles. The three groups operated independently, with variations in donors, sample collection procedures, separation strategies, and data acquisition and analysis. As a result, different numbers of proteins were identified by each group (Table 1). In total, the three research teams submitted 173 811 peptide and 68 789 protein identifications. These produced 11 592 distinct peptide sequences that matched 2153 distinct protein accession numbers (ACs) (1682 parotid, 1623 SM/SL) (Supplemental Tables 1 and 2 in Supporting Information). Overall, the integration process yielded 1166 nonredundant clusters, with each cluster containing at least one peptide that was distinguished from peptides used in other clusters; 642 contained a single AC, 524 contained more than one AC, and the cluster with the maximum number contained 47 ACs (Supplemental Table 3 in Supporting Information).
Table 1.
Overview of the Data Sets Submitted by the Research Groups of the Human Salivary Proteome Consortiuma
| group | Sample Source | MS instruments | program | database | confidence | distinct peptide Ids | distinct protein Ids | no. of clusters (IPI v.3.24) |
|---|---|---|---|---|---|---|---|---|
| TSRI/UR | Parotid | LCQ Deca, LCQ DecaXP, LTQ, LTQ Orbitrap | SEQUEST | IPI v.3.01 | Peptide FPR ≤0.35%, Protein FPR ≤3% | 7187 | 1277 | 855 |
| SM/SL | 7550 | 1189 | 848 | |||||
| Total | 10713 | 1690 | 1102 | |||||
| UCSF | Parotid | QSTAR QTOF 4800 TOF/TOF | Mascot | IPI v.3.15 | Peptide FPR ≤3% | 585 | 409 | 197 |
| SM/SL | 702 | 427 | 205 | |||||
| Total | 893 | 489 | 239 | |||||
| UCLA | Parotid | QSTAR QTOF | Mascot | IPI v.3.03 | Protein FPR ≤2% | 1524 | 777 | 401 |
| SM/SL | 1010 | 668 | 309 | |||||
| Total | 1858 | 910 | 453 | |||||
| Total | Parotid | 7734 | 1682 | 914 | ||||
| SM/SL | 7899 | 1623 | 917 | |||||
| Total | 11248 | 2153 | 1166 |
The table presents the major elements of the pipeline that the three groups used, together with a summary of the protein identifications. FPR, false-positive rate.
Following data integration, a representative protein was chosen from each cluster that contained isoforms, fragments, or homologues. In general, the representatives were picked because they were confirmed by the most research groups and conformed to the rules described in Materials and Methods. Of a total of 1166 representative proteins, 914 were identified from parotid saliva, and 917 were identified from SM/SL saliva. Fifty-seven percent of the protein identifications were shared between the two glandular secretions (Figure 1).
Figure 1.
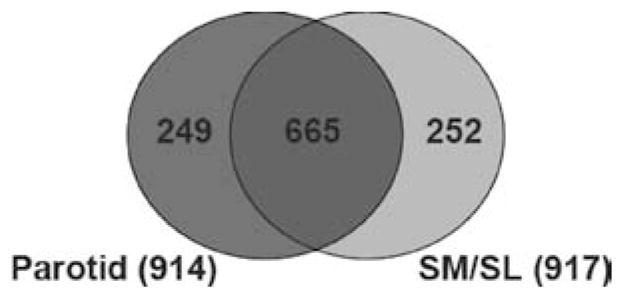
Human parotid and SM/SL salivas share many of the same protein components. The protein composition of each ductal fluid is presented as a Venn diagram. Fifty-seven percent of proteins were found in both parotid and submandibular/sublingual (SM/SL) salivas, whereas 27% were unique to the gland(s) of origin.
To maximize interrogation of the salivary proteome, the three research groups used different experimental approaches resulting in different numbers of proteins identified: University of California—San Francisco, 197 parotid and 205 SM/SL; The Scripps Research Institute/University of Rochester, 855 parotid and 848 SM/SL; and University of California—Los Angeles/University of Southern California, 401 parotid and 309 SM/SL. In total, 152 parotid and 139 SM/SL proteins were identified by all three research groups (Figure 2). These proteins constituted a minimal set of the salivary proteins reflecting differences in the number of experiments performed and experimental approaches that were the least susceptible to variations in experimental approach. We termed these proteins the “core minimally overlapping proteome.” An additional 235 parotid and 167 SM/SL proteins were identified by at least two groups. The 527 parotid and 611 SM/SL proteins that were identified by only one group are probably the salivary components that reflect saliva donor and/or methodological variations.
Figure 2.

Venn diagrams illustrating the number of parotid (left) and SM/SL (right) proteins that were identified at each research site. Areas of overlap depict the number of proteins that were contributed by more than one group, with the core proteome—proteins that were identified by all three groups—shown in the center. Areas where the circles diverge show the number of protein identifications that were site-specific. Differences are likely attributable to the different methodologies employed by each research team.
We hypothesized that the most abundant proteins would be among those identified by all three groups. To test this theory, we classified components of the salivary proteome based on several criteria: (A) the research group that identified the protein, (B) the salivary gland where it was produced, and (C) the sequence coverage (high, intermediate, or low). The classified proteins were listed in a matrix, and the matrix was displayed using Array Viewer software (Figure 3). The Array Viewer was created using the Genesis software tool.46
Figure 3.

Array view of the minimally overlapping human salivary proteome. Data were grouped according to the research team that identified each protein, the salivary gland where it was produced, and percent sequence coverage. The extent of coverage positively correlated with the number of research groups that identified a particular protein. The core proteome included one SM/SL-specific protein. Blue, protein identified; yellow, protein not identified; red, high-sequence coverage; yellow, intermediate-sequence coverage; and green, low-sequence coverage.
