

Abstract
Background:
The clinical phenotype of DYT6 consists mainly of primary craniocervical dystonia. Recently, the THAP1 gene was identified as the cause of DYT6, where a total of 13 mutations have been identified in Amish-Mennonite and European families.
Methods:
We sequenced the THAP1 gene in a series of 362 British, genetically undetermined, primary dystonia patients (78 with focal, 186 with segmental, and 98 with generalized dystonia) and in 28 dystonia-manifesting DYT1 patients and 176 normal control individuals.
Results:
Nine coding mutations were identified in the THAP1 gene. Two were small deletions, 2 were nonsense, and 5 were missense. Eight mutations were heterozygous, and 1 was homozygous. The main clinical presentation of cases with THAP1 mutations was early-onset (<30 years) dystonia in the craniocervical region or the limbs (8 of 9 patients). There was phenotypic variability with laryngeal or oromandibular dystonia present in 3 cases. Four of 9 THAP1 cases developed generalized dystonia.
Conclusions:
The number of THAP1 mutations has been significantly expanded, indicating an uncommon but important cause of dystonia. Coding mutations account for 9 of 362 dystonia cases, indicating a mutation frequency of 2.5% of dystonia cases in the population that we have screened. The majority of cases reported here with THAP1 mutations had craniocervical- or limb-onset segmental dystonia, but we also identified 1 homozygous THAP1 mutation, associated initially with writer's dystonia and then developing segmental dystonia. Three of our patients had a nonsense or frameshift THAP1 mutation and the clinical features of laryngeal or oromandibular dystonia. These data suggest that early-onset dystonia that includes the involvement of the larynx or face is frequently associated with THAP1 mutations.
GLOSSARY
- mRNA
= messenger RNA;
- SNP
= single nucleotide polymorphism;
- THAP1
= Thanatos-associated-domain containing, apoptosis-associated protein 1.
Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures and movements.1–3 The underlying cause of dystonia may be acquired, developmental, or genetic.4 To date, 16 genetic loci have been associated with dystonia, most of which are inherited as an autosomal dominant trait with reduced penetrance. The DYT1, DYT6, DYT7, and DYT13 genes are associated with primary or pure dystonia.5,6
DYT6 is a dominantly inherited dystonia that causes an early-onset primary torsion dystonia with a sex independent penetrance of 60%.7 Originally defined in 2 Amish-Mennonite families, DYT6 is equally likely to start in the craniocervical region as in the legs, and in the majority of cases spreads to involve both body parts. The age at onset in DYT6 is later than in DYT1 families (18.9 vs 13.6 years).8 Recently, the DYT6 gene was identified as the Thanatos-associated-domain containing, apoptosis-associated protein 1 (THAP1) gene.9 In total, 12 mutations have so far been identified in American Amish-Mennonite and European families.9–11 The THAP1 gene is part of a family of THAP proteins that bind specific DNA sequences and regulate cell proliferation through the pRB/E2F cell cycle target genes,12,13 a pathway recently proposed to be involved in cell death in Parkinson disease.14
To assess the frequency and phenotype of mutations in the DYT6 gene, we screened a large series of 362 adult-onset dystonia cases. The THAP1 gene was also examined in 28 DYT1 dystonia-manifesting patients to look for modifying factors.
METHODS
Standard protocol approvals, registrations, and patient consents.
All patients gave informed consent, and ethics approval was obtained from the joint medical and ethics committee at The National Hospital for Neurology and Neurosurgery to perform this clinical and genetic study (University College London Hospital ethical approval 06/N076).
Patients.
Patients were assessed and followed up by movement disorder specialists (K.P.B. and Professor N.P. Quinn, MD). All cases analyzed here were cases of primary dystonia where the genetic etiology was unknown.
