

Abstract
Pulmonary edema occurs when fluid flux into the lung interstitium exceeds its removal, resulting in hypoxemia and even death. Noncardiogenic pulmonary edema (NPE) generally results when microvascular and alveolar permeability to plasma proteins increase, one possible etiology being oxidant injury. Reactive oxygen and nitrogen species (RONS) can modify or damage ion channels, such as epithelial sodium channels, which alters fluid balance. Experimental systems in which either RONS are increased or protective antioxidant mechanisms are decreased result in alterations of epithelial sodium channel activity and support the hypothesis that RONS are important in NPE. Both basic and clinical studies are needed to critically define the RONS–NPE connection and the capacity of antioxidant therapy (either alone or as a supplement to β-agonists) to improve patient outcome.
Keywords: antioxidants, free radicals, pulmonary edema, reactive oxygen and nitrogen species, RONS, sodium transport
Pulmonary edema occurs when the fluid flux into the lung interstitium exceeds the rate of removal by the lymphatics. As the extravascular lung fluid accumulates, alveolar flooding occurs, resulting in impairment in gas exchange. The magnitude of impairment can lead to hypoxemia, and may ultimately result in respiratory failure and even death. Patients that present with pulmonary edema are a heterogeneous group as pulmonary edema can be the end result of many different diseases and disorders. The underlying etiologies can be generally classed into either of two broad groups: cardiogenic or noncardiogenic (‘permeability’). Cardiogenic pulmonary edema (CPE) results when capillary hydrostatic pressure increases or oncotic capillary pressure decreases, leading to increased fluid flux into the interstitial space; in this case, the fluid is an ultrafiltrate of plasma. Noncardiogenic pulmonary edema (NPE) generally results when microvascular and alveolar permeability to plasma proteins increase, most likely due to the oxidant injury to the alveolar and endothelial cells. Collectively, diseases that lead to pulmonary edema are among the most common causes of respiratory failure among intensive care patients.
There is a growing body of literature implicating reactive oxygen and nitrogen species (RONS) in many diverse kinds of pulmonary damage. There are many different possible scenarios in which underlying disease states can increase RONS, directly increasing damage and/or upregulating inflammatory responses in the lung, which in turn can further increase RONS production and lead to significant pathology. Teasing out cause and effect can be challenging, particularly in pathological conditions such as pulmonary edema, which can have different initiating events and/or which may be the end result of multiple disease processes. Modification or damage to ion channels may be particularly detrimental as they have a key role in maintaining fluid balance in the lung. Of equal importance in the response to RONS are endogenous antioxidants that prevent much of the ‘first-pass’ damage that would otherwise occur in the lung. While the antioxidant defense system of the lung is complex, one of the key cellular defenses is the thiol tripeptide glutathione (GSH). This brief review will present an overview of key RONS known to be produced in the lung, will examine the literature linking RONS production and endogenous antioxidant defenses to pulmonary edema, and will specifically focus on known and potential modifications of ion transport by RONS, which may be broadly responsible for pulmonary edema etiology and/or progression.
Cardiogenic pulmonary edema
Cardiogenic pulmonary edema results from increased capillary pressure, most often due to congestive heart failure. Generally, it is a manifestation of the left ventricle’s inability to adequately pump the blood it is receiving from the lungs. This leads to an increase in pressure in the left atrium, followed by increased pressure in the pulmonary veins and capillaries, ultimately causing fluid to be exuded through the capillary walls into the air sacs. Major causes for left ventricular failure include: coronary artery disease; mitral or aortic valve disease; myocarditis; cardiomyopathies; severe tachycardia or bradycardia; or other arrhythmias; hypertensive crisis; and fluid overload resulting from intravenous therapy or liver or kidney failure. Cardiogenic edema may also result from liver injury leading to decreased albumin plasma concentration and oncotic pressure. These insults generally do not result in increased reactive oxygen species (ROS) production; hence, this review will focus primarily on NPE, while still including the literature that links cardiogenic edema and increased production of reactive nitrogen species (RNS).
Noncardiogenic pulmonary edema
Noncardiogenic pulmonary edema arises from damage to the alveolar and microvascular compartments, leading to increased permeability to plasma proteins. The precipitating damage to the lung can include both direct physical injury as well as injury resulting from biochemical reactions occurring in the lungs in response to environmental stressors. These reactions often involve the production of RONS. Most are the result of some form of direct alveolar injury. These include the inhalation of toxic gases such as chlorine [1,2], direct trauma to the chest resulting in a pulmonary contusion [3–5] and re-expansion injury following a pneumothorax or other causes of lung deflation [5–7]. Reperfusion injury may also produce pulmonary edema. Reperfusion pulmonary edema (RPE) may occur following a lung transplant or following massive blood loss and subsequent transfusions [8,9]. Indeed, transfusion-related acute lung injury (ALI) is the leading cause of transfusion-related morbidity and mortality in the USA [9]. Clinically, sepsis is a leading cause of pulmonary edema. The pathogenesis of sepsis involves hyper-function or dysfunction of immune cells such as macro-phages or neutrophils [10]. Increased production of ROS and RNS by these cells can impact on the dysfunction of endothelial cells and pulmonary epithelial cells in the progression of sepsis. The final consequence of many of the forms of ALI outlined above is the development of acute respiratory distress syndrome (ARDS), which is marked by permeability pulmonary edema, hypoxemia and, depending on the severity, respiratory distress.