We also created a histogram that displays the sequence coverage of individual proteins according to the number of research groups that made a particular identification (Figure 4A). There was a strong positive correlation between the sequence coverage and membership in the core proteome. Proteins that were identified by a single research group had low sequence coverage, on average 16%, whereas proteins that were identified by all three groups had an average sequence coverage of 62%. However, there were notable exceptions: a few proteins at the low end of sequence coverage were detected by all three groups. This subproteome included the highly glycosylated SM/SL protein MUC5b, illustrating the fact that post-translational modifications and/or unusual amino acid sequences limit peptide generation and/or MS detection. We also compared a protein’s predicted cellular location with the number of research groups making the identification (Figure 4B). Overall, the entire salivary proteome was enriched in extracellular proteins as compared to the IPI human reference data set. Proteins that are well-known components of salivary secretions, such as α-amylase, cystatin, histatin, proline-rich proteins, and mucins, were prominently represented in the core proteome. Interestingly, proteins identified by a single group were more likely to be associated with the nucleus, cytoplasm, or plasma membrane than were proteins reported by all three groups. However, exceptions were noted. For example, the core proteome included one protein that was specific to SM/SL saliva: Golgi phosphoprotein 2.
Figure 4.

Sequence coverage and cellular origin of salivary proteins identified by single or multiple groups. (A) Histogram of the sequence coverage for proteins that were identified in parotid and SM/SL salivas. The shaded areas depict the number of proteins that were reported by one (gray), two (dark gray), or all three (black) research groups. (B) Comparison of the cellular location of salivary proteins identified by one, two, or all three research groups with a reference data set containing all human IPI database entries. Ratios of protein associations with the extracellular, plasma membrane, cytoplasm, or nuclear subcellular compartments were calculated using Ingenuity software. In some cases, this information was not known.
As expected, the salivary protein components reported in this study covered a wide range of molecular masses and pIs (Figure 5). With regard to molecular mass distribution, 46.6% of the human salivary proteins identified were ≤40 kDa, 44.6% were between 40 and 120 kDa, and 8.7% were ≥120 kDa. The human urine proteome has a similar molecular mass distribution.47 As with the urine study, the distribution of pIs of salivary proteins was broad, ranging from 3.38 to 12.56. However, the majority of salivary proteins had a pI between 4 and 8, indicative of a large number of acidic species (pI 4–5) that play an important role in buffering this biological fluid. The maintenance of a neutral pH in the oral cavity is critical to oral health and taste sensation.
Figure 5.

Molecular mass and pI distribution of the entire human salivary proteome. (A) The distribution was skewed to relatively low-molecular-mass components. (B) In contrast, the salivary proteins had a relatively broad range of pIs.
To obtain a functional overview, we searched the salivary proteome against gene ontology and protein pathway databases using slim definitions. The results showed that 568 nonredundant parotid and 578 SM/SL proteins mapped to the cellular components, 647 parotid and 658 SM/SL proteins had molecular functions, and 582 parotid and 594 SM/SL proteins were associated with biological processes (Supplemental Table 4 in Supporting Information). Given their nearly identical composition, we expected that parotid and SM/SL proteins would be similarly distributed across the gene ontology slim categories. With regard to location, a high proportion of the parotid and SM/SL proteins mapped to the extracellular region; others localized to the plasma membrane, cytoplasm, organelles, or cytoskeleton or formed protein complexes (Figure 6A). With regard to molecular functions, salivary proteins had binding, catalytic, structural, and enzymatic activities (Figure 6B). With regard to biological processes, parotid and SM/SL protein constituents had the highest distribution in metabolic and regulatory pathways (Figure 6C).
Figure 6.

Relative allocation of the proteins identified in parotid and SM/SL saliva according to their gene ontology annotations. Components of both fluids were similarly distributed with regard to cellular locations (A), molecular functions (B), and biological processes (C). A high percentage of salivary proteins are predicted to be extracellular components or to reside within organelles (A). Salivary proteins were commonly involved in binding and catalysis (B). Parotid and SM/SL proteins had the highest distribution in metabolic and regulatory pathways (C).
About a quarter of the salivary proteins reported in this study were classified as hypothetical and, as such, lacked annotations (Figure 6, “unknown”). To provide insights into their possible functions, sequence similarities to proteins with known functions were assessed using the PSI-BLAST search engine, which enables template modeling. The results showed that a number of these hypothetical proteins showed sequence similarities to immunoglobulins and extracellular matrix components.
The salivary proteins were also searched against protein pathway databases, revealing 434 entries in BioCarta and 887 entries in KEGG. As expected, these proteins were involved in a number of metabolic processes involving amino acids (123 entries), carbohydrates (157 entries), energy utilization (80 entries), glycans (78 entries), lipids (31 entries), secondary metabolites (17 entries), biodegradation of xenobiotics (21 entries), and cofactors/vitamins (16 entries) (Supplemental Table 5 in Supporting Information). This analysis also showed that some salivary proteins functioned in the complement cascade and coagulation, a possible sign of plasma leakage. The presence of other proteins that play a role in cell adhesion and communication, cell cycle progression, and regulation of the actin cytoskeleton could be attributable to cellular debris arising from degradative processes such as apoptosis or breakage during prolonged secretion. Interestingly, a number of the salivary proteins mapped to pathways involved in neurode-generative conditions (Alzheimer’s, Huntington’s, and Parkinson’s diseases), cancers (breast, colorectal, and pancreatic), or type I/II diabetes.
We were also interested in comparing the human salivary proteome with that of human plasma and tears. To address the possible origin of plasma proteins, we made correlative comparisons between the two proteomes. The results showed that 192 of 657 plasma proteins were found in human saliva, including the most abundant species, which is possible evidence of vascular leakage or the contribution of fluid from the interstitial compartment (Figure 7). Given the fact that the parotid gland is composed entirely of serous acini and that the SM/SL glands also contain these elements, we investigated the degree to which the salivary proteome resembles that of other serous glands. For comparison, we chose tear fluid, which is produced by the lacrimal gland, a serous structure (Figure 7).43 In total, 259 proteins identified as components of either parotid or SM/SL saliva were present in lacrimal gland secretions, representing approximately 55% of the available tear proteome. Confirming earlier results, individual protein species that were common to all the glands included cystatins B, C, SA, S, and SN, zinc-alpha-glycoprotein, and prolactin-inducible protein.48 In addition, our work showed that all glands contained dermcidin, MUC5b, MUC7, and cathepsins B and D. There were also notable differences. For example, as previously reported, secretion of histatins and acidic, basic, and glycosylated proline-rich proteins, with the exception of PRP4 and PRP1, was specific to salivary glands.49 In addition, we found that cystatins A and D, calnexin, PDI, and cathepsins H and S were specific to salivary gland secretions. Finally, protein components unique to tear fluid included lactadherin, aspartyl aminopeptidase, and lupus La protein.