We sequenced the THAP1 gene in a series of 362 British patients with clinically diagnosed dystonia who were genetically undetermined (for details, see appendix e-1 on the Neurology® Web site at www.neurology.org). Where there was a family history of dystonia, only the proband was analyzed. The proband in family J had previously been reported.15 We also examined a series of 28 DYT1 dystonia-manifesting carriers, to look for possible modifying factors in these cases. The patients were all based in the United Kingdom, with a high frequency in and around London. We estimate that our British, mainly London population includes 15% of dystonia cases that were originally from other countries, mainly Europe or India. Overall, the dystonia population included 78 with primary focal, 186 with segmental, and 98 with a predominantly generalized dystonia. In this group, 272 had an age at onset less than 30 years. The control series that were sequenced consisted of 176 healthy United Kingdom white individuals who were older than 50 years and neurologically normal. We also sequenced the THAP1 gene in 40 North London Jewish controls (entire gene) and analyzed 68 Indian and 95 Northern European control individuals for the specific mutations identified in patients with the respective ancestries.
Genetics.
The DYT1 deltaGAG TOR1A gene had been screened in all cases. All 3 exons and flanking regions of the THAP1 gene were sequenced. Mutations were confirmed by analyzing in a duplicate DNA sample from the proband. None of the coding mutations were identified in the control individuals described in the previous section. The c.-237_-236GA>TT and c.71+126bpT>C noncoding variants were seen in cases and controls, but the 3 other intronic variants that were identified in cases were not seen in controls. The difference in frequency of polymorphisms between cases and controls was analyzed using the χ2 statistical analysis applying the Yates correction for continuity and also the χ2 test for independence.
RESULTS
A total of 9 coding mutations were identified (table 1, figure, and figure e-1). Eight mutations were heterozygous, and 1 homozygous mutation was identified. The mutations were mainly located in exons 1 (n = 5) and 2 (n = 3). The mutations were all private changes and not previously reported. The frameshift mutations in families K (F58fs72X) and LH (T79fs119X) led to a premature stop codon and a short messenger RNA (mRNA) species. Families F (Q3X) and G (Y50X) had nonsense mutations; by their nature, these also lead to a short mRNA species. Families E (S6F), S (Y8C), M (P26R), C (N136S), and J (R169Q) all had missense mutations (table 1 and figure). The mutation segregated with the disease in the 3 other affected cases, but not in 1 unaffected sibling in family S. In family E, the mutation was present in the affected mother and daughter.
Table 1THAP1 mutations and intronic variants identified in dystonia cases
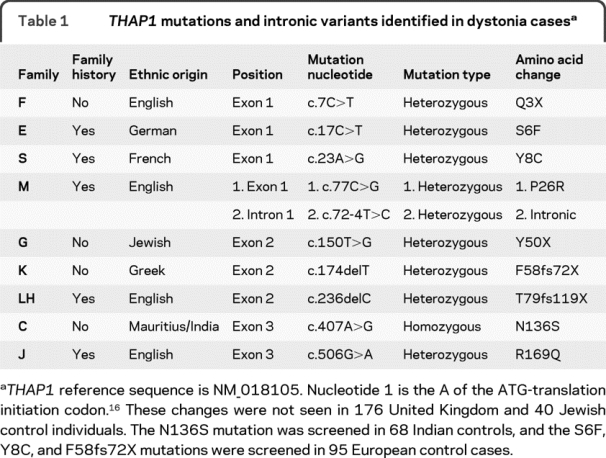

Figure THAP1 protein with the mutation types and positions identified here and the THAP1 protein domains
The start ATG (codon 1) is indicated in the figure, and all mutations are labeled from this codon up to the stop codon TAA at amino acid 213. Reference sequence is NM_018105, and protein is NP_060575.
