

Abstract
Metastatic colorectal cancer (mCRC) continues to be counted as a major health problem. The introduction of newer cytotoxics, irinotecan and oxaliplatin, has achieved a significant improvement in survival rates. Novel targeted therapies (bevacizumab, and cetuximab) in combination with most efficient chemotherapy regimens have pushed the median survival beyond the 2-year mark and increased the proportion of patients which could benefit from resection of metastatic lesions. In addition, several studies have proved that the CRC mutation profiles should influence patient selection or stratification in prospective trials. KRAS mutational status represents a paradigm for biomarker development in the era of molecular targeted therapies. The present article is an overview of the most important studies in the development of biomarkers for the optimization of anti-epidermal growth factor receptor (anti-EGFR) treatment in mCRC, beyond KRAS mutations, which is a work in progress. The aim will be to identify molecular markers that might be used to select patients with a higher probability of response to anti-EGFR monoclonal antibodies. Overall the accumulating evidence of the molecular biology of CRC has substantially changed the approach to mCRC treatment and has given clinicians more rational options for treating this illness.
Keywords: Colorectal cancer, Epidermal growth factor receptor protein, Monoclonal antibodies, KRAS, BRAF, PIK3CA, Mutation
INTRODUCTION
Now, more than ever, clinical oncologists are struggling to optimize treatment in cancer patients. With the use of molecular targeted agents and the incorporation of pharmacogenetics and pharmacogenomics in basic cancer treatment, a meaningful relationship between genotype (polymorphisms and mutations), gene expression profiles (level of gene expression of all or of target genes in the genome) and phenotype is being established, aimed at interpreting the variability among individuals in terms of response, resistance and toxicity to different drugs[1,2].
Pharmacogenetics (e.g. toxicity, age, comorbidities) commonly refers to the effects of a limited number of genes most often associated with drug metabolism, whereas pharmacogenomics (e.g. activity/resistance, gene expression level of all or targeted genes) involves the study of multigene patterns and pathways within the genome[3]. Genetic polymorphisms (variants in individual genomes, present in more than 1.5% of the population), somatic mutations in key target genes and differences in gene copy numbers may be responsible for different functional molecular roles and contribute to variability in drug pharmacokinetic and pharmacodynamic processes, altered drug metabolism or activation[4]. In colorectal cancer (CRC), as well as in other types of cancer, it has long been recognized that the same medications cause different responses in different patients. Genetic variations in drug targets and genes affecting target signal transduction can have a profound effect on drug efficacy and toxicity. This information could help to identify patients who are at increased risk of toxicity and select those likely to respond to specific agents, so that a more patient-specific treatment approach can be initiated[5].
The epidermal growth factor receptor (EGFR) belongs to the erbB receptor tyrosine kinase family which consists of 4 related transmembrane receptors: erbB1 (EGFR or HER1), erbB2 (HER2/neu), erbB3 (HER3) and erbB4 (HER4). Upon ligand binding, EGFR homo- or hetero-dimerizes with other erbB family members and initiates signaling through 2 main intracellular cascades which are mostly involved in cell survival, proliferation and motility. On one side, membrane localization of the lipid kinase PIK3CA counteracts PTEN and promotes AKT1 phosphorylation, and on the other, KRAS activates BRAF, which in turn triggers the mitogen-activated protein kinases[6]. EGFR is found to be overexpressed in various human malignancies, including CRC, lung, head and neck cancers and, as was initially hypothesized, therapeutic strategies designed to disrupt EGFR function could have anti-tumor activity[7] (Figure 1).
Figure 1.
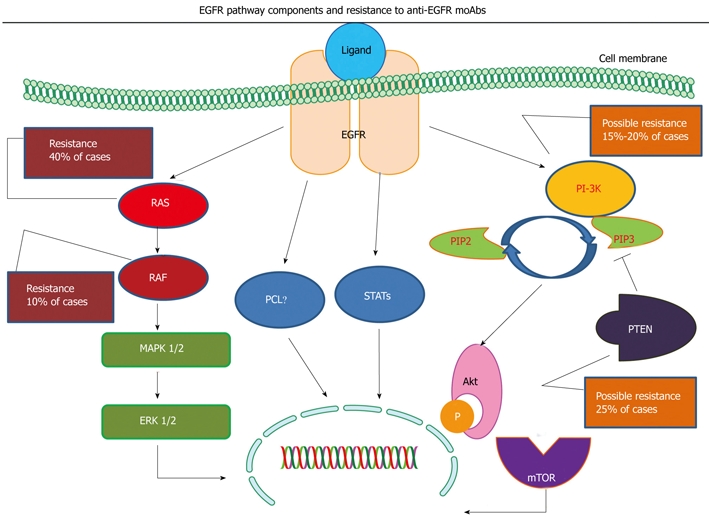
Simplified illustration of the epidermal growth factor receptor (EGFR) pathway with the RAS/MAPK and PIK3CA/PTEN cascades. Specific components of the pathway are correlated with resistance to anti-EGFR monoclonal antibodies (moAbs). As shown, KRAS and BRAF mutations are correlated with resistance to anti-EGFR moAbs, while further evaluation is required for PIK3CA mutations and PTEN loss of expression.
Two monoclonal antibodies (moAbs) targeting EGFR, the chimeric IgG1 moAb cetuximab and the fully humanized IgG2 moAb panitumumab, have recently entered clinical practice in the metastatic CRC (mCRC) setting. Both bind to the extracellular domain of the EGFR, thus leading to inhibition of its downstream signaling and have been found to provide a modest clinical benefit in pretreated patients[8-10]. Although they were initially registered for patients whose tumors were found to express the EGFR protein in immunohistochemistry, subsequently, it was clearly demonstrated that this methodology was not adequate to predict treatment efficacy[11]. Only the development of a skin rash was consistently associated with an increased response rate and progression-free survival in patients treated with anti-EGFR moAbs[8,10].
Although anti-EGFR therapies are active in some patients, the disease eventually becomes refractory to therapy in nearly all patients. As clinical parameters seem to be inadequate for patient selection, a major challenge is the identification of specific biomarkers that are likely to predict which patients will achieve the best response to such a treatment. EGFR gene status, as it is evaluated by fluorescent or chromogenic in situ hybridization (FISH or CISH), the absence or presence of mutations in genes downstream of EGFR and the presence of germline polymorphisms are implicated in response to anti-EGFR treatment and can independently impair or enhance its efficacy[12-15]. As most available data has come from retrospective studies, validation in prospective trials is imperative.
MECHANISMS OF RESISTANCE
Mutations
KRAS mutations: KRAS proto-oncogene encodes K-ras G-protein which plays a critical key role in the Ras/mitogen-activated protein kinase (MAPK) signaling pathway located downstream of many growth factor receptors including EGFR and which is involved in CRC carcinogenesis. K-ras recruitment by the activated EGFR is responsible for the activation of a cascade of serine-threonine kinases from the cell surface to the nucleus. KRAS mutations (in exon 2, codons 12 and 13) are present in more than one third of CRC patients and lead to the activation of one of the most important pathways for cell proliferation, the Ras/MAPK pathway, by inducing cyclin D1 synthesis. Consequently, in the presence of a KRAS mutation this pathway activation cannot be significantly inhibited by an anti-EGFR moAb (cetuximab or panitumumab) which acts upstream of the K-ras protein[13] (Figure 1).