There are also additional rare triggers of NPE. These include unusual responses to medications, complications resulting from an upper airway obstruction [11], and a rare disorder associated with rapid high altitude ascent known as high altitude pulmonary edema (HAPE).
High altitude pulmonary edema is one of the most clearly defined forms of noncardiogenic pulmonary edema. It is an acute and potentially lethal pulmonary disease caused by rapid ascent to high altitudes in nonacclimatized individuals [12]. The onset of symptoms occur between 2–3 days after acute exposure to altitudes above 2500 m [13]. Defining characteristics include increased pulmonary arterial pressure, increased vascular permeability and hypoxemia. Clinical investigations have identified that a sub-set of the population may be acutely sensitive to HAPE [14–17]. Collectively, studies to date have found that:
Increased peripheral arterial pressure with precapillary vasoconstriction can lead to edema in HAPE patients
The pathogenesis of HAPE is attributed to hydrostatic mechanisms leading to capillary leak and subsequent pulmonary edema
Inflammation/reactive species may play a role in both the development and the complications arising from HAPE [12,17]
A thorough understanding of HAPE may help to delineate the targets in other forms of pulmonary edema and thus define its biochemical basis. Recent studies in HAPE have focused on damage to sodium transporters as an initiating event in fluid imbalance. As RONS and redox imbalance have also been shown to damage sodium transporters, this may signify a common underlying mechanism for the involvement of RONS in NPE.
Reactive species in pulmonary edema etiology & progression
Overview of reactive oxygen/nitrogen species
Reactive oxygen species and RNS, sometimes collectively referred to as RONS, include both free radicals and other bioreactive compounds. Free radicals are compounds that contain one or more unpaired electrons in their valence shell, which contributes to their chemical reactivity. Key free radicals in the lung are the ROS superoxide anion (O2•−) and the hydroxyl radical (OH•), while key RNS include nitric oxide (NO•) and peroxynitrite (ONOO−). Hydrogen peroxide (H2O2), although not a free radical because it lacks an unpaired electron, is a strong oxidizer (a ROS) and can still cause pulmonary damage when it is produced at supra-physiological levels, that is, at concentrations beyond which the natural endogenous removal mechanisms can function. While this list is not exhaustive, these RONS have been shown to be directly or indirectly involved in producing pulmonary damage in diseases and disorders of the lung including those culminating in pulmonary edema. Figure 1 summarizes the main mechanisms of formation and the fates of key RONS.
Figure 1. Formation of reactive oxygen and nitrogen species.
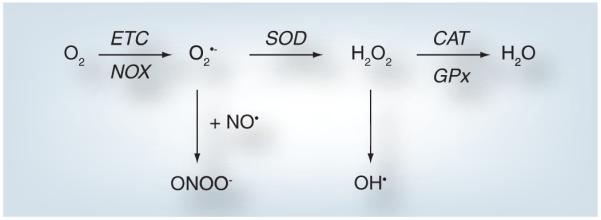
This figure summarizes the formation pathways and some of the fates of the reactive oxygen and nitrogen species discussed in the text. Molecular oxygen can be converted into O2•− via the NOX proteins in a controlled, regulated enzymatic process. Proton leak from the mitochondrial ETC may also contribute to the formation of O2•− from molecular oxygen in an unregulated fashion. O2•− can spontaneously react with NO• to form ONOO−. Alternatively, SOD1, −2 and −3 may dismutate O2•− to H2O2. This reaction also occurs spontaneously. H2O2 may be converted to water by either CAT or GPx. Alternatively, H2O2 can be a source of OH• leading to cellular damage. Not discussed in this review are the roles H2O2 can play in regulating gene expression via activation of redox-sensitive cell signaling cascades.
CAT: Catalase; ETC: Electron transport chain; GPx: Glutathione peroxidase; H2O2: Hydrogen peroxide; NO : Nitric oxide; O2•−: Superoxide anion; OH•: Hydroxyl radical; ONOO−: Peroxynitrite; SOD: Superoxide dismutase.
Superoxide
The superoxide anion radical (O2•−) is formed by the monovalent reduction of molecular oxygen either by electrons generated by the mitochondrial electron transport chain (ETC), by the NOX proteins (also known as the NADPH oxidases), or by the enzyme xanthine oxidase.
| (1) |
Intracellularly, superoxide may have several fates. Although reactive, it is a relatively short-lived intermediate that will rapidly react with nearby targets. In the presence of Fe3+ it may react to produce Fe2+ and O2. Fe2+, in turn, in the presence of H2O2 may produce even more damaging radicals (equations 3 & 4). Superoxide may also react directly with NO• to form ONOO− (discussed in more detail in the section on ‘Peroxynitrite’), or it may be converted to H2O2 via mechanisms discussed next.