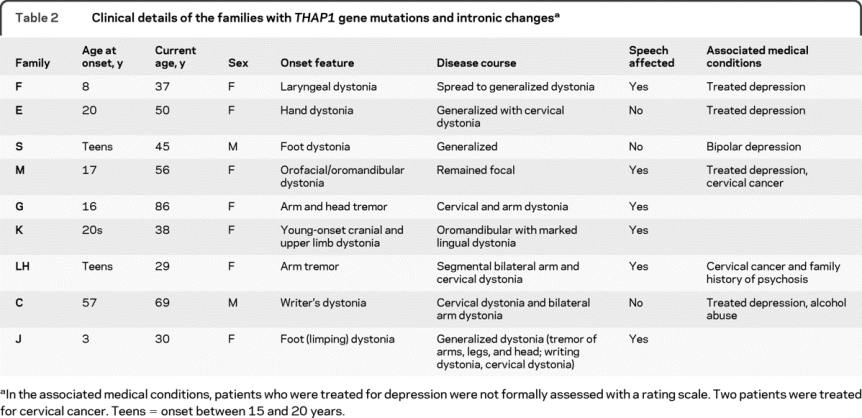
All coding mutations are highly conserved in species (figure e-1) except the R169Q mutation, which is conserved through mammals and fish but not in the 2 frog species analyzed. The mutation in family C was a homozygous N136S missense change; like the other mutations, this is conserved in species and not present in controls. In family M, a heterozygous missense coding mutation was identified, and an exon 2–4 bp intronic change was seen. This intronic change is unlikely to be pathogenic because it did not affect splicing using the SpliceView program (http://bioinfo.itb.cnr.it/oriel/splice-view.html), NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html), and FSPLICE 1.0 (http://linux1.softberry.com/berry.phtml). Two other intronic changes (c.71+87G>C and c.268-24A>G) that we identified were also not predicted to affect splicing. The clinical phenotype of the dystonia families with THAP1 mutations is given in table 2 and discussed further in the Discussion.
Table 2 Clinical details of the families with THAP1 gene mutations and intronic changes
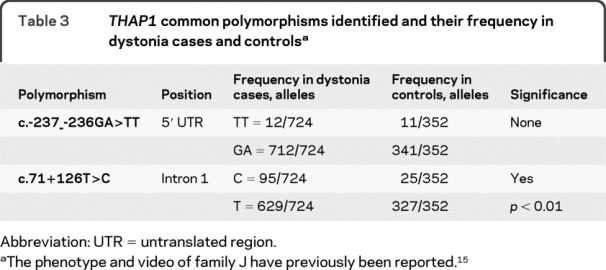
Two THAP1 gene polymorphisms were identified (c.-237_-236GA>TT and c.71+126T>C) and seen in dystonia cases and controls (table 1), but no other intronic or exonic changes were seen in the 176 control individuals (352 control chromosomes) sequenced (table 3). The THAP1 gene was also sequenced in 40 Jewish control individuals (exon 2); the N136S mutation was screened in 68 Indian controls; and the S6F, Y8C, and F58fs72X mutations were screened in 95 European control cases. The c.-237_-236GA>TT polymorphism was not significantly different from controls: χ2 analysis, p = 0.18 (Yeats) and p = 0.11 (Pearson). In the c.71+126T>C polymorphism, the frequency of the C allele was 13% (found in 95 of 362 patients with dystonia) as compared with 7% (found in 25 of 352 healthy controls): χ2 analysis, p < 0.01 (Yeats and Pearson). The frequency of the c.71+126T>C polymorphism (86 of 618, 14%) in a white population gave a result similar to that of the diverse population when analyzed.
Table 3THAP1 common polymorphisms identified and their frequency in dystonia cases and controls
DISCUSSION
The THAP1 coding mutations identified here indicate an overall mutation frequency of 2.5% in the British population (table 1 and figure) as compared with the German dystonia population, where the mutation frequency was 1%.11 In American (mainly German, Irish, or Italian ancestry) non-DYT1 multiplex dystonia families where at least 1 person had nonfacial involvement with an age at onset less than 22 years, the frequency was 25%.10 This indicates that THAP1 mutations are significantly less frequent than the most common genetic cause of primary torsion dystonia, DYT1.
The polymorphic c.-237_-236GA>TT change was not associated with dystonia in this study; the TT allele was previously seen in significant excess in German patients with dystonia.16 The C allele of the c.71+126T>C polymorphism was seen at a significantly higher frequency in dystonia cases. This suggests that this THAP1 allele could be a risk factor for developing dystonia, but this association needs extending to include other single nucleotide polymorphisms (SNPs) in and around the THAP1 gene. We have only analyzed this association in 1 population of cases and controls, and although the cases are predominantly from the United Kingdom, this group consists of patients with a number of ancestral backgrounds, even when only the white individuals are analyzed. It will be important for other groups to investigate these 2 THAP1 polymorphisms and other SNPs in the THAP1 region to confirm or not confirm this association.