In 2005, Moroni et al[16] assessed, in a small retrospective study, the mutation status of EGFR downstream intracellular effectors KRAS, BRAF and PIK3CA, and for the first time a trend towards higher response was seen in cetuximab-treated CRC patients whose tumors were of wild-type (WT) KRAS status. Subsequently, in 2006 in a study by Lièvre et al[13], KRAS mutations were found in 13 out of 30 tumors tested (43%) and this finding was significantly associated with the absence of response to cetuximab (KRAS mutation in 0% of the 11 responders vs 68.4% of the 19 non-responders; P = 0.0003). The overall survival (OS) of patients without KRAS mutation in their tumor was significantly higher compared with those patients with a mutation in the tumor (P = 0.016; median OS, 16.3 mo vs 6.9 mo) (Table 1).
Table 1.
Significance of KRAS mutations in retrospective single arm studies and randomized prospective trials
| Published studies | n | % KRAS mutations | Significant correlations |
| Retrospective single arm studies | |||
| Moroni et al[16] | 31 | 32 | None |
| Lièvre et al[13] | 30 | 43 | RR, mOS |
| Lièvre et al[18] | 89 | 27 | RR, PFS, mOS |
| De Roock et al[17] | 66 | 40 | RR, mOS |
| Randomized prospective trials | |||
| Amado et al[12] | 427 | 43 | RR, PFS, mOS |
| Tol et al[22] | 256 | 38 | RR, PFS |
| Van Cutsem et al[19] | 277 | 38 | RR, PFS, mOS |
| Bokemeyer et al[20] | 233 | 42 | RR, PFS |
| Hecht et al[21] | 865 | 40 | PFS, mOS |
| Douillard et al[23] | 1096 | 40 | RR, PFS |
RR: Response rate; mOS: Median overall survival; PFS: Progression-free survival.
When the results of the 2 above-mentioned studies were analyzed together, the predictive value of the KRAS mutation remained significant with a KRAS mutation frequency of 52.5% in non-responders compared with 9.5% in responders (P = 0.001). Thus, the probability of no response to cetuximab was 91.3% in the presence of KRAS mutation whereas as in the absence of such a mutation the probability of being a responder was 50%. The relative risk for a response to cetuximab was 10-fold higher for non-mutated patients compared with that of patients with the KRAS mutation [hazard ratio (HR), 10.5; 95% CI: 2.1-51.1]. Accordingly, in 2008, 3 studies, one with panitumumab[14] and 2 with cetuximab[17,18], confirmed the importance of KRAS mutations in the mCRC setting. In the study by Amado et al[12], KRAS mutation status was assessed in tumor samples from mCRC patients who were enrolled in the randomized phase III trial comparing panitumumab plus best supportive care (BSC) with BSC only after failure in 5-fluorouracil (5-FU)-, oxaliplatin- and irinotecan-based chemotherapy[10]. KRAS status was ascertained in 427 (92%) of 463 patients (208 panitumumab, 219 BSC). KRAS mutations were found in 43% of patients. The treatment effect on progression-free survival (PFS) in the WT KRAS group (HR, 0.45; 95% CI: 0.34-0.59) was significantly greater (P = 0.0001) than in the mutation group (HR, 0.99; 95% CI: 0.73-1.36). Median PFS in the WT KRAS group was 12.3 wk for panitumumab and 7.3 wk for BSC. Response rates to panitumumab were 17% and 0%, for the WT and mutant groups, respectively. WT KRAS patients had longer overall survival (HR, 0.67; 95% CI: 0.55-0.82; treatment arms combined). No significant differences in toxicity were observed between the WT KRAS group and the overall population[12]. Lièvre et al[18] assessed KRAS status by allelic discrimination in 89 mCRC patients treated with cetuximab in 6 different institutions. KRAS mutations were present in 27% of the patients and were associated with resistance to cetuximab (0% vs 40% of responders among the 24 mutated and 65 nonmutated patients, respectively; P < 0.001) and a poorer outcome (median PFS, 10.1 wk vs 31.4 wk in patients without mutation; P = 0.0001; median OS, 10.1 mo vs 14.3 mo in patients without mutation; P = 0.026). When these 89 patients were analyzed together with the 30 patients from the previous study[13], the multivariate analysis showed that KRAS status was an independent prognostic factor associated with OS and PFS. In a combined analysis, median OS for patients with 2, one, or no favorable prognostic factors (severe skin toxicity and absence of KRAS mutation) was 15.6, 10.7, and 5.6 mo, respectively. Lastly, De Roock et al[17] studied the KRAS mutation status in 113 irinotecan-refractory mCRC patients treated with cetuximab in 4 institutions and similar results were observed. Objective responses were detected in 27 of 66 WT KRAS patients vs 0 of 42 KRAS mutants. Median OS was significantly better in WT KRAS versus mutants (43.0 wk vs 27.3 wk; P = 0.020). In this study an additional association with radiologic response was made and it was found that the benefit was even more pronounced in patients with an early radiological response. The decrease in tumor size was significantly larger at all time points in WT patients. WT KRAS patients with an initial relative decrease of tumor size > 9.66% at week 6 had a significantly better median OS compared with all other patients (74.9 wk vs 30.6 wk; P = 0.0000025). Among WT KRAS patients OS was significantly better in patients with an initial decrease compared with those without (median OS, 74.9 wk vs 30.6 wk; P = 0.00000012). KRAS WT status was associated with survival benefit in cetuximab-treated mCRC[17]. An objective response was not observed in patients with mutant tumors treated with cetuximab or panitumumab monotherapy.
The predictive significance of KRAS mutations was also retrospectively analyzed in 5 prospective randomized trials. The CRYSTAL[19] trial was the first randomized trial which proved that the addition of cetuximab to a standard chemotherapy regimen (FOLFIRI) improved the response rate and PFS. Despite the statistically significant decrease in the risk of disease progression (HR, 0.85), the absolute benefit was modest (0.9 mo). Subsequently, when a patients’ subpopulation was analyzed according to the KRAS mutation status, the benefit from the addition of cetuximab was greater (HR, 0.68) in patients with WT primary tumors. In contrast, patients with KRAS mutant primary tumors experienced no benefit from the addition of the moAb[19]. Similar results have been reported from subgroup analysis in 3 other randomized trials, OPUS[20], PACE[21] and CAIRO2[22], in which different combination regimens were used with anti-EGFR moAbs. Finally, the first prospective analysis of a randomized trial (PRIME) has been recently reported[23]. The patients were randomized to receive FOLFOX4 or FOLFOX4 plus panitumumab and the KRAS mutation status was determined in 93% of the enrolled patients. Significant differences were observed in terms of RR (55% vs 48%) and PFS (9.6 mo vs 8.0 mo; P = 0.0234) in favor of the addition of panitumumab in the WT group. In contrast, a detrimental effect was recorded in the KRAS mutant group with the addition of the moAb to FOLFOX4 (7.3 mo vs 8.8 mo; P = 0.0227)[23] (Table 1). In all these trials, the objective RR were comparable between patients with KRAS mutant and KRAS WT tumors treated with chemotherapy alone, indicating that KRAS mutations are not predictive of the response to chemotherapy. No studies have been published comparing the impact of the 7 specific KRAS mutations on the response to anti-EGFR moAbs.