Figure 7.

Human parotid and SM/SL gland secretions share many protein components with human tears and plasma. (A) Areas of overlap depict the number of proteins that were identified in both tears and parotid (top) or SM/SL saliva (bottom). (B) Similarly, parotid (top) and SM/SL gland secretions (bottom) contained a substantial number of proteins that are also found in plasma.
In additional experiments, we used an immunoblot approach to confirm the presence in saliva of a portion of the proteins that were identified by MS techniques. Figure 8A shows the electrophoretic profile, visualized by staining the gels with Coomassie brilliant blue, of the nine individual samples of ductal saliva that were analyzed in most of the experiments. In general, we selected for validation salivary components outside the core proteome that had low sequence coverage and for which well-characterized antibodies existed. Overall, the results of these experiments showed that proteins in the relatively low confidence end of the salivary proteome were found in at least a subset of the ductal or whole saliva samples that were analyzed. For example, a mAb that specifically recognized a complex glycopeptide epitope carried by CEA (CD66) reacted with a band that corresponded to the estimated molecular mass (~180 kDa)50 of CEA, primarily in several SM/SL samples (Figure 8B). Kallikrein 1 was detected as several immunoreactive species, most prominent in parotid saliva, in the range of the predicted molecular mass (29–38 kDa) spanned by this proteinase (Figure 8C). A goat polyclonal antibody against apoE reacted with multiple bands. In parotid saliva, the 34-kDa species corresponded to the predicted molecular mass of the apoE monomer (Figure 8D, left). The electrophoretic mobility of the lower-molecular-mass band was consistent with previously published data regarding the major product of cathepsin D processing of the full-length protein.51,52 The higher-molecular-mass immunoreactive proteins may be heteromeric complexes of apoE and apoAII (Figure 8D, apo[AII-E] and apo[AII-E-AII]), which have been detected in cerebrospinal fluid.53 In SM/SL saliva, immunoreactive bands with the molecular mass of apoE/apoAII complexes were also observed along with higher-molecular-mass species that corresponded to the relative electrophoretic mobility of apoE/β-amyloid heterodimers.54 With regard to TIMP-1, immunore-active bands that were higher than the predicted molecular mass (29 kDa)55 were detected only in SM/SL saliva (Figure 8E). This molecule’s ability to form complexes with matrix metalloproteinase (MMP) 2 (72 kDa)56,57 is a likely explanation, whereas association with an MMP that had lost its hemopexin domain58 or activated MMP-7 (19–21 kDa)59 could explain the ~50-kDa immunoreactive band that was present in some samples. We were also interested in whether antigens could be detected in whole saliva, the most likely fluid to be employed in diagnostic tests. Accordingly, nitrocellulose replicas of electrophoretically separated whole saliva samples were probed with anti-HLA-G.45 Most of the samples contained an immunoreactive band of 50 kDa, which corresponds to the glycosylated form of this molecule (Figure 8F). Figure 8G shows that both parotid and SM/SL salivas contain a protein of the expected molecular mass (45 kDa)60 that reacts with anti-human prostatic acid phosphatase. Finally, some samples contained the largest subunit of calpain-1 in its latent (~80 kDa) and active (75–78 kDa) forms;61 the 58-kDa form may be a proteolytic fragment of this autolytic molecule.62
Figure 8.

Detection of novel salivary proteins and those with low sequence coverage. An immunoblot approach was used to confirm the presence in saliva of a portion of the proteins that were identified by using MS approaches. Most of the analyses shown employed parotid and SM/SL saliva samples collected as the ductal secretions from 5 females (lanes 1–5) and 4 males (lanes 6–9). As shown in panel F, whole saliva samples were collected from 10 females (lanes 1, 4–12) and 2 males (lanes 2–3). Proteins were selected for validation based on the number of research groups that reported the identification and the extent of sequence coverage. In general, emphasis was placed on the analysis of novel components and proteins with the least MS evidence of their presence in ductal saliva. (A) Samples were separated on 4–12% BIS-TRIS gradient gels under reducing conditions, and proteins were visualized by staining with Coomassie brilliant blue. Immunoblots detected the following: (B) CEA (IPI00027486, 6% sequence coverage, 1 group reporting), (C) kallikrein-1 (IPI00304808, 89% sequence coverage, 2 groups reporting), (D) apoE (IPI00021842, 21% sequence coverage, 1 group reporting), (E) TIMP-1 (SM/SL only, IPI00032292, 67% sequence coverage, 2 groups reporting), (F) HLA-G alpha chain, a nonclassical class I histocompatibility antigen (whole saliva, IPI00015988; a single peptide was sequenced by 1 research group), (G) prostatic acid phosphatase (IPI00396434, 8% sequence coverage, 1 group reporting), and (H) calpain-1 catalytic subunit (IPI00011285, 15% sequence coverage, 1 group reporting). E, apoE; AII-E, a complex of apoAII and apoE; AII-E-AII, AII-E with an additional apoAII; E-β-amyloid, a complex of apoE and β-amyloid.