All mutations identified here in the THAP domain are highly conserved (figure e-1) except the R169Q, which is partially conserved in species. The N136S and the R169Q mutations are located in the low-complexity proline-rich and the coiled-coil domains, respectively. The N136S mutation is a homozygous change indicating recessive inheritance; this has not previously been reported. This patient presented with writer's dystonia progressing to segmental dystonia in the neck and arms. The family contained only 1 affected case with dystonia; both parents were said to be unaffected, but we could not examine them because they were dead. The homozygous change is highly conserved; the mutation results in a change to a much less hydrophobic serine amino acid and will likely affect solubility and membrane interactions. Given the location of this change outside the THAP domain, to prove pathogenicity would require functional investigation, although perhaps a less severe mutation outside the THAP1 domain would be expected in a recessive case.
The clinical presentation of our DYT6 patients was that of young-onset focal or segmental dystonia (table 2). This phenotype was similar to previous mutation reports.9–11 Our series had an equal number of patients that presented with onset of dystonia in the craniocervical region or in the limbs. The ages at onset in our cases were less than 30 years except for 1 case (family C), who had an age at onset of 57 years. The age at onset of less than 30 years is consistent with previous reports, with the exception of 2 reported cases with onset at 38 and 49 years. Interestingly, 1 early-onset patient presented with laryngeal dystonia, which spread to become generalized (family F). In our series, a further patient had orofacial dystonia at onset (family M), and a third developed oromandibular dystonia with marked lingual dystonia (family K). These cases with laryngeal or oromandibular dystonia indicate that this is a frequent clinical feature in patients with THAP1 mutations. Involvement of the larynx and oromandibular region was previously reported in patients with THAP1 mutations.11 Bressman et al.10 found that 18% of cases presented with this form of dystonia, and in 68% of cases speech was affected.
The comparison between the clinical features of the cases with THAP1 mutations and patients with DYT1 shows that in contrast to DYT1 dystonia, where speech involvement is rare, 6 of our patients had dysarthria or spasmodic dysphonia due to oromandibular, tongue, or laryngeal involvement. Tumors are common in the general population; however, it is interesting to note that 4 of our patients had tumors or a family history, in view of the role of THAP1 in or links to leukemia, prostate cancer, and apoptosis.
In the previous 3 reports of THAP1 mutations, no genotype phenotype effect was observed.9–11 In our series, we did not observe an overall difference in THAP1 mutation position and phenotype, but 2 of 3 of the cases with either laryngeal or oromandibular dystonia had a nonsense or a frameshift mutation (tables 1 and 2). This mutation type associated with laryngeal dystonia was also seen in 2 German families, suggesting a possible correlation.11 The frequent involvement of the larynx, oromandibular region, and face and the common occurrence of speech problems in THAP1-associated dystonia suggest that this group of patients will be a particularly important group to target for genetic screening.
ACKNOWLEDGMENT
The authors thank the patients and families for their essential contribution to this research.
STUDY FUNDING
This work was undertaken at University College London Hospitals/University College London, which received a portion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centers funding scheme.