In conclusion, the present data in the international literature suggest that KRAS mutations are a predictor of resistance to anti-EGFR moAb therapy and are associated with a worse prognosis and a shorter survival. Approximately 40% of mCRC patients (those with mutated KRAS) could be selected to avoid costly and potentially toxic treatment. WT KRAS status identifies mCRC patients who are likely to respond to such a treatment and, thus, have a longer OS. Prospective randomized studies are needed to validate these results which introduce a new era in mCRC targeted therapy.
BRAF mutations: The role of KRAS has been extensively analyzed. However, KRAS mutations account for only 30%-40% of patients unresponsive to anti-EGFR moAbs treatment, suggesting that additional genetic determinants of resistance must exist. The RAS-RAF-MAPK kinase pathway mediates cellular responses to growth signals (Figure 1). The 3 RAF genes encode for cytoplasmic serine-threonine kinases that are the principal effectors of KRAS and are regulated by binding to it[24]. The single substitution missense mutation V600E, located within the kinase domain of BRAF (one of the 3 RAF genes), is the most common oncogenic mutation in cancer, accounting for more than 80%. The highest frequency is detected in melanomas (about 65%), the BRAF V600E mutation is also found at lower frequencies in a wide range of human cancers, such as CRC (10%), gliomas, ovarian and others. The V600E amino acid change results in constitutive activation of the BRAF kinase and promotes cell transformation[25,26]. KRAS and BRAF mutations are mutually exclusive in CRC[27,28].
Di Nicolantonio et al[26] retrospectively analyzed 113 mCRC tumors from cetuximab or panitumumab treated patients for KRAS and BRAF mutations and correlated the results with response, time to progression (TTP) and OS. KRAS mutations were present in 30% of the patients and were associated with resistance to cetuximab or panitumumab (P = 0.011). The BRAF V600E mutation was detected in 11 of 79 patients with WT KRAS. None of the BRAF-mutated patients responded to treatment, whereas none of the responders carried BRAF mutations (P = 0.029). BRAF-mutated patients had significantly shorter PFS (P = 0.011) and OS (P < 0.0001) than WT patients, meaning that the BRAF V600E mutation was inversely associated with response to anti-EGFR MoAb therapy and correlated with a worse prognosis. In CRC cell lines, the introduction/presence of the BRAF V600E allele impaired the therapeutic potential of cetuximab and panitumumab. Pharmacologic inhibition of BRAF, as initially hypothesized, restored sensitivity to anti-EGFR MoAbs in the CRC cell lines carrying the BRAF V600E mutation. The clinically approved small-molecule kinase BRAF inhibitor sorafenib when administered in combination with cetuximab slightly affected proliferation compared with sorafenib alone, whereas it showed a prominent proapoptotic effect. Thus, in the clinic the therapeutic effect of anti-EGFR MoAbs could be restored by 2-hit approaches aimed at blocking the EGFR pathway in multiple locations[26].
In the same frame as the Di Nicolantonio et al[26] study, Souglakos et al[29] sought to determine retrospectively the predictive value of the BRAF (exon 15), KRAS (exon 2) and PIK3CA (exons 9 and 20) point mutations with respect to clinical outcomes and response to active agents in 168 mCRC patients treated in the USA and Greece with 5-FU-based first-line chemotherapy (71% in combination with oxaliplatin and 34% with irinotecan and 58% with the addition of bevacizumab). In this study population, KRAS, BRAF and PIK3CA mutations were present in 62 (37%), 13 (8%) and 26 (15%) cases, respectively. Multivariate analysis uncovered BRAF mutation as an independent prognostic factor for decreased survival (HR, 3.6; 95% CI: 1.7-7.3). However, patients with BRAF-mutant tumors had significantly lower PFS (HR, 1.9; 95% CI: 1.03-3.5; P < 0.0001) than those whose primary tumors carried only WT BRAF. Of 100 patients treated with cetuximab and chemotherapy (8 in first-line and 92 as salvage treatment), the KRAS mutation predicted lack of response (P = 0.001) and shorter PFS (P = 0.015), in accordance with the international literature. BRAF mutations also correlated with reduced PFS in response to second-line use of cetuximab (P < 0.001). The likelihood of a response between patients with BRAF-mutant or BRAF-WT tumors was 0% vs 17%, and PFS with cetuximab-based therapy was significantly lower when tumors carried mutations in any of the 3 examined genes. BRAF mutations conferred a higher risk of relapse (HR, 3.9; P = 0.0005) after treatment with cetuximab-containing salvage combinations. These results underscore the potential of mutational profiling to help identify CRCs with different natural history or differential response to particular therapies. Lack of a cetuximab response observed with KRAS-mutant tumors may extend to other oncogene mutations, especially BRAF. The adverse significance of BRAF mutations should guide patient selection and stratification in future clinical trials[29].
In CRC tumors, BRAF mutations are reported to occur more frequently in those cases characterized by the presence of defective DNA mismatch repair (dMMR)[28]. Although the etiology is still ill-defined, in subsequent studies these mutations were found to occur almost exclusively in tumors showing the involvement of the hMLH1 gene (one of the genes involved in MMR) due to promoter hypermethylation. Current studies suggest that the BRAF V600E mutation occurs in 10% of tumors that are proficient in the MMR pathway (microsatellite stable - MSS/low microsatellite instability - MSI-L) and in > 50% of tumors that have dMMR (high microsatellite instability - MSI-H) due to promoter hypermethylation of the hMLH1 gene. BRAF mutations rarely, if ever, occur in tumors with dMMR because of the presence of germ-line mutations[30,31]. Thus, BRAF V600E is tightly associated with dMMR due to hMLH1 promoter hypermethylation and not with dMMR due to germ-line alterations.
In conclusion, it seems that the natural history and treatment response of BRAF-mutant tumors differ markedly from all others implying that the BRAF mutation does not simply substitute for KRAS activation in a linear signaling pathway, but likely confers additional or distinct properties, with ominous consequences. The current evidence supports that KRAS and BRAF mutations are mutually exclusive events. Of course, all these findings need to be formally confirmed prospectively in randomized clinical trials but if they are, then patients with the BRAF V600E mutation might justify foregoing approved treatments in favor of investigational therapy.
PIK3CA mutations: PIK3CA is one of the 2 most frequently mutated oncogenes in human tumors. Most of the reported mutations in the PIK3CA cluster are in conserved regions within the region coding for the helical and kinase domains of p110α. These mutations constitutively activate its kinase activity and, thus, make this enzyme an ideal target for drug development[32].