Finally, we noticed that a high proportion of the salivary identifications were annotated as protein precursors; however, in many cases, these were sequences that included a signal peptide that is co-translationally cleaved in the endoplasmic reticulum. Therefore, this was simply the result of selecting representative proteins for an individual cluster that had the longest sequence.40 Another contributing factor could be the inherent characteristics of the IPI database, which was created using the same strategy we used to pick a prototypical representative from a group of homologous proteins with highly related sequences. Additionally, the annotations are derived from mRNA sequences, cDNA libraries, and automatic gene prediction programs.41 To determine if precursor forms of proteins were actually secreted, we extracted the location of signal peptides of salivary proteins from the UniProt database and looked for evidence of peptides that contained the corresponding sequences. Of the 250 salivary proteins that contained an N-terminal signal peptide, we found evidence for only 7 such peptides. Finally, a manual check of the IPI database showed that all the amylase entries were annotated in the IPI database as precursors. Therefore, we concluded that the salivary proteins annotated as precursors are most likely secreted as the mature forms of the proteins.
Discussion
A consortium of three research groups was formed to produce a catalogue of the human salivary proteome with maximal coverage. This work provides a framework to consolidate other MS-based analyses of saliva. Previously published studies are summarized in Table 2 and reviewed in several publications.63–67 Many of these investigators employed samples of whole saliva, which, in addition to secretions from the parotid and the SM/SL glands, contains the protein products of the cells that make up the oral cavity, minor gland and nasal secretions, gingival crevicular fluid, and contributions from microorganisms.68–73 Other investigators focused on subsets of salivary secretions. For example, 2-D SDS-PAGE separation followed by MS identification was used to characterize parotid, SM, SL, and/or SM/SL salivas collected as the ductal secretions.36,74,75 Other investigators targeted the salivary glycoproteome for analysis.76,77 Still others focused on pellicle components, the subset of proteins that adhere to the tooth surface.78,79 Finally, it will be interesting to compare the salivary gland transcriptomes and proteomes, an effort that has already begun, with this secretion-based proteome.80
Table 2.
Summary of Published LC-MS/MS Data in Terms of the Protein Components of Whole Saliva
The results of the work reported here also highlight the impact of methodological differences on the number of protein identifications that were made. For example, the groups at University of California—San Francisco and University of California—Los Angeles/University of Southern California used a larger sample size of donors than The Scripps Research Institute/University of Rochester group, who wanted to probe more deeply into the proteome. In other words, the objective of their coordinated fractionation/mass spectrometry approach was to improve coverage of the salivary proteome and access low-copy proteins. In addition, the workflow at The Scripps Research Institute/University of Rochester illustrates the fact that high-coverage analysis and detection of the proteome is possible if high-copy-number proteins are purified away from low-copy-number components and if a sufficient quantity of starting sample is employed. Such large-scale protein purification methods coupled with high-throughput LC-MS technology identified 914 and 917 proteins in parotid and SM/SL salivas, respectively. Purification of microgram quantities of low-copy-number proteins with an abundance of 1 in 100 000 required considerable amounts of starting material. Considering a purification yield of 10%, this objective required 10 μg of low-copy-number proteins and 106 μg of total starting saliva sample. Collection and fractionation of a total quantity of salivary proteins at an order of magnitude above this 1-g target were required to achieve substantial coverage of the salivary proteome. To this end, nearly 3 L of saliva was obtained and dialyzed to yield a dry weight of 10 g of lyophilized material. Dialysis was essential to remove a large quantity of low-molecular-mass components. Because salivary isoform distribution was expected to vary across the population, repeated collections of salivary samples were pooled from only one donor before a large-scale purification effort was initiated. In addition, parotid saliva was collected, pooled, and fractionated separately from SM/SL saliva. MS analysis was conducted on the starting material and fractions from chromatographic runs. Multidimensional chromatographic steps were also conducted to facilitate the fractionation of low-copy-number proteins from abundant species. Some Golgi enzymes with low-copy numbers are known to be secreted or shed into saliva and other fluids at very low rates.81 However, extensive protein fractionation and large-scale protein purification were required to detect this class of low-copy-number proteins; GALNT-1, -2, -5, -6, -7 (and other glycosyltransferases) were, in fact, detected with spectral counts ranging from 2 to 6 only in the large-scale saliva preparations (Supplemental Table 2 in Supporting Information).
Ultimately, the value of a compiled proteome depends on whether the mass spectrometric identifications, particularly those at the low end of the confidence spectrum, are accurate, for example, HLA-G, which was not included in the final protein catalogue because only a single peptide was identified by one group. To begin to address this question, we used an immunoblot approach and ductal saliva samples collected by the University of California—San Francisco group to verify the presence of novel components and proteins that were identified by one research team, which in most cases was The Scripps Research Institute/University of Rochester. The results of these experiments showed that, in the vast majority of cases, we detected the immunoreactive bands that corresponded to either (a) the predicted molecular mass of the antigen of interest or (b) complexes that contained the relevant protein. Category (a) included CEA, kallikrein, HLA-G, prostatic acid phosphatase, and calpain. Category (b) contained apoE and TIMP-1. In the latter cases, the band patterns corresponded to the published results of other groups that have characterized protein interactions involving these antigens. ApoE forms complexes with apoAII and β-amyloid;54 TIMP-1 binds to MMP-2 and -7.56–59 The results of these experiments led us to infer that other proteins that were not detected with the MS approaches were also in saliva, for example, β-amyloid and MMP-7. Other immunoblot experiments, for example, probing nitrocellulose blots of electrophoretically transferred ductal saliva samples with an antibody that specifically reacted with the transmembrane tyrosine kinase receptor ephrin A1, revealed a band of the expected molecular mass, predominantly in SM/SL saliva. However, most of the immunoreactivity was associated with very high molecular mass material that barely entered the separating gel (data not shown). The same situation occurred in the HLA-E analyses (data not shown). Of the 10 proteins we chose for verification, only one, MUC16, was not detected using an immunoblot approach.