Hydrogen peroxide
Hydrogen peroxide is not a free radical but it can still cause significant cellular damage when it is produced at higher-than-normal concentrations. H2O2 is formed when superoxide dismutase (SOD) dismutates two molecules of superoxide to generate equimolar concentrations of H2O2 and oxygen:
| (2) |
There are three forms of SOD: SOD1 (cytosolic, CuZnSOD), SOD2 (mitochondrial, MnSOD) and SOD3 (extracellular, ecSOD). Some H2O2 is also formed by the spontaneous reduction of superoxide within the cell. H2O2 can be subsequently reduced by catalase to yield a 2:1 ratio of water to oxygen, or can be reduced by some members of the GSH peroxidase (GPx) family. It is noteworthy that H2O2 is also known to function as an endogenous cell signaling molecule [18,19], thus it also has physiological functions within the cell [19]. In some respects, H2O2is even more potentially cytotoxic than superoxide because its lifetime is sufficient to allow for its diffusion into almost any cellular compartment where it may act as a source of OH•.
Hydroxyl radical
The hydroxyl radical is extremely reactive; in theory, it is primarily formed via Fenton chemistry (equations 3 & 4), although there is some continuing debate as to the extent to which these reactions occur in vivo.
| (3) |
| (4) |
Polyunsaturated fatty acids (PUFA) are particularly vulnerable to attack and modification by OH• once they are formed. This can result in the formation of many reactive lipid species, which may have damaging effects of their own. In the lung, the lipid component of pulmonary surfactant is approximately 11% phosphatidylglycerol and is a key source of PUFA; alveolar epithelial Type I and Type II cell lipid membranes also contain PUFA. OH• lipid peroxidation in turn leads to lipid hydroperoxide formation. Lipid hydroperoxides have two main potential fates: they may either be reduced to their corresponding alcohols by GPx enzymes using GSH as a reducing equivalent (discussed in the section ‘Pulmonary antioxidants: GSH’) or they may be converted into other, more deleterious, compounds, which include the hydroxyl alkenals. These include 4-hydroxyl-2-nonenal (4-HNE) and 4-hydroxyl-2-hexanal (4-HHE). Relative to the OH• that initiated these reactions, these lipid peroxidation products are much more stable, can diffuse within and beyond the cell, and act as electrophilic compounds that can damage other cellular components, including proteins, via covalent adduct formation. Indeed, formation of stable HNE adducts in cells in culture has been shown to cause constitutive activation of cell signaling pathways [20].
Nitric oxide
Nitric oxide is generated from the oxidative deamination of the amino acid l-arginine by the enzyme nitric oxide synthase (NOS). There are three isoforms of this enzyme: endothelial NOS (NOS3) is constitutively active and is generally expressed in the endothelium where it plays a pivotal role in vasodilation; inducible NOS (iNOS; NOS2) is predominantly expressed in the immune and cardiovascular systems where it plays a critical role in host defense; and neuronal NOS (NOS1) is broadly expressed in both peripheral and central nervous systems. Potential sources of NO• in the lung mainly result from increased activity of iNOS in activated macrophages and neutrophils, alveolar Type II cells and other airway cells [21]. Increased levels of iNOS have been reported in the lungs of patients with ARDS and other inflammatory pulmonary diseases [22–24]. The biological impact of NO• depends on its concentration, potential targets and the presence of other radicals. NO• may bind to the heme group of guanylate cyclase, resulting in increases in cellular cGMP levels and the activation of PKC [25]. In the presence of an electron acceptor, it may react with thiols to produce nitrosothiols [26] which, in the context of pulmonary inflammation, have been shown to attenuate pulmonary damage [27,28]. NO• also reacts with superoxide in an extremely rapid diffusion-limited reaction (6.7 × 109 M−1·s−1) to produce ONOO− [29]; However, at physiological levels the reactivity and toxicity of NO• is mild, and it is important to remember that it is also an essential component of many cellular processes and functions. Indeed, most of the toxicity of NO• has been attributed to either ONOO− or to higher oxides of nitrogen.
Peroxynitrite
Peroxynitrite is formed in a reaction between superoxide and NO• (equation 5). Hence, the relative balance of these two radicals within the cell can have serious implications for pulmonary health.
| (5) |
Peroxynitrite is a potent oxidizing and nitrating agent, which oxidizes thiols at rates at least 1000-times faster than H2O2. It also causes iron-dependent peroxidation of lipids and it can nitrate or oxidize proteins. Because of this diverse reactivity, ONOO− has been shown to damage a wide range of biological targets including DNA, the protein components of the mitochondrial ETC, lung ion channels and the pulmonary surfactant system [30] Furthermore, ONOO−, owing to its high chemical reactivity, will modify/damage biological targets even in the presence antioxidants [31].