The PIK3CA gene encodes a lipid kinase that regulates alongside KRAS signaling pathways downstream of the EGFR. In addition, the p110α subunit of phosphatidylinositol-3-kinase (PI3K) which is encoded by PIK3CA, can be activated by interactions with the RAS proteins[33] (Figure 1). The PIK3CA gene is found mutated in approximately 20% of CRCs and the majority of the relevant mutations are located in the “hotspots” of exon 9 (E542K, E545K) and exon 20 (H1047R)[34]. PI3K-initiated signaling is normally inhibited by PTEN (phosphate and tensin homologue deleted on chromosome ten). In vitro it has been shown that cell lines with activating PIK3CA mutations or loss of PTEN expression (PTEN null) were more resistant to cetuximab than WT PIK3CA/PTEN-expressing cell lines (14% ± 5.0% vs 38.5% ± 6.4% growth inhibition, mean ± SE; P = 0.008). Consistently, PIK3CA mutant isogenic HCT116 cells showed increased resistance to cetuximab compared with WT PIK3CA controls. Furthermore, cell lines that were PIK3CA mutant/PTEN null and RAS/BRAF mutant were highly resistant to cetuximab compared with those without dual mutations/PTEN loss (10.8% ± 4.3% vs 38.8% ± 5.9% growth inhibition, respectively; P = 0.002), indicating that constitutive and simultaneous activation of the RAS and PIK3CA pathways confers maximal resistance to this agent[35]. In addition, in vivo, Frattini et al[36] have shown that loss of PTEN expression, which occurs in approximately 30% of sporadic CRC cases, may be associated with lack of response to cetuximab.
Sartore-Bianchi et al[37] analyzed 110 mCRC patients treated with anti-EGFR MoAbs for mutations of the PIK3CA and KRAS genes along with PTEN expression. Fifteen PIK3CA (13.6%) and 32 KRAS (29.0%) mutations were present. PIK3CA mutations were significantly associated with clinical resistance to panitumumab or cetuximab. None of the mutated patients achieved an objective response (P = 0.038) and when only WT KRAS tumors were analyzed, the statistical correlation was even stronger (P = 0.016). Patients with PIK3CA mutations displayed a worse clinical outcome also in terms of PFS (P = 0.035). The authors conclude that these results indicate that PIK3CA mutations can independently hamper the therapeutic response to panitumumab or cetuximab in mCRC. When the molecular status of the PIK3CA/PTEN and KRAS pathways are concomitantly ascertained, up to 70% of mCRC patients unlikely to respond to EGFR MoAbs can be identified[37]. In the study by Souglakos et al[29], PIK3CA mutations were also found to be associated with reduced PFS (P = 0.06), in response to second-line use of cetuximab. In addition, PIK3CA mutations conferred a higher risk of relapse (HR, 2.1; P = 0.01) after treatment with cetuximab-containing salvage combinations. However, regarding the response to first-line therapy, PFS was similar between patients whose tumors carried mutant or wild-type and PIK3CA[29].
In conclusion, the sum of the published data in the international literature imply that patients with KRAS-, BRAF- or PIK3CA-mutant tumors may all derive little benefit from treatment with anti-EGFR MoAbs. BRAF or PIK3CA mutations may account for about 1/3 of patients whose WT KRAS tumors do not respond to cetuximab[29]. A priori screening of CRC tumors for RAS/BRAF/PIK3CA mutations could help stratify patients likely to benefit from anti-EGFR MoAbs therapy.
Fcγ-RIIa exon 4 131G>A, Fcγ-RIIIa exon 5 158T>G single nucleotide polymorphisms (SNPs)
As has been shown recently, antibody-dependent cell-mediated cytotoxicity (ADCC) mediated through Fc receptors plays an important role in the anti-tumor activity of IgG1 antibodies. ADCC is an immunological mechanism which involves the interaction between Fc receptors carried on the surface of immune cells such as macrophages and natural killer (NK) cells and the Fc fragment of moAbs which are bound on tumor cells[38]. This way, moAbs may exert an indirect antitumor activity by recruiting cytotoxic host effector cells, such as monocytes and NK cells[39]. One group of IgG Fc receptors, FcγRs are expressed on leukocytes and are composed of 3 distinct classes: FcγRI, FcγRII (FcγRIIa and FcγRIIb), FcγRIII (FcγRIIIa and FcγRIIIb). The receptors are also distinguished by their affinity for IgG. FcγRI exhibits high affinity for IgG, whereas FcγRII and FcγRIII show a weaker affinity. FcγRIIa and FcγRIIIa are activating FcγRs which are expressed on monocytes/macrophages and monocytes/macrophages/NK cells, respectively, and can trigger cytotoxicity of human targets[39].
Cetuximab, an IgG1 moAb, competes with the natural ligands of EGFR, EGF and transforming growth factor-α (TGF-α). When binding to cancer cells, it inhibits EGFR dimerization and downstream signaling, thus inhibiting proliferation and inducing apoptosis[40]. In experimental models, it has been shown that another mechanism of action of cetuximab against cancer cells is mediated via ADCC. The effectiveness of ADCC may depend on the degree of activation of effector cells after FcγRIIa and FcγRIIIa engagement[41].
The binding affinity of the FcγRs is under the influence of germline genetic polymorphisms detected on genes encoding for FcγRIIa and FcγRIIIa. The SNP 131G>A (or H131R) in position 131 of exon 4 of FcγRIIa gene which leads to the substitution of an arginine with an histidine and the SNP 158Τ>G (or V158F) in position 158 of FcγRIIIa gene, which leads to the substitution of a phenylalanine with a valine, are shown to affect the receptors’ affinities for the Fc fragment of antibodies and probably ADCC efficiency[42,43].
In a publication of Zhang et al[44] in 39 patients with mCRC treated with cetuximab, TTP was statistically significantly better for patients with the FcγRIIa H131R SNP (P = 0.037) and for the FcγRIIIa V158F (P = 0.055). In a study by Bibeau et al[45], in 69 mCRC patients treated with the combination of irinotecan and cetuximab, it was observed that the patients who were homozygous for the H/Η allele of SNP FcγRIIa H131R and/or for the V/V allele of SNP FcγRIIIa V158F had a greater TTP compared with those who carried the R or F alleles (3.2 mo, 2.8 mo, respectively, P = 0.015). Nevertheless, the above-mentioned correlations could not be confirmed in other studies[46,47].