Interestingly, there were noticeable differences in the antibody reactivity among donors and secretions, suggesting that the composition of the primary salivary proteome is gland-specific and may differ among individuals; formation of protein complexes is another source of sample-to-sample variation. These findings are in agreement with the concept of population proteomics, the subdiscipline that explores protein diversity within and across populations.82 It will be important to explore this heterogeneity in the context of oral health and disease, as it is likely that individual differences are biologically relevant. Thus, we expect that the variations we observed and the myriad more that are likely to emerge from subsequent analyses of the various salivary proteomes that have been assembled may eventually lead to the development of clinically useful biomarkers.
Compilation of the salivary proteome is the first step in identifying aberrations in the protein and/or peptide composition of saliva that track with disease processes. These aberrations are potential diagnostic and/or prognostic markers that could be used in a variety of nanotechnology-based formats being devised by another NIDCR-funded initiative (reviewed by Wong).66 With regard to oral diseases, IL-1 polymorphisms appear to track with periodontal disease;83,84 MUC7 levels in elderly patients predict Streptococcus mutans titers,85 and proteolytic processing of parotid salivary proteins diverges among individuals with different caries experiences.86 Sjögren’s syndrome, which leads to hyposalivation, is associated with a rise in the concentration of several major salivary proteins.87 Oral cancer is also associated with alterations in salivary composition. For example, PCR using saliva samples correlated p53 mutations with oral squamous cell carcinoma lesions;88 the presence of IgG and IgA antibodies against this cell cycle regulator has been used for the same purpose.89 It is envisioned that the generation of a comprehensive catalogue of parotid and SM/SL salivary proteins will provide an opportunity to evaluate the utility of a number of novel candidate biomarkers (e.g., MMP-9, TIMP-1, serpin B12, and kallikreins 1 and 11) for evaluating oral diseases.
In addition, salivary constituents are being used to monitor systemic conditions.8,90–93 For example, α-amylase levels correlate with catecholamine activity, a measure of stress.94 Hormone levels are particularly useful for a variety of purposes, including the assessment of ovarian function.95 The use of saliva samples for HIV testing is now common,96 and the diagnosis of Helicobacter pylori infections is possible.97 With regard to nonoral cancers, salivary CA125 expression and epidermal growth factor levels have diagnostic value in ovarian cancer and breast cancer, respectively.98,99 Our data suggest that a number of other markers of systemic disease may also be present in saliva. A recent example is the discovery of a panel of IFN-regulated proteomic and genomic biomarkers in whole saliva that are highly discriminatory of patients with primary Sjögren’s syndrome.100
Finally, we note the difficulties inherent in determining when any given proteome nears completion. For example, polymorphic isoforms, post-translational modifications, unique splice variants, deletions, and truncations are extremely difficult to characterize. Furthermore, large-scale proteomics methods based on bottom-up strategies rarely detect unknown isoforms and may misinterpret the presence or absence of documented isoforms, particularly in cases where assignments are made on the basis of low sequence coverage. We note that in this analysis these methods were used to collect as many protein entries as possible for the initial edition of the secreted salivary protein catalogue. Thus, more sophisticated methods that target specific questions involving post-translational modifications, splice variants, protein quantification, and other issues must be used to fill in the gaps. It is likely that top-down proteomic strategies will be able to address the question of polymorphic isoforms, as molecular weight measurements of intact proteins can yield this type of information.101 Although current top-down proteomics strategies are not amenable to high-throughput measurements and thus are used to answer highly focused questions, approaches such as top-down and de novo sequencing may become the methods of choice to address the complexity inherent in biological fluids such as human saliva.
Supplementary Material
Acknowledgments
We acknowledge the expert technical assistance of Lauren Jensen, Gene Watson, Jennifer Hryhorenko, Eric Johansen, Jon Sargent, Lindsay Thomas, Mattheus Dahlberg, Heidi Schmidt, Alan Jew, Albert Jew, Matthew Gormley, and Scott Dixon. This work was supported by the National Institute of Dental and Craniofacial Research (U01 DE 016274 to UCSF, U01 DE016267 to TSRI-UR, and U01 DE016275 to UCLA-USC).