Pathophysiology of pulmonary fluid homeostasis
Mechanisms of fluid control in the normal lung
For optimal gas exchange, the alveoli of adult mammalian lungs must remain open and free from fluid. The alveolar epithelial cells of mammalian lungs actively transport sodium (Na+) and chloride (Cl−) ions and these processes are critical to preventing a build-up of fluid in the lungs. Na+ ions enter the apical membrane both alveolar epithelial Type I and Type II cells through sodium-selective cation channels and cyclic nucleotide-gated channels, and are extruded across the basolateral membrane by an energy-dependent Na/K-ATPase. This vectorial transport of Na+ ions and the concomitant movement of Cl− ions to maintain electroneutrality create an osmotic force that leads to the reabsorption of fluid across both normal and damaged lungs. Figure 2 depicts the movement of the ions across alveolar epithelial cells that are involved in the maintenance of fluid homeostasis. Pulmonary stress, either via direct damage to the epithelial cells, or damage mediated by RONS generated in close proximity to the alveolar epithelial cells by activated inflammatory cells, can modulate the activity of sodium channels via a number of redox-regulated mechanisms [32–34]. These include altering signal transduction pathways [34], which may lead to changes in gene expression with resultant downstream changes in the proteome and hence cell function, and/or direct oxidative or other posttranslational modifications of sodium channel proteins or related proteins, such as their chaperones [32,33]. This situation can be further exacerbated as pulmonary stresses that increase oxidant formation deplete key pulmonary antioxidant defenses such as GSH and may damage other protective proteins, such as the SODs or γ-glutamyltransferase. The following section will focus on the roles of RONS and the cellular defenses that protect against RONS-mediated damage in the etiology and progression of pulmonary edema.
Figure 2. Ion transport across alveolar epithelial cells.

Na+ and Cl− ions enter the apical membranes of alveolar epithelial cells (both Type I and Type II) via ENaC and Cl− channels such as CFTR located in the apical face of the cell. The Na+ ions are driven by an electrochemical gradient created by the energy-dependent Na/K-ATPase. K+ ions, brought in by the Na/K-ATPase pump in exchange for the Na+ ions, leave the cell passively through K+ channels located in the basolateral membrane.
CFTR: Cystic fibrosis transport regulator; ENaC: Epithelial sodium channel.
Disruption of normal fluid homeostasis: the role of RONS
A number of studies reported in the literature have established that active Na+ transport plays an important role in limiting the degree of pulmonary edema in adult mammalian lungs following acute or chronic injury to the alveolar epithelium. Patients with ALI who are still able to concentrate alveolar protein as a result of active Na+ reabsorption have a better prognosis than those who cannot [35, 36]. In animal studies, rats exposed to hyperoxia who also received intratracheal instillation of a sodium channel blocker exhibited increased extravascular lung fluid compared with shaminstilled controls [37]. Furthermore, intratracheal instillation of adenoviral vectors expressing the Na/K-ATPase genes increased survival in rats exposed to hyperoxia.
Additional observational evidence from humans comes from mountaineers who develop pulmonary edema. A recent study has reported that decreased sodium transport predisposes mountaineers to pulmonary edema [15]. Nasal transepithelial potential difference (which is a marker of the transepithelial sodium and water transport in the distal airways) was reduced at low altitudes in subjects known to be susceptible to HAPE [15,16]. These studies also established that β-adrenergic agonists (which increase Na+ uptake) can decrease pulmonary edema in human patients. Previous studies had established that β-adrenergic agonists increased alveolar fluid clearance, and attenuated pulmonary edema in animal models [38] and in resected human lungs [39] but not in human patients. The β-adrenergic agonist salmeterol was given prophylactically to subjects prone to HAPE; pretreatment with the drug caused a significant decrease in the development of HAPE [15]. This study provided direct evidence for the role of impaired Na+ transport in HAPE-prone individuals. Collectively, these findings in vitro and in vivo attest to the importance of Na+ transport in attenuating fluid accumulation in conditions of pulmonary disease and stress [15,38,39].
Other studies have investigated the possible association between RONS and Na+ transport across the alveolar epithelium both in animals with ALI and in patients with edema. Several of these have been linked to increased NO• production and/or its metabolites. In rats with prolonged hemorrhagic shock, reabsorption of isotonic fluid (secondary to vectorial transport of Na+ ions) is inhibited [40]. However, this inhibition was reversed by instillation of an iNOS inhibitor (aminoguanidine) and fluid reabsorption was restored to normal levels [40]. This suggests that either NO• or one of its metabolites contributed to the downregulation of fluid reabsorption. Zhu et al. also showed that increased levels of nitrate and nitrite (the stable by-products of NO• and other RONS) correlated with decreased rates of alveolar fluid clearance in the lungs of patients with ALI (Figure 3) [41]. A smaller but significant increase was also found in patients with cardiogenic pulmonary edema [41]. Additional evidence for an effect of RNS on ion channels comes from in vitro studies of ion transport. Alveolar epithelial cells exposed to micromolar concentrations of ONOO− exhibited significantly decreased 22Na+ uptake across freshly isolated rabbit ATII cells [30]. Treatment with amilioride, a specific inhibitor of the apical sodium transporter (epithelial sodium channel [ENaC]), inhibited uptake indicating that ONOO− was impacting the apical transport of Na+. In another study, SIN-1 (a generator of ONOO−) profoundly inhibited the amilioride-sensitive whole-cell current in Xenopus oocytes expressing ENaC [42]. ONOO− may exert its effects by oxidizing, nitrating or nitrosylating critical amino acids either on ENaC or on other structural proteins necessary for the proper functioning of the channel. Both the extracellular and transmembrane segments of the three ENaC subunits contain domains rich in cysteine residues. Cysteine residues that are not involved in disulfide formation remain particularly redox labile, and thus may become oxidized or nitrosylated and alter ENaC function.