Other polymorphisms
EGFR intron-1 (CA)n repeat polymorphisms: A highly polymorphic sequence of (CA)n repeats (n = 15-22) is located in intron 1 of the EGFR gene. Allele 16 (with 16 CA repeats) is seen more frequently (42%), followed by allele 20 (26%) and 18 (20%)[48]. In vivo and in vitro studies have shown that transcriptional activity of the gene is affected, as a result of a variable impact on DNA binding sites, in such a way that the greater number of CA repeats reflects lower EGFR mRNA levels and protein expression[49,50]. In a study by Amador et al[51], from 19 patients with mCRC treated with gefitinib, those who had a small number of CA repeats more frequently manifested dermatologic toxicity (84% of the patients with ≤ 35 CA repeats and only 33% of those with > 35). In a study by Graziano et al[46] in 110 patients with mCRC who underwent irinotecan-cetuximab salvage therapy after disease progression during or after oxaliplatin-based first-line and irinotecan-based second-line chemotherapy, a small number of intron 1 CA repeats (< 17) was correlated, in a multivariate analysis, with favorable OS (HR, 0.41; 95% CI: 0.21-1.78; P = 0.006), treatment response (P = 0.008) and more frequent grade 2 and 3 dermatologic toxicity (P = 0.001) compared with a larger number of CA repeats (≥ 17). Although the above-mentioned study did not have a control arm and thus the predictive role of the polymorphic repeats could not be determined, it is probable that the higher EGFR expression in patients with a small number of CA repeats could also reflect a greater effect of cetuximab[46].
EGFR exon 13 497G>A (or R521K) SNP
Another SNP, a G>A substitution (rs11543848) in codon 521 (previously described as codon 497) in exon 13, which encodes a part of the extracellular region of the EGFR, has been described and results in an amino acid substitution of an arginine (R) with a lysine (K). This is located at the boundary between EGFR domain III (the direct interaction site with cetuximab) and domain IV[52]. This amino acid substitution has been shown to significantly reduce TGF-α binding and ligand-induced EGFR signaling, which could make the cell even more sensitive to targeted receptor inhibition through cetuximab, for example[52,53].
In a study by Gonçalves et al[52], tumor tissue samples from patients with mCRC treated with irinotecan/cetuximab were analyzed and the EGFR exon 13 variant (R521K) was associated with better PFS and OS. Indeed, the above-mentioned SNP was observed in 11 of the 21 patients who achieved an objective response or stable disease and in only 1 of the 11 patients who had disease progression (P = 0.02). In addition, in a third study it has been correlated with longer OS in stage II and III patients after surgery[54]. Nevertheless, in the study by Graziano et al[46], in mCRC patients treated with irinotecan/cetuximab, this SNP was not found to be associated with response to treatment or OS.
Epidermal growth factor (EGF) 5’-UTR 61A>G SNP
EGF is one of the natural ligands of EGFR and upon binding it may activate DNA synthesis and cellular proliferation and it has been shown to stimulate mitosis in epidermal cells. The EGF protein is encoded by the EGF gene which is located on chromosome 4q25-27 and contains 24 exons[55]. The only functional polymorphism of the EGF gene was identified in 2002 and is located 61 base pairs (bp) downstream of the EGF promoter, in the 5’-untranslated region of the gene. It consists of a substitution of guanine (G) for adenine (A), (61Α>G), it modulates the transcription of EGF and it has been correlated in vitro and in vivo with elevated serum levels[56,57]. Primarily, it has been studied in patients with melanoma and glioblastoma multiforme, but it has also been detected in 44% of the European white population[56,57].
In the study by Graziano et al[46], in 110 patients with mCRC treated with irinotecan and cetuximab, the EGF 61G/G allele was associated with a greater OS (HR, 0.44; 95% CI: 0.23-0.84, P = 0.01) but not with greater response rate, PFS and skin toxicity. The exact mechanism by which this SNP is associated with greater survival is not yet known, but, in experimental models and with different concentrations, EGF has been shown to induce apoptosis and growth inhibition rather than the usual growth-promoting effect[58]. In addition, in another study with 133 mCRC patients treated with cetuximab monotherapy the EGF 61G/G allele was associated with greater PFS (P = 0.04)[47]. In contrast, in the Zhang et al[59] study with mCRC treated with cetuximab, the EGF 61A/A allele was correlated favorably with an increased OS (median OS 15 mo for EGF 61A/A, 2.3 mo for EGF 61G/G and 5.7 mo for the heterozygote EGF 61Α/G).
Cyclin D1 exon 4 870A>G SNP
Cyclin D1 is a cell cycle regulatory protein whose upregulation has been associated with increased proliferation and poor clinical outcome in a number of neoplasms including CRC[60]. Cyclin D1 is a key element in the downstream EGFR signaling pathway; EGFR inhibition results in cyclin D1 downregulation, leading cells into the G1 phase and subsequently to apoptosis[61]. The 870Α>G SNP in exon 4 of the cyclin D1 gene (A to G substitution) influences cyclin D1 mRNA splicing in the border between exon 4 and intron 4 resulting in 2 different mRNA transcripts, a and b. The G allele encodes transcript a, whereas the A allele encodes transcript b, which results in a longer half-life cyclin-D1[61,62].
Zhang et al[59] in a pilot study of 39 mCRC patients under treatment with cetuximab monotherapy, found that both the cyclin-D1 870Α>G SNP, as well as the EGF 61Α>G SNP could be used as predictive molecular markers of cetuximab therapy. More specifically, the cyclin-D1 870Α>G SNP was statistically significantly correlated with OS. Patients with the Α/Α genotype had a very short median OS of 2.3 mo, whereas, patients with at least one G (A/G or G/G) had a median OS of 8.7 mo (P = 0.019). Furthermore, when combined with EGF 61Α>G, the cyclin-D1 870Α>G shows an even further significant association with OS. Patients with the favorable genotypes (at least one A for the first one and one G for the second) had a median OS of 12 mo, in contrast with 4.4 mo in the patients with unfavorable genotypes[59]. Unfortunately, Nagashima et al[47] and Graziano et al[46] did not find similar correlations in their studies.
Rare SNPs
The G765C SNP of cyclooxygenase-2 (COX-2) which has been correlated in vitro with reduced promoter activity and the T251A SNP of interleukin-8 which has been correlated with increased interleukin-8 production, have not been associated with response to cetuximab treatment in mCRC patients. However, the COX-2 G765C SNP has been weakly correlated with skin toxicity (P = 0.15)[59]. A schematic representation of the above-mentioned SNPs is shown in Table 2.
Table 2.
Single nuclear polymorphism (SNP) analysis in patients treated with anti-EGFR moAbs
| Publication | SNP | n | Variable | Significance |
| Zhang et al[44] | FcγRIIa H131R, V158F | 39 | TTP | 0.037 |
| Bibeau et al[45] | FcγRIIa H131R, V158F | 69 | TTP | 0.015 |
| Graziano et al[46] | FcγRIIa H131R, V158F | 110 | RR, OS | NS |
| Nagashima et al[47] | FcγRIIa H131R, V158F | 98 | PFS | NS |
| Graziano et al[46] | EGFR in1 (CA)n repeats | 110 | RR, OS | 0.006, 0.008 |
| Gonçalves et al[52] | EGFR ex 13 R521K | 32 | PFS | 0.020 |
| Graziano et al[46] | EGF 61G/G | 110 | OS | 0.001 |
| Zhang et al[44] | cyclin-D1 870Α>G | 39 | TTP | 0.019 |
| Graziano et al[46] | cyclin-D1 870Α>G | 110 | RR, OS | NS |
| Nagashima et al[47] | cyclin-D1 870Α>G | 98 | PFS | NS |
anti-EGFR: Anti-epidermal growth factor receptor; NS: Not significant.