Footnotes
Supporting Information Available: Detailed information about the methods that each group used and tables summarizing peptide and protein evidence and cluster composition, as well as gene ontology, KEGG, and BioCarta annotations of the salivary proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Levine MJ. Ann NY Acad Sci. 1993;694:11–16. doi: 10.1111/j.1749-6632.1993.tb18337.x. [DOI] [PubMed] [Google Scholar]
- 2.Lamkin MS, Oppenheim FG. Crit Rev Oral Biol Med. 1993;4:251–259. doi: 10.1177/10454411930040030101. [DOI] [PubMed] [Google Scholar]
- 3.Mandel ID. J Dent Res. 1987;66:623–627. doi: 10.1177/00220345870660S203. [DOI] [PubMed] [Google Scholar]
- 4.Humphrey SP, Williamson RT. J Prosthet Dent. 2001;85:162–169. doi: 10.1067/mpr.2001.113778. [DOI] [PubMed] [Google Scholar]
- 5.Dodds MW, Johnson DA, Yeh CK. J Dent. 2005;33:223–233. doi: 10.1016/j.jdent.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Lukacs JR, Largaespada LL. Am J Hum Biol. 2006;18:540–555. doi: 10.1002/ajhb.20530. [DOI] [PubMed] [Google Scholar]
- 7.Tabak LA. Pediatr Dent. 2006;28:110–117. discussion 192–118. [PubMed] [Google Scholar]
- 8.Puy C. Med Oral Patol Oral Cir Bucal. 2006;11:E449–455. [PubMed] [Google Scholar]
- 9.Lendenmann U, Grogan J, Oppenheim FG. Adv Dent Res. 2000;14:22–28. doi: 10.1177/08959374000140010301. [DOI] [PubMed] [Google Scholar]
- 10.Henkin RI, Martin BM, Agarwal RP. Am J Med Sci. 1999;318:380–391. doi: 10.1097/00000441-199912000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Shimada T. J Chem Ecol. 2006;32:1149–1163. doi: 10.1007/s10886-006-9077-0. [DOI] [PubMed] [Google Scholar]
- 12.Wahl SM, McNeely TB, Janoff EN, Shugars D, Worley P, Tucker C, Orenstein JM. Oral Dis. 1997;3(Suppl 1):S64–69. doi: 10.1111/j.1601-0825.1997.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 13.Roth G, Calmes R. Oral Biology. CV Mosby; St. Louis, MO: 1981. pp. 196–236. [Google Scholar]
- 14.Tandler B, Gresik EW, Nagato T, Phillips CJ. Anat Rec. 2001;264:121–145. doi: 10.1002/ar.1108. [DOI] [PubMed] [Google Scholar]
- 15.Yahiro J, Nagato T. Arch Oral Biol. 2002;47:631–635. doi: 10.1016/s0003-9969(02)00056-0. [DOI] [PubMed] [Google Scholar]
- 16.Kutta H, May J, Jaehne M, Munscher A, Paulsen FP. J Anat. 2006;208:609–619. doi: 10.1111/j.1469-7580.2006.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kutta H, May J, Jaehne M, Paulsen FP. Laryngorhinootologie. 2006;85:903–908. doi: 10.1055/s-2006-925296. [DOI] [PubMed] [Google Scholar]
- 18.Eng J, McCormack A, Yates JI. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 19.Sadygov RG, Cociorva D, Yates JR. Nat Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 20.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 21.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, Yates JR., III Anal Chem. 1997;69:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 22.Yates JR, III, Gilchrist A, Howell KE, Bergeron JJ. Nat Rev Mol Cell Biol. 2005;6:702–714. doi: 10.1038/nrm1711. [DOI] [PubMed] [Google Scholar]
- 23.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 24.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 25.Washburn MP, Wolters D, Yates JR., III Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 26.Lim H, Eng J, Yates JR, III, Tollaksen SL, Giometti CS, Holden JF, Adams MW, Reich CI, Olsen GJ, Hays LG. J Am Soc Mass Spectrom. 2003;14:957–970. doi: 10.1016/s1044-0305(03)00144-2. [DOI] [PubMed] [Google Scholar]
- 27.Lashley K. J Exp Psychol. 1916;1:461–493. [Google Scholar]
- 28.Block P, Brotman S. NY State Dent J. 1962;28:116–118. [Google Scholar]
- 29.Yates JR, Cociorva D, Liao L, Zabrouskov V. Anal Chem. 2006;78:493–500. doi: 10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
- 30.Bern M, Goldberg D, McDonald WH, Yates JR., III Bioinformatics. 2004;20(Suppl 1):I49–I54. doi: 10.1093/bioinformatics/bth947. [DOI] [PubMed] [Google Scholar]
- 31.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 32.Tabb DL, McDonald WH, Yates JR., III J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolff A, Begleiter A, Moskona D. J Dent Res. 1997;76:1782–1786. doi: 10.1177/00220345970760111001. [DOI] [PubMed] [Google Scholar]
- 34.Elias JE, Haas W, Faherty BK, Gygi SP. Nat Methods. 2005;2:667–675. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 35.Gillece-Castro BL, Prakobphol A, Burlingame AL, Leffler H, Fisher SJ. J Biol Chem. 1991;266:17358–17368. [PubMed] [Google Scholar]
- 36.Hardt M, Thomas LR, Dixon SE, Newport G, Agabian N, Prakobphol A, Hall SC, Witkowska HE, Fisher SJ. Biochemistry. 2005;44:2885–2899. doi: 10.1021/bi048176r. [DOI] [PubMed] [Google Scholar]
- 37.Robinson S, Niles R, Witkowska H, Rittenbach K, Nichols R, Sargent J, Dixon S, Prakobphol A, Hall S, Fisher S, Hardt M. Proteomics. 2008;8:435–445. doi: 10.1002/pmic.200700680. [DOI] [PubMed] [Google Scholar]