Figure 3. Increased levels of nitrate/nitrite in the edema fluid of patients with acute respiratory distress syndrome correlate with impaired alveolar fluid clearance.
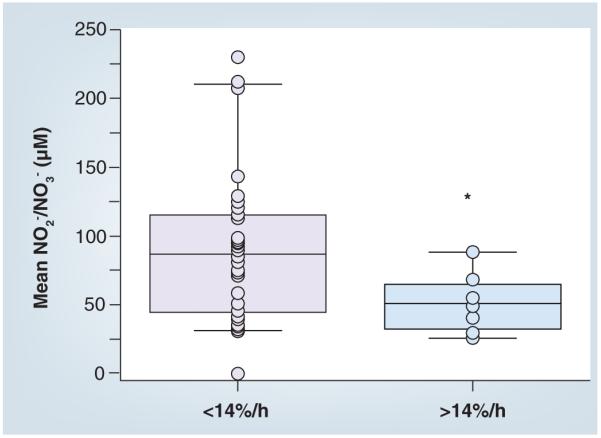
This box-whisker plot graph shows the mean interval nitrate + nitrite concentration of pulmonary edema fluid in two groups of patients with different alveolar fluid clearance (AFC): <14%/h (or impaired/submaximal) and >14%/h (or maximal). The horizontal line represents the median and the box encompasses the 25th to the 75th percentile. AFC was significantly impaired in patients with increased nitrate + nitrite concentrations in edema fluid. Adapted with permission from [41].
Reactive oxygen species have also been shown to impair apical sodium uptake [43]. The xanthine/xanthine oxidase (X/XO) experimental system produces superoxide, from which H2O2 and ultimately OH• can be derived. ROS produced by the X/XO system were shown to decrease amilioride-sensitive (ENaC) currents across ventral toad skin [43]. Changes in current only occurred when X/XO was added to the apical side, indicating that the ROS were impacting ENaC and subsequently sodium uptake [43]. By contrast, in thrombin and oleic acid injury, the reactive oxygen–nitrogen intermediates that are generated have also been shown to decrease Na/K-ATPase activity in ATII cells. This appears to occur via upregulation of endocytosis from the basolateral membrane via a PKCζ-dependent mechanism [44,45].
Although the studies outlined above separately connect edema and increased RONS to altered Na+ transport, increased RONS production has been directly documented in some forms of edema [46]. For example, RPE is characterized by diffuse inflammation in the lung parenchyma and the development of permeability pulmonary edema. In RPE, it has been shown that unilateral lung injury is initiated by cytotoxic oxygen metabolites that are formed upon the reoxygenation of the collapsed lung [47]. Temporally, this correlates with the influx of poly-morphonuclear neutrophils, which may further contribute to the generation of RONS [47]. Others have reported that high altitude exposure results in increased ROS generation leading to enhanced oxidative damage of lipids, proteins and DNA [48]. Experiments with hypoxia-treated rats strongly suggest that the increase in ROS observed in human patients with HAPE is a causal factor in the disease [49]. In rats exposed to hypobaric hypoxia (280 mmHg) for increasing lengths of time a significant increase in ROS in the lungs, as measured by dichlorofluorescin fluorescence, and a concomitant decrease in GPx activity were observed, indicating that as ROS increased, a key antioxidant defense decreased.
Key pulmonary defenses & edema prevention/attenuation
Pulmonary antioxidants: GSH
When the production of RONS increase, one mechanism to compensate is to increase the activity of cellular defenses. GSH (γ-l-glutamyl-l-cysteinyl-glycine) is the most abundant nonprotein thiol in the cell and a key pulmonary antioxidant. It is a tripeptide composed of glutamate, cysteine and glycine, and is a critical component of the pulmonary protection arsenal. GSH has many cellular and extracellular functions including the metabolism of endogenous and exogenous compounds (e.g., reactive oxygen, nitrogen and lipid-based species), it plays an ill-defined role in the regulation of cell proliferation and modulates the rate of gene expression [50,51]. Most importantly is its role as a cofactor (i.e., reductant) in the removal and/or modification of compounds by two enzyme families, the GSH S-transferases (GST) and GPx. The GST proteins may be found in every cell fraction, and in the liver can account for up to 10% of the total cytosolic proteins. Members of this superfamily, which have varying substrate specificities, catalyze the addition of GSH, via its sulfhydryl group, to electrophilic carbon centers on various xenobiotic compounds. This process may both detoxify the compound (i.e., biotransformation) and may also mark it for export from the cell. The GPx proteins, similar to the GST proteins, have varying substrate specificities, and are produced in specific cell types, but they all function by catalytically using GSH to reduce hydroperoxides, including lipid and alkyl hydroperoxides, to their corresponding alcohol. This reducing of peroxides decreases the oxidative stress burden on the cell. For a thorough review of the role of GSH as a reductant in enzyme-based defenses, see [52].