Gene copy numbers and EGFR ligands mRNA expression: As initially reported in a cohort study, the objective tumor response to the EGFR-targeted moAbs, cetuximab and panitumumab, in mCRC occurred in a fraction of patients whose tumors had an increased EGFR gene copy number (GCN), as assessed by FISH[16]. Subsequently, in further studies, the predictive role of EGFR GCN was evaluated and an association with objective tumor response[63] and OS[63] was demonstrated.
Lièvre et al[13], using CISH instead of FISH, confirmed the results of Moroni et al[16], but both studies were inconclusive probably due to the limited number of patients tested and the non-homogeneous treatments they received. In the study by Lenz et al[63], the EGFR GCN was evaluated with polymerase chain reaction (PCR) and although no association was detected with objective responses and PFS, increased GCN was significantly positively correlated with OS. The discrepancies between these studies could be a result of different techniques or sample limitations, but the association of EGFR GCN with OS could also reflect its role as an independent prognostic variable[13,16,63].
In an attempt to test EGFR GCN in a larger and more homogeneous patient population and clarify its predictive role in terms of OR, PFS and OS Sartore-Bianchi et al[14] analyzed mCRC patients’ tumors from the randomized phase III trial comparing panitumumab plus BSC with BSC only after failure in 5-FU-, oxaliplatin- and irinotecan-based chemotherapy[10]. In this study EGFR GCN was assessed by FISH and its status was evaluated as the mean value of EGFR gene copies/nucleus, as the mean value of EGFR gene/CEP7 (α-centromeric probe of chromosome 7) and as the percentage of chromosome 7 polysomy (≥ 3 signals per nucleus) scoring 200 tumor cells. A statistically significant positive correlation between increase in mean EGFR GCN and probability of response to panitumumab (odds ratio, 5.62; 95% CI: 1.506-20.974) was found with 98.1% specificity (95% CI: 78.5%-96%). In addition, the best cut-off value of mean EGFR GCN, ≥ 2.47 EGFR gene copies/nucleus, as assessed by ROC analysis, to discriminate responders from nonresponders, had a sensitivity of 100% (95% CI: 54.1%-100%), since no objective response was observed when the EGFR GCN was less than this value. Because of the non-homogeneous pattern of EGFR GCN in the tumors, the percentage of cells displaying chromosome 7 polysomy and/or EGFR gene amplification was also calculated. In the same way, an increase in the percentage of chromosome 7 polysomy was also significantly associated with the probability of objective response (odds ratio, 1.04; 95% CI: 1.007-1.074), with a specificity of 100% (95% CI: 93.2%-100%) and a negative predictive value of 89.7% (95% CI: 78.8%-96.1%). In other words, in patients treated with panitumumab, a mean EGFR GCN of less than 2.5/nucleus or less than 40% of tumor cells displaying chromosome 7 polysomy within the tumor predicted a shorter PFS (P = 0.039 and P = 0.029, respectively) and OS (P = 0.015 and P = 0.014, respectively), thus generating the hypothesis that these tumors were probably not driven by the EGFR pathway. None of the treated patients with mean EGFR GCN of less than 2.47/nucleus or less than 43% of tumor cells displaying chromosome 7 polysomy obtained an objective response compared with 6 of 20 and 6 of 19 patients with values greater than these cut-off limits (P = 0.0009 and P = 0.0007, respectively). Evaluation of BSC-treated patients showed no correlation between EGFR GCN or chromosome 7 polysomy status and PFS. Interestingly, in the tumors of patients from the BSC only arm, no correlation was found between EGFR GCN and PFS, thus indicating more its predictive, rather than its prognostic role[14].
In an exploratory clinical trial conducted by Khambata-Ford et al[64], a large prospective human cohort uniformly treated with cetuximab was exploited in an attempt to systematically identify biomarkers associated with disease control to anti-EGFR MoAb treatment. Transcriptional profiling was conducted on RNA from tumor cells in order to identify genes whose expression correlated with best clinical response. EGFR GCN was detected at a frequency of 6% by quantitative PCR but its increase within the disease control group was not statistically significant. In addition, gene expression profiles showed that tumors that express high levels of the EGFR ligands epiregulin (EREG) and amphiregulin (AREG) are more likely to show disease control with cetuximab (EREG, P = 0.000015; AREG, P = 0.000025) and have significantly longer PFS than patients with low expression (EREG: P = 0.0002; HR, 0.47; median PFS, 103.5 d vs 57 d, respectively; AREG: P = 0.0001; HR, 0.44; median PFS, 115.5 d vs 57 d, respectively)[64]. Lately, these results were confirmed in another patients’ cohort treated with cetuximab and irinotecan in combination[65]. The authors found that overexpression of EREG and AREG in KRAS WT patients was associated with a decreased risk of progression (HR, 0.41 and 0.43, respectively) and death (HR, 0.42 and 0.4, respectively)[65].
Lastly, in a study by Scartozzi et al[66], the role of nuclear factor-κB (NF-κB) was investigated. NF-κB plays a role in the activation of the EGFR downstream signaling pathway and was shown to be responsible for resistance to antineoplastic agents. EGFR can induce NF-κB, and high levels of EGFR expression are essential for EGFR-mediated NF-κB activation[67]. In the above-mentioned study, NF-κB and EGFR expression were evaluated retrospectively by immunohistochemistry and the results were correlated with response rate, TTP and OS in mCRC patients receiving irinotecan-cetuximab treatment. The response rate was 10% (4 partial responses) vs 48% (12 partial responses; P = 0.0007) in NF-κB-positive and NF-κB-negative tumors, respectively. Median TTP in NF-κB-positive patients was 3 vs 6.4 mo in the remaining patients (P = 0.021). Median OS was 9.5 mo vs 15.8 mo for NF-κB-positive and NF-κB-negative patients, respectively (P = 0.036). The difference in median TTP, OS and response rate could mean that NF-κB may play a role in predicting the efficacy of irinotecan-cetuximab therapy in the mCRC setting[66].
CONCLUSION
The CRC mutation profiles should influence patient selection or stratification in prospective trials. KRAS mutational status represents a paradigm for biomarker development in the era of molecular targeted therapies. As a result, KRAS testing is now mandatory at the presentation of metastatic disease in patients with CRC. In total, almost 50% of mCRC patients’ tumors harbor either the KRAS (40%) or BRAF (10%) mutation and are not candidates for anti-EGFR moAb therapy. Patients with KRAS mutations may benefit from combination chemotherapy and anti-vascular endothelial growth factor moAbs (such as bevacizumab). Patients with KRAS mutations and resistance or relapse to chemotherapy and/or bevacizumab have limited treatment options and could be candidates for clinical trials with investigational agents, such as mTOR or extracellular-signal-regulated kinase inhibitors. The BRAF V600E mutation identifies a subgroup (less than 10%) of patients with an exceptionally unfavorable prognosis. These patients might justify forego approved treatments in favor of investigational therapy, such as molecules that inhibit the WT (sorafenib) and/or the mutant (PXL 4032) BRAF allele, either alone or in combination with chemotherapy. In addition, PIK3CA mutations appear to be useful predictors for response to anti-EGFR moAbs, but definitive conclusions should be based on the analysis of larger cohorts of patients in randomized trials that include patients who have not been exposed to anti-EGFR targeted therapies.