- 38.Carr S, Aebersold R, Baldwin M, Burlingame A, Clauser K, Nesvizhskii A. Mol Cell Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Bradshaw RA, Burlingame AL, Carr S, Aebersold R. Mol Cell Proteomics. 2006;5:787–788. doi: 10.1074/mcp.E600005-MCP200. [DOI] [PubMed] [Google Scholar]
- 40.Adamski M, Blackwell T, Menon R, Martens L, Hermjakob H, Taylor C, Omenn GS, States DJ. Proteomics. 2005;5:3246–3261. doi: 10.1002/pmic.200500186. [DOI] [PubMed] [Google Scholar]
- 41.Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 42.States DJ, Omenn GS, Blackwell TW, Fermin D, Eng J, Speicher DW, Hanash SM. Nat Biotechnol. 2006;24:333–338. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 43.de Souza GA, Godoy LM, Mann M. GenomeBiology. 2006;7:R72. doi: 10.1186/gb-2006-7-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prakobphol A, Boren T, Ma W, Zhixiang P, Fisher SJ. Biochemistry. 2005;44:2216–2224. doi: 10.1021/bi0480180. [DOI] [PubMed] [Google Scholar]
- 45.McMaster M, Zhou Y, Shorter S, Kapasi K, Geraghty D, Lim KH, Fisher S. J Immunol. 1998;160:5922–5928. [PubMed] [Google Scholar]
- 46.Sturn A, Quackenbush J, Trajanoski Z. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 47.Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. Genome Biology. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schenkels LC, Rathman WM, Veerman EC, Nieuw Amerongen AV. Biol Chem Hoppe-Seyler. 1991;372:325–329. doi: 10.1515/bchm3.1991.372.1.325. [DOI] [PubMed] [Google Scholar]
- 49.Schenkels LC, Veerman EC, Nieuw Amerongen AV. Crit Rev Oral Biol Med. 1995;6:161–175. doi: 10.1177/10454411950060020501. [DOI] [PubMed] [Google Scholar]
- 50.Liebig B, Brabletz T, Staege MS, Wulfanger J, Riemann D, Burdach S, Ballhausen WG. Cancer Lett. 2005;223:159–167. doi: 10.1016/j.canlet.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 51.Caruso A, Motolese M, Iacovelli L, Caraci F, Copani A, Nicoletti F, Terstappen GC, Gaviraghi G, Caricasole A. J Neurochem. 2006;98:364–371. doi: 10.1111/j.1471-4159.2006.03867.x. [DOI] [PubMed] [Google Scholar]
- 52.Zhou W, Scott SA, Shelton SB, Crutcher KA. Neuroscience. 2006;143:689–701. doi: 10.1016/j.neuroscience.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 53.Yamauchi K, Tozuka M, Nakabayashi T, Sugano M, Hidaka H, Kondo Y, Katsuyama T. J Neurosci Res. 1999;58:301–307. [PubMed] [Google Scholar]
- 54.Yamauchi K, Tozuka M, Hidaka H, Hidaka E, Kondo Y, Katsuyama T. Clin Chem. 1999;45:1431–1438. [PubMed] [Google Scholar]
- 55.Kim TD, Song KS, Li G, Choi H, Park HD, Lim K, Hwang BD, Yoon WH. BMC Cancer. 2006;6:211. doi: 10.1186/1471-2407-6-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Visse R, Nagase H. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 57.Howard E, Banda M. J Biol Chem. 1991;266:17972–17977. [PubMed] [Google Scholar]
- 58.Overall CM. Mol Biotechnol. 2002;22:51–86. doi: 10.1385/MB:22:1:051. [DOI] [PubMed] [Google Scholar]
- 59.Kivela-Rajamaki M, Maisi P, Srinivas R, Tervahartiala T, Teronen O, Husa V, Salo T, Sorsa T. J Periodontal Res. 2003;38:583–590. doi: 10.1034/j.1600-0765.2003.00688.x. [DOI] [PubMed] [Google Scholar]
- 60.Seki K, Miyakoshi S, Lee GH, Matsushita H, Mutoh Y, Nakase K, Ida M, Taniguchi H. Am J Surg Pathol. 2004;28:1384–1388. doi: 10.1097/01.pas.0000132743.89349.35. [DOI] [PubMed] [Google Scholar]
- 61.Melloni E, Michetti M, Salamino F, Minafra R, Pontremoli S. Biochem Biophys Res Commun. 1996;229:193–197. doi: 10.1006/bbrc.1996.1779. [DOI] [PubMed] [Google Scholar]
- 62.Evans JS, Turner MD. J Neurochem. 2007;103:849–859. doi: 10.1111/j.1471-4159.2007.04815.x. [DOI] [PubMed] [Google Scholar]
- 63.Drake RR, Cazare LH, Semmes OJ, Wadsworth JT. Expert Rev Mol Diagn. 2005;5:93–100. doi: 10.1586/14737159.5.1.93. [DOI] [PubMed] [Google Scholar]
- 64.Amado FM, Vitorino RM, Domingues PM, Lobo MJ, Duarte JA. Expert Rev Proteomics. 2005;2:521–539. doi: 10.1586/14789450.2.4.521. [DOI] [PubMed] [Google Scholar]
- 65.Hu S, Loo JA, Wong DT. Proteomics. 2006;6:6326–6353. doi: 10.1002/pmic.200600284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong DT. J Am Dent Assoc. 2006;137:313–321. doi: 10.14219/jada.archive.2006.0180. [DOI] [PubMed] [Google Scholar]
- 67.Tan S, Liang CRMY, Yeoh KG, So J, Hew CL, Chung MCM. Proteomics: Clin Appl. 2007;1:820–833. doi: 10.1002/prca.200700169. [DOI] [PubMed] [Google Scholar]
- 68.Hirtz C, Chevalier F, Centeno D, Egea JC, Rossignol M, Sommerer N, de Periere D. J Physiol Biochem. 2005;61:469–480. doi: 10.1007/BF03168453. [DOI] [PubMed] [Google Scholar]
- 69.Wilmarth PA, Riviere MA, Rustvold DL, Lauten JD, Madden TE, David LL. J Proteome Res. 2004;3:1017–1023. doi: 10.1021/pr049911o. [DOI] [PubMed] [Google Scholar]
- 70.Vitorino R, Lobo MJ, Ferrer-Correira AJ, Dubin JR, Tomer KB, Domingues PM, Amado FM. Proteomics. 2004;4:1109–1115. doi: 10.1002/pmic.200300638. [DOI] [PubMed] [Google Scholar]