DISCLOSURE
Dr. Houlden receives research support from the Medical Research Council (MRC) UK, the Michael J Fox Foundation, the Brain Research Trust (BRT), the National Organization for Rare Disorders (NORD), Ataxia UK, and the British Medical Association (Vera Down Award). Dr. Schneider receives support from Novartis (Stipend for Therapeutic Research) and from the Brain Research Trust (JJ Astor Prize Studentship). Dr. Paudel and Dr. Melchers report no disclosures. Dr. Schwingenschuh received an Austrian Science Fund FWF and Erwin Schrödinger Grant. Dr. Edwards receives royalties from publication of Oxford Specialist Handbook of Parkinson's Disease and Other Movement Disorders (Oxford University Press, 2008) and receives research support from a National Institute for Health Research (NIHR) grant where he is the PI. Dr. Hardy serves as a consultant to Eisai Inc.; and receives research support from the Medical Research Council UK, the Bachmann Strauss Foundation, the Michael J Fox Foundation, and the Brain Research Trust. Dr. Bhatia has received funding for travel from GlaxoSmithKline, Orion Corporation, Ipsen, and Merz Pharmaceuticals, LLC; serves on the editorial boards of Movement Disorders and Therapeutic Advances in Neurological Disorders; receives royalties from the publication of Oxford Specialist Handbook of Parkinson's Disease and Other Movement Disorders (Oxford University Press, 2008); has received speaker honoraria from GlaxoSmithKline, Ipsen, Merz Pharmaceuticals, LLC, and Sun Pharmaceutical Industries Ltd.; and has received research support from Ipsen and from the Halley Stewart Trust through Dystonia Society UK.
Supplementary Material
Address correspondence and reprint requests to Dr. Henry Houlden, University College London Institute of Neurology, Queen Square, London WC1N 3BG, England h.houlden@ion.ucl.ac.uk or Prof. Bhatia at k.bhatia@ion.ucl.ac.uk
Supplemental data at www.neurology.org
Disclosure: Author disclosures are provided at the end of the article.
Received May 15, 2009. Accepted in final form January 12, 2010.
REFERENCES
- 1.Albanese A. Dystonia: clinical approach. Parkinsonism Relat Disord 2007;13(suppl 3):S356–S361. [DOI] [PubMed] [Google Scholar]
- 2.Klein C, Ozelius LJ. Dystonia: clinical features, genetics, and treatment. Curr Opin Neurol 2002;15:491–497. [DOI] [PubMed] [Google Scholar]
- 3.Ozelius LJ. Update on the genetics of primary torsion dystonia loci DYT6, DYT7, and DYT13 and the dystonia-plus locus DYT12. Adv Neurol 2004;94:109–112. [PubMed] [Google Scholar]
- 4.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci 2008;9:222–234. [DOI] [PubMed] [Google Scholar]
- 5.Bressman SB, de Leon D, Raymond D, et al. The role of the DYT1 gene in secondary dystonia. Adv Neurol 1998;78:107–115. [PubMed] [Google Scholar]
- 6.Bressman SB, Raymond D, Wendt K, et al. Diagnostic criteria for dystonia in DYT1 families. Neurology 2002;59:1780–1782. [DOI] [PubMed] [Google Scholar]
- 7.Saunders-Pullman R, Raymond D, Senthil G, et al. Narrowing the DYT6 dystonia region and evidence for locus heterogeneity in the Amish-Mennonites. Am J Med Genet A 2007;143A:2098–2105. [DOI] [PubMed] [Google Scholar]
- 8.Almasy L, Bressman SB, Raymond D, et al. Idiopathic torsion dystonia linked to chromosome 8 in two Mennonite families. Ann Neurol 1997;42:670–673. [DOI] [PubMed] [Google Scholar]
- 9.Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 2009;41:286–288. [DOI] [PubMed] [Google Scholar]
- 10.Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol 2009;8:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djarmati A, Schneider SA, Lohmann K, et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol 2009;8:447–452. [DOI] [PubMed] [Google Scholar]
- 12.Cayrol C, Lacroix C, Mathe C, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood 2007;109:584–594. [DOI] [PubMed] [Google Scholar]
- 13.Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Girard JP. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc Natl Acad Sci USA 2005;102:6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoglinger GU, Breunig JJ, Depboylu C, et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc Natl Acad Sci USA 2007;104:3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards M, Russo N, Summers B, Morton J, Peake D, Bhatia K. An unusual family with multiple movement disorders. J Neurol 2003;250:793–796. [DOI] [PubMed] [Google Scholar]
- 16.den Dunnen JT, Antonarakis SE. Mutation nomenclature. Curr Protoc Hum Genet 2003;chap 7:unit 7 13. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.