GSH is produced by most cell types for direct use, and in a few cells type it is also produced for export to the surrounding milieu. Type II alveolar epithelial cells, as an example, produce and export GSH to the epithelial lining fluid, where it accumulates to millimolar concentrations. A few cells can take-up GSH directly from the surrounding fluid, but most depend on de novo synthesis to maintain adequate levels. It is vital to understand that the content of GSH within a cell or the concentration within a fluid (such as the ELF) is a function, or balance, of both the rate of production (or influx) and the rate of consumption by enzymes such as GST and GPx. The regulation of de novo synthesis is well studied, and will be outlined only briefly in this review.
Biosynthesis of GSH occurs via two sequential ATP-dependent reactions. In the first and rate-limiting reaction, the enzyme glutamate cysteine ligase (GCL) catalyzes the addition of glutamate to cysteine via an unusual γ-linkage to form γ-glutamylcysteine; the γ-linkage is the reason this tripeptide is resistant to normal cellular proteases. A second ATP-dependent reaction catalyzed by GSH synthase adds glycine to the γ-glutamylcysteine intermediate to form reduced GSH (i.e., γ-l-glutamyl-l-cysteinyl-glycine). GCL is a heterodimeric enzyme composed of one catalytic (GCLC) or heavy subunit, and one modulatory (GCLM) or light subunit. In vivo it is generally believed that only the holoenzyme contributes significantly to GSH production. The cellular content of one or both subunits may be upregulated in response to oxidative stress, and this upregulation of protein content resides almost exclusively at the level of gene expression. A remarkably large number of compounds, including those that form GSH conjugates and those that generate ROS, have been shown to induce GSH biosynthesis through increased transcription of the Gcl genes (for a thorough review see [53]). Factors that control the export of GSH from the cell to the surroundings remain equivocal. Thus, increased production of RONS can impact on both the production and the consumption of GSH. While other antioxidant defenses (most notably the SOD proteins) may be important in modulating pulmonary edema, in this short review we will focus on the changes in GSH homeostasis.
Role of GSH in pulmonary edema
As GSH levels are acutely sensitive to oxidant stress, any changes in GSH may be indicative of increased RONS production. The impact of pulmonary edema on GSH homeostasis appears to be somewhat dependent on the model system and when it is measured, relative to the initial insult. In one study, the serum GSH levels of human patients presenting for treatment for HAPE were significantly lower than those of healthy controls [54], although it is not known whether the lower basal GSH levels contributed to the increased susceptibility to HAPE, or if it was rather a consequence of the disease. However, given that HAPE presents after a hypoxic insult and hypoxia increases RONS production, it is likely that the decreased serum GSH was a consequence of the hypoxia. A recent study in which healthy young men were progressively exposed to higher altitudes (starting from 1320 m to 3600 m by air and finally to 4580 m by foot) found a dramatic increase in oxidized GSH coupled with a decrease in reduced GSH in the subjects when they returned to the starting elevation [55]. GSH metabolism, as evidenced by increased GSH reductase and increased GPx, was also altered. This study provides further confirmation that high altitude exposure alters GSH metabolism and homeostasis, and in general renders this system less effective following exposure.
In a gas (phosgene) inhalation mouse model of pulmonary edema, the development and resolution of pulmonary edema correlated with changes in the GSH redox system [56]. At the earliest time point (1 h postexposure) there was no change in total GSH. However, with some insults, depletion of GSH can occur in 30 min or less; feedback inhibition of de novo synthesis is removed by rapid depletion and basal levels may be restored within an hour [57,58]. Subsequently, GSH levels will increase if the stress continues. In the phosgene-inhalation study, total GSH levels were significantly increased 4 h postexposure and were maintained until at least 72 h postexposure. Levels had returned to baseline 1 week later. This study suggested that the edematous lung has the capacity to repair itself in part by re-establishing a functional GSH redox system.
In other model systems (i.e., chronic ethanol exposure) the depletion of pulmonary GSH has been shown to impair alveolar epithelial barrier function [59]. This suggests that GSH levels may also be relevant to the prevention or attenuation of pulmonary edema, and that any of the initiating events that also deplete GSH or impair normal GSH metabolism may result in more profound damage.