In addition, the expression of AREG and EREG was consistently associated with the outcome of cetuximab and panitumumab combination chemotherapy. The results of SNPs and GCN are premature and controversial, and thus need to be explored in a more systematic approach.
The development of biomarkers for the optimization of anti-EGFR treatment in mCRC, beyond KRAS mutations, is a work in progress. The aim will be to identify molecular markers that might be used to select patients with a higher probability of response to anti-EGFR moAbs.
Footnotes
Peer reviewers: Dr. Inti Zlobec, PhD, Institute for Pathology, University Hospital Basel, Schoenbeinstrasse 40, Basel, CH-4031, Switzerland; Toshiyuki Ishiwata, Associate Professor, Department of Pathology, Integrative Oncological Pathology, Nippon Medical School, 1-1-5 Sendagi, Bunkyo-ku, Tokyo 113-8602, Japan
S- Editor Wang YR L- Editor Cant MR E- Editor Ma WH
References
- 1.Chabner BA. Cytotoxic agents in the era of molecular targets and genomics. Oncologist. 2002;7 Suppl 3:34–41. doi: 10.1634/theoncologist.7-suppl_3-34. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen H, Tran A, Lipkin S, Fruehauf JP. Pharmacogenomics of colorectal cancer prevention and treatment. Cancer Invest. 2006;24:630–639. doi: 10.1080/07357900600896281. [DOI] [PubMed] [Google Scholar]
- 3.Yan L, Beckman RA. Pharmacogenetics and pharmacogenomics in oncology therapeutic antibody development. Biotechniques. 2005;39:565–568. [PubMed] [Google Scholar]
- 4.Araújo A, Ribeiro R, Azevedo I, Coelho A, Soares M, Sousa B, Pinto D, Lopes C, Medeiros R, Scagliotti GV. Genetic polymorphisms of the epidermal growth factor and related receptor in non-small cell lung cancer--a review of the literature. Oncologist. 2007;12:201–210. doi: 10.1634/theoncologist.12-2-201. [DOI] [PubMed] [Google Scholar]
- 5.Evans WE, McLeod HL. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 6.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roda JM, Joshi T, Butchar JP, McAlees JW, Lehman A, Tridandapani S, Carson WE 3rd. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res. 2007;13:6419–6428. doi: 10.1158/1078-0432.CCR-07-0865. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 9.Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–1208. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 10.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 11.Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–1810. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 12.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 13.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 14.Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, Sobrero A, Barone C, Cascinu S, Colucci G, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25:3238–3245. doi: 10.1200/JCO.2007.11.5956. [DOI] [PubMed] [Google Scholar]
- 15.Spano JP, Milano G, Vignot S, Khayat D. Potential predictive markers of response to EGFR-targeted therapies in colorectal cancer. Crit Rev Oncol Hematol. 2008;66:21–30. doi: 10.1016/j.critrevonc.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–286. doi: 10.1016/S1470-2045(05)70102-9. [DOI] [PubMed] [Google Scholar]
- 17.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 18.Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 19.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 20.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 21.Hecht JR, Mitchell E, Chidiac T, Scroggin C, Hagenstad C, Spigel D, Marshall J, Cohn A, McCollum D, Stella P, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–680. doi: 10.1200/JCO.2008.19.8135. [DOI] [PubMed] [Google Scholar]
- 22.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 23.Douillard J, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel ME, Humblet Y, Cunningham D, Wolf M, Gansert JL. Randomized phase 3 study of panitumumab with FOLFOX4 compared to FOLFOX4 alone as 1st-line treatment (tx) for metastatic colorectal cancer (mCRC): the PRIME trial. Eur J Cancer Suppl. 2009;7:9. [Google Scholar]
- 24.Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001;93:53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- 25.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 26.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 27.Frattini M, Balestra D, Suardi S, Oggionni M, Alberici P, Radice P, Costa A, Daidone MG, Leo E, Pilotti S, et al. Different genetic features associated with colon and rectal carcinogenesis. Clin Cancer Res. 2004;10:4015–4021. doi: 10.1158/1078-0432.CCR-04-0031. [DOI] [PubMed] [Google Scholar]
- 28.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 29.Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, Silver J, Ogino S, Hooshmand S, Kwak E, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer. 2009;101:465–472. doi: 10.1038/sj.bjc.6605164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, Westra J, Frebourg T, Espín E, Armengol M, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–668. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–5212. [PubMed] [Google Scholar]
- 32.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 34.Samuels Y, Diaz LA Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97:1139–1145. doi: 10.1038/sj.bjc.6604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 38.Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1:118–129. doi: 10.1038/35101072. [DOI] [PubMed] [Google Scholar]
- 39.Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood. 1990;76:2421–2438. [PubMed] [Google Scholar]
- 40.Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002;94:1593–1611. doi: 10.1002/cncr.10372. [DOI] [PubMed] [Google Scholar]
- 41.Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M, et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res. 2007;13:1552–1561. doi: 10.1158/1078-0432.CCR-06-1726. [DOI] [PubMed] [Google Scholar]
- 42.Niwa R, Hatanaka S, Shoji-Hosaka E, Sakurada M, Kobayashi Y, Uehara A, Yokoi H, Nakamura K, Shitara K. Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 Is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res. 2004;10:6248–6255. doi: 10.1158/1078-0432.CCR-04-0850. [DOI] [PubMed] [Google Scholar]