- 71.Huang CM. Arch Oral Biol. 2004;49:951–962. doi: 10.1016/j.archoralbio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 72.Hu S, Xie Y, Ramachandran P, Loo RO, Li Y, Loo JA, Wong DT. Proteomics. 2005;5:1714–1728. doi: 10.1002/pmic.200401037. [DOI] [PubMed] [Google Scholar]
- 73.Guo T, Rudnick PA, Wang W, Lee CS, Devoe DL, Balgley BM. J Proteome Res. 2006;5:1469–1478. doi: 10.1021/pr060065m. [DOI] [PubMed] [Google Scholar]
- 74.Hu S, Denny P, Denny P, Xie Y, Loo JA, Wolinsky LE, Li Y, McBride J, Ogorzalek Loo RR, Navazesh M, Wong DT. Int J Oncol. 2004;25:1423–1430. [PubMed] [Google Scholar]
- 75.Walz A, Stuhler K, Wattenberg A, Hawranke E, Meyer HE, Schmalz G, Bluggel M, Ruhl S. Proteomics. 2006;6:1631–1639. doi: 10.1002/pmic.200500125. [DOI] [PubMed] [Google Scholar]
- 76.Ramachandran P, Boontheung P, Xie Y, Sondej M, Wong DT, Loo JA. J Proteome Res. 2006;5:1493–1503. doi: 10.1021/pr050492k. [DOI] [PubMed] [Google Scholar]
- 77.Savitski MM, Nielsen ML, Zubarev RA. Mol Cell Proteomics. 2006;5:935–948. doi: 10.1074/mcp.T500034-MCP200. [DOI] [PubMed] [Google Scholar]
- 78.Vitorino R, de Morais Guedes S, Ferreira R, Lobo MJ, Duarte J, Ferrer-Correia AJ, Tomer KB, Domingues PM, Amado FM. Eur J Oral Sci. 2006;114:147–153. doi: 10.1111/j.1600-0722.2006.00328.x. [DOI] [PubMed] [Google Scholar]
- 79.Yao Y, Berg EA, Costello CE, Troxler RF, Oppenheim FG. J Biol Chem. 2003;278:5300–5308. doi: 10.1074/jbc.M206333200. [DOI] [PubMed] [Google Scholar]
- 80.Hu S, Li Y, Wang J, Xie Y, Tjon K, Wolinsky L, Loo RR, Loo JA, Wong DT. J Dent Res. 2006;85:1129–1133. doi: 10.1177/154405910608501212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tomana M, Zikan J, Kulhavy R, Bennett JC, Mestecky J. Mol Immunol. 1993;30:277–286. doi: 10.1016/0161-5890(93)90056-h. [DOI] [PubMed] [Google Scholar]
- 82.Nedelkov D, Kiernan UA, Niederkofler EE, Tubbs KA, Nelson RW. Mol Cell Proteomics. 2006;5:1811–1818. doi: 10.1074/mcp.R600006-MCP200. [DOI] [PubMed] [Google Scholar]
- 83.Kornman KS, Crane A, Wang HY, di Giovine FS, Newman MG, Pirk FW, Wilson TG, Jr, Higginbottom FL, Duff GW. J Clin Periodontol. 1997;24:72–77. doi: 10.1111/j.1600-051x.1997.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 84.Socransky SS, Haffajee AD, Smith C, Duff GW. J Clin Periodontol. 2000;27:810–818. doi: 10.1034/j.1600-051x.2000.027011810.x. [DOI] [PubMed] [Google Scholar]
- 85.Baughan LW, Robertello FJ, Sarrett DC, Denny PA, Denny PC. Oral Microbiol Immunol. 2000;15:10–14. doi: 10.1034/j.1399-302x.2000.150102.x. [DOI] [PubMed] [Google Scholar]
- 86.Ayad M, Van Wuyckhuyse BC, Minaguchi K, Raubertas RF, Bedi GS, Billings RJ, Bowen WH, Tabak LA. J Dent Res. 2000;79:976–982. doi: 10.1177/00220345000790041401. [DOI] [PubMed] [Google Scholar]
- 87.Rujner J, Socha J, Barra E, Gregorek H, Madalinski K, Wozniewicz B, Giera B. Acta Paediatr. 1996;85:814–817. doi: 10.1111/j.1651-2227.1996.tb14157.x. [DOI] [PubMed] [Google Scholar]
- 88.Liao PH, Chang YC, Huang MF, Tai KW, Chou MY. Oral Oncol. 2000;36:272–276. doi: 10.1016/s1368-8375(00)00005-1. [DOI] [PubMed] [Google Scholar]
- 89.Warnakulasuriya S, Soussi T, Maher R, Johnson N, Tavassoli M. J Pathol. 2000;192:52–57. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH669>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 90.Streckfus C, Bigler L, O’Bryan T. Gerontology. 2002;48:282–288. doi: 10.1159/000065250. [DOI] [PubMed] [Google Scholar]
- 91.Lawrence HP. J Can Dent Assoc. 2002;68:170–174. [PubMed] [Google Scholar]
- 92.Forde MD, Koka S, Eckert SE, Carr AB, Wong DT. Int J Prosthodont. 2006;19:43–52. [PubMed] [Google Scholar]
- 93.Koka S, Forde MD, Khosla S. Int J Prosthodont. 2006;19:53–60. [PubMed] [Google Scholar]
- 94.Chatterton RT, Jr, Vogelsong KM, Lu YC, Ellman AB, Hudgens GA. Clin Physiol. 1996;16:433–448. doi: 10.1111/j.1475-097x.1996.tb00731.x. [DOI] [PubMed] [Google Scholar]
- 95.Lu Y, Bentley GR, Gann PH, Hodges KR, Chatterton RT. Fertil Steril. 1999;71:863–868. doi: 10.1016/s0015-0282(99)00093-x. [DOI] [PubMed] [Google Scholar]
- 96.Franco-Paredes C, Tellez I, del Rio C. Curr HIV/AIDS Rep. 2006;3:169–175. doi: 10.1007/s11904-006-0012-3. [DOI] [PubMed] [Google Scholar]
- 97.Jiang C, Li C, Ha T, Ferguson DA, Jr, Chi DS, Laffan JJ, Thomas E. Dig Dis Sci. 1998;43:1211–1218. doi: 10.1023/a:1018847522200. [DOI] [PubMed] [Google Scholar]
- 98.Navarro MA, Mesia R, Diez-Gibert O, Rueda A, Ojeda B, Alonso MC. Breast Cancer Res Treat. 1997;42:83–86. doi: 10.1023/a:1005755928831. [DOI] [PubMed] [Google Scholar]
- 99.Chen DX, Schwartz PE, Li FQ. Obstet Gynecol. 1990;75:701–704. [PubMed] [Google Scholar]
- 100.Hu S, Wang J, Meijer J, Leong S, Xie Y, Yu T, Zhou H, Vissink A, Pijpe J, Kallenberg C, Elashoff D, Loo J, Wong D. Arthritis Rheum. 2007;56:3588–3600. doi: 10.1002/art.22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kelleher NL. Anal Chem. 2004;76:197A–203A. [PubMed] [Google Scholar]
- 102.Xie H, Rhodus NL, Griffin RJ, Carlis JV, Griffin TJ. Mol Cell Proteomics. 2005;4:1826–1830. doi: 10.1074/mcp.D500008-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.