Summary
In this short review, it is not possible to summarize all of the possible mechanisms whereby increased RONS formation may either be causal or contribute to the development of pulmonary edema. This review has focused predominantly on the potential impact of RONS on fluid reabsorption and changes in the key cellular antioxidant GSH that may increase the vulnerability of the Na+ transporters to damage by RONS. As pulmonary edema results from a persistent imbalance between the forces that drive water into the airspace and the physiologic mechanisms that remove it, it is not unreasonable to presume that RONS may also impact the mechanisms that drive water out (including increased leak). This is one of the many areas that require further study.
In general, the role of RONS in pulmonary edema is still being defined. While experimental evidence and patient data suggest that the development of some forms of pulmonary edema correlate with increased RONS production and dysregulation of both apical and basolateral Na+ transport, further studies are needed to define the precise mechanisms whereby this occurs.
Expert commentary
Ion transport is a seminal feature of airway and distal lung epithelial cells. The vectorial absorption of sodium through distal lung epithelial cells generates an oncotic gradient that plays an important role in the reabsorption of alveolar fluid in pulmonary edema (both cardiogenic and permeability types). This process is essential in facilitating gas exchange across the lung. In this review, we discussed how RONS, which are common constituents of environmental pollutants and bioreactive gases (such as nitrogen dioxide, chlorine, ozone, cigarette smoke, and so on) and which may also be generated by activated inflammatory cells, can damage ENaC located on the apical surfaces of lung epithelial cells. We also discussed the importance of antioxidants, reviewing the roles of both low-molecular-weight scavengers such as GSH, and enzymatic defenses such as SOD and catalase, which limit the oxidant-induced injury to the ion channel ENaC under physiological conditions. Furthermore, we hypothesized that it is the inability of these same systems, or perhaps only specific components of these systems, to limit oxidant-induced injury to ENaC and thus play a significant role in the etiology of edema, as the pathophysiological manifestation of ENaC damage. Currently, there is considerable interest in agents (such as β-agonists) that increase sodium transport (and thus fluid reabsorption) across the distal lung spaces in patients with pulmonary edema secondary to ALI and in those suffering from high altitude-induced pulmonary edema. Prophylactic or postexposure administration of antioxidants may also have beneficial effects (especially coupled with β-agonists) by reducing the steady-state concentrations of reactive species and thus preventing this injury from occurring. Understanding the molecular bases for ENaC dysfunction resulting from RONS-mediated damage is quite likely to yield promising new therapies that will significantly lessen the impact of pulmonary edema, a major source of morbidity and mortality in ALI.
Five-year view
Collectively, diseases that lead to pulmonary edema are among the most common causes of respiratory failure among intensive care patients. Therefore, continued growth in the understanding of the mechanisms underlying the development of pulmonary edema is an ongoing priority in pulmonary research. Definitive studies on patients suffering from HAPE have established the involvement of ROS in this form of pulmonary edema [12,13,15-17]. Another seminal study has correlated increased RNS with severity of disease in patients with pulmonary edema [41]. It is anticipated that future studies will apply these findings to other clinical and experimental forms of pulmonary edema and attempt to define common underlying RONS-mediated mechanisms.
Although key experimental and clinical studies have linked the production of RONS to pulmonary edema, the source of these species remains largely undefined. Future studies that delineate the cellular source(s) of these species are critical as they will help to further develop therapeutic modalities. Such studies could easily be performed in vitro with the wide range of targeted probes that can now delineate whole-cell versus organelle-specific (i.e., mitochondrial) production of reactive oxygen and some RNS.
It is also critical to determine which RONS are being formed in the initiation, development and progression of pulmonary edema. It can be argued that many of the current methods (i.e., redox sensitive dyes) do not specifically identify which RONS are being produced. We are currently developing spin trapping/electron spin resonance methods to identify specific RONS species produced in experimental models of pulmonary edema. While these directions may provide invaluable mechanistic information in the experimental setting, they may not be applicable to patients given that many of these species are short-lived and highly reactive. Both the technology and the sensitivity for identifying compounds in exhaled breath condensate (EBC) has advanced tremendously in the last decade. It is postulated that these technologies might be applied (i.e., the measurement of exhaled NO•) and further developed to assess the severity of pulmonary edema and outcome based on EBC.
Clearly, more work, both at the bench and the bedside, needs to be carried out in this area, but there is the potential for several new exciting and potentially diagnostic and therapeutic options to be developed in the next 5 years.
Key issues.
Pulmonary edema occurs when the rate of fluid flux into the lung interstitium exceeds the rate of removal by the lymphatics, leading to extravascular lung fluid accumulation and eventual alveolar flooding, resulting in impairment in gas exchange, hypoxemia and even death.
Noncardiogenic pulmonary edema (NPE) arises from damage to the pulmonary system and results in permeability pulmonary edema, which is the end result of many forms of acute lung injury.
Reactive oxygen and nitrogen species (RONS) are known to be involved in many diverse kinds of pulmonary damage, although their direct role in NPE etiology/progression remains relatively unstudied.
Underlying disease states can increase RONS production, directly increasing damage and/or upregulating inflammatory responses in the lung, which in turn can further increase RONS production and lead to significant pathology.