- 43.van Royen-Kerkhof A, Sanders EA, Wijngaarden S, van Roon JA, Voorhorst-Ogink M, Walraven V, Gerritsen A, van Dijk MA, Kuis W, Rijkers GT, et al. Flow cytometric determination of FcgammaRIIa (CD32) polymorphism. J Immunol Methods. 2004;294:135–144. doi: 10.1016/j.jim.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, Chang HM, Borucka E, Lurje G, Sherrod AE, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25:3712–3718. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- 45.Bibeau F, Crapez E, Di Fiore F, Thezenas S, Sabourin J, Lamy A, Frebourg T, Michel P, Ychou M, Boissiere-Michot F. Association of FcγRIIa and FcγRIIa polymorphisms with clinical outcome in metastatic colorectal cancer patients (mCRC) treated with cetuximab and irinotecan. J Clin Oncol. 2008;26:(May 20 suppl; abstr 11004). Available from: http://www.asco.org/ASCOv2/Meetings/Abstracts?&vmview=abst_detail_view&confID=55&abstractID=32263. [Google Scholar]
- 46.Graziano F, Ruzzo A, Loupakis F, Canestrari E, Santini D, Catalano V, Bisonni R, Torresi U, Floriani I, Schiavon G, et al. Pharmacogenetic profiling for cetuximab plus irinotecan therapy in patients with refractory advanced colorectal cancer. J Clin Oncol. 2008;26:1427–1434. doi: 10.1200/JCO.2007.12.4602. [DOI] [PubMed] [Google Scholar]
- 47.Nagashima F, Zhang W, Gordon M, Chang HM, Lurje G, Borucka E, Yang D, Ladner R, Rowinsky E, Lenz HJ. EGFR, Cox-2, and EGF polymorphisms associated with progression-free survival of EGFR-expressing metastatic colorectal cancer patients treated with single-agent cetuximab (IMCL-0144) J Clin Oncol. 2007;25:18S (June 20 Supplement). doi: 10.1200/JCO.2006.08.8021. Available from: http://www.asco.org/ASCOv2/Meetings/Abstracts?&vmview=abst_detail_view&confID=47&abstractID=30766. [DOI] [PubMed] [Google Scholar]
- 48.Chi DD, Hing AV, Helms C, Steinbrueck T, Mishra SK, Donis-Keller H. Two chromosome 7 dinucleotide repeat polymorphisms at gene loci epidermal growth factor receptor (EGFR) and pro alpha 2 (I) collagen (COL1A2) Hum Mol Genet. 1992;1:135. doi: 10.1093/hmg/1.2.135. [DOI] [PubMed] [Google Scholar]
- 49.Etienne-Grimaldi MC, Pereira S, Magné N, Formento JL, Francoual M, Fontana X, Demard F, Dassonville O, Poissonnet G, Santini J, et al. Analysis of the dinucleotide repeat polymorphism in the epidermal growth factor receptor (EGFR) gene in head and neck cancer patients. Ann Oncol. 2005;16:934–941. doi: 10.1093/annonc/mdi189. [DOI] [PubMed] [Google Scholar]
- 50.Gebhardt F, Zänker KS, Brandt B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J Biol Chem. 1999;274:13176–13180. doi: 10.1074/jbc.274.19.13176. [DOI] [PubMed] [Google Scholar]
- 51.Amador ML, Oppenheimer D, Perea S, Maitra A, Cusatis G, Iacobuzio-Donahue C, Baker SD, Ashfaq R, Takimoto C, Forastiere A, et al. An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res. 2004;64:9139–9143. doi: 10.1158/0008-5472.CAN-04-1036. [DOI] [PubMed] [Google Scholar]
- 52.Gonçalves A, Esteyries S, Taylor-Smedra B, Lagarde A, Ayadi M, Monges G, Bertucci F, Esterni B, Delpero JR, Turrini O, et al. A polymorphism of EGFR extracellular domain is associated with progression free-survival in metastatic colorectal cancer patients receiving cetuximab-based treatment. BMC Cancer. 2008;8:169. doi: 10.1186/1471-2407-8-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moriai T, Kobrin MS, Hope C, Speck L, Korc M. A variant epidermal growth factor receptor exhibits altered type alpha transforming growth factor binding and transmembrane signaling. Proc Natl Acad Sci USA. 1994;91:10217–10221. doi: 10.1073/pnas.91.21.10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang WS, Chen PM, Chiou TJ, Liu JH, Lin JK, Lin TC, Wang HS, Su Y. Epidermal growth factor receptor R497K polymorphism is a favorable prognostic factor for patients with colorectal carcinoma. Clin Cancer Res. 2007;13:3597–3604. doi: 10.1158/1078-0432.CCR-06-2601. [DOI] [PubMed] [Google Scholar]
- 55.Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990;265:7709–7712. [PubMed] [Google Scholar]
- 56.Bhowmick DA, Zhuang Z, Wait SD, Weil RJ. A functional polymorphism in the EGF gene is found with increased frequency in glioblastoma multiforme patients and is associated with more aggressive disease. Cancer Res. 2004;64:1220–1223. doi: 10.1158/0008-5472.can-03-3137. [DOI] [PubMed] [Google Scholar]
- 57.Shahbazi M, Pravica V, Nasreen N, Fakhoury H, Fryer AA, Strange RC, Hutchinson PE, Osborne JE, Lear JT, Smith AG, et al. Association between functional polymorphism in EGF gene and malignant melanoma. Lancet. 2002;359:397–401. doi: 10.1016/S0140-6736(02)07600-6. [DOI] [PubMed] [Google Scholar]
- 58.Zhao X, Dai W, Zhu H, Zhang Y, Cao L, Ye Q, Lei P, Shen G. Epidermal growth factor (EGF) induces apoptosis in a transfected cell line expressing EGF receptor on its membrane. Cell Biol Int. 2006;30:653–658. doi: 10.1016/j.cellbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Zhang W, Gordon M, Press OA, Rhodes K, Vallböhmer D, Yang DY, Park D, Fazzone W, Schultheis A, Sherrod AE, et al. Cyclin D1 and epidermal growth factor polymorphisms associated with survival in patients with advanced colorectal cancer treated with Cetuximab. Pharmacogenet Genomics. 2006;16:475–483. doi: 10.1097/01.fpc.0000220562.67595.a5. [DOI] [PubMed] [Google Scholar]
- 60.Le Marchand L, Seifried A, Lum-Jones A, Donlon T, Wilkens LR. Association of the cyclin D1 A870G polymorphism with advanced colorectal cancer. JAMA. 2003;290:2843–2848. doi: 10.1001/jama.290.21.2843. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ, Lowell AM, Golub T, Meyerson M, Tenen DG, Shapiro GI, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006;66:11389–11398. doi: 10.1158/0008-5472.CAN-06-2318. [DOI] [PubMed] [Google Scholar]
- 62.Bélanger H, Beaulieu P, Moreau C, Labuda D, Hudson TJ, Sinnett D. Functional promoter SNPs in cell cycle checkpoint genes. Hum Mol Genet. 2005;14:2641–2648. doi: 10.1093/hmg/ddi298. [DOI] [PubMed] [Google Scholar]
- 63.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, Mirtsching B, Cohn AL, Pippas AW, Azarnia N, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–4921. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 64.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 65.Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27:5068–5074. doi: 10.1200/JCO.2008.21.3744. [DOI] [PubMed] [Google Scholar]
- 66.Scartozzi M, Bearzi I, Pierantoni C, Mandolesi A, Loupakis F, Zaniboni A, Catalano V, Quadri A, Zorzi F, Berardi R, et al. Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J Clin Oncol. 2007;25:3930–3935. doi: 10.1200/JCO.2007.11.5022. [DOI] [PubMed] [Google Scholar]
- 67.Nishi H, Neta G, Nishi KH, Akers LM, Rikiyama T, Proctor KN, Murphy BA, Johnson AC. Analysis of the epidermal growth factor receptor promoter: the effect of nuclear factor-kappaB. Int J Mol Med. 2003;11:49–55. [PubMed] [Google Scholar]