Modification or damage to ion channels, notably epithelium sodium channels, are considered to be particularly detrimental as they have a key role in maintaining the fluid balance in the lung.
Experimental systems in which RONS are deliberately increased, or in which protective antioxidant mechanisms are decreased, have been shown to result in alterations of epithelium sodium channel activity, supporting the hypothesis that RONS are important in NPE.
Further studies, both basic and clinical, are warranted to firmly determine the role of RONS in NPE etiology/progression, and the capacity of antioxidant therapy (either alone or as a supplement to β-agonists) to improve prognosis in patients with lung injury.
Acknowledgements
This article is dedicated to the memory of Charles A Bosworth, PhD.
This work was supported by grants U01 ES015676 and U54 ES017218 from the NIH to Sadis Matalon.
The authors thank Teri Potter for her superb editorial assistance. No other writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Contributor Information
Karen E Iles, Department of Anesthesiology, University of Alabama at Birmingham, 901 19th Street South, 304 BMR II, Birmingham, AL 35294-2172, USA, Tel.: +1 205 975 2761, Fax: +1 205 934 7447, kiles@uab.edu and Department of Environmental Health Sciences, School of Public Health, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Weifeng Song, Department of Anesthesiology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
David W Miller, Department of Anesthesiology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Dale A Dickinson, Department of Environmental Health Sciences, School of Public Health, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Sadis Matalon, Department of Anesthesiology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA and Department of Environmental Health Sciences, School of Public Health, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
References
Papers of special note have been highlighted as: • of interest
- 1.Lehavi O, Leiba A, Dahan Y, et al. Lessons learned from chlorine intoxications in swimming pools: the challenge of pediatric mass toxicological events. Prehosp. Disaster Med. 2008;23:90–95. doi: 10.1017/s1049023x00005641. [DOI] [PubMed] [Google Scholar]
- 2.Leustik M, Doran S, Bracher A, et al. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295:L733–L743. doi: 10.1152/ajplung.90240.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor G, Miller H, Shulman H, DeLacy J, Maggisano R. Controversies in the management of pulmonary contusion. Can. J. Surg. 1982;25:167–170. [PubMed] [Google Scholar]
- 4.Reske A, Seiwerts M, Reske A, Gottschaldt U, Schreiter D. Early recovery from posttraumatic acute respiratory distress syndrome. Clin. Physiol. Funct. Imaging. 2006;26:376–379. doi: 10.1111/j.1475-097X.2006.00702.x. [DOI] [PubMed] [Google Scholar]
- 5.Tariq S, Sadaf T. Images in clinical medicine. Reexpansion pulmonary edema after treatment of pneumothorax. N. Engl. J. Med. 2006;354:2046. doi: 10.1056/NEJMicm050849. [DOI] [PubMed] [Google Scholar]
- 6.Sohara Y. Reexpansion pulmonary edema. Ann. Thorac. Cardiovasc. Surg. 2009;14:205–209. [PubMed] [Google Scholar]
- 7.Conen A, Joos L, Bingisser R. Ipsilateral reexpansion pulmonary edema after drainage of a spontaneous pneumothorax: a case report. J. Med. Case Reports. 2007;1:107. doi: 10.1186/1752-1947-1-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akindipe O, Fernandez-Bussy S, Staples E, Baz M. Late unilateral pulmonary edema in single lung transplant recipients. J. Heart Lung Transplant. 2008;27:1055–1058. doi: 10.1016/j.healun.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Triulzi D. Transfusion-related acute lung injury: current concepts for the clinician. Anesth. Analg. 2009;108:770–776. doi: 10.1213/ane.0b013e31819029b2. [DOI] [PubMed] [Google Scholar]
- 10.Biswal S, Remick D. Sepsis: redox mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2007;9:1959–1961. doi: 10.1089/ars.2007.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bisinotto F, Cardoso RP, Abud T. Acute pulmonary edema associated with obstruction of the airways. Case report. Rev. Bras. Anestesiol. 2008;58:165–171. doi: 10.1590/s0034-70942008000200009. [DOI] [PubMed] [Google Scholar]
- 12.Bärtsch P, Mairbäurl H, Maggiorini M, Swenson E. Physiological aspects of high-altitude pulmonary edema. J. Appl. Physiol. 2005;98:1101–1110. doi: 10.1152/japplphysiol.01167.2004. [DOI] [PubMed] [Google Scholar]
- 13.Hopkins S, Garg J, Bolar D, Balouch J, Levin D. Pulmonary blood flow heterogeneity during hypoxia and high-altitude pulmonary edema. Am. J. Respir. Crit. Care Med. 2005;171:83–87. doi: 10.1164/rccm.200406-707OC. [DOI] [PubMed] [Google Scholar]
- 14.Knowles M, Carson J, Collier A, Gatzy J, Boucher R. Measurements of nasal transepithelial electric potential differences in normal human subjects in vivo. Am. Rev. Respir. Dis. 1981;124:484–490. doi: 10.1164/arrd.1981.124.4.484. [DOI] [PubMed] [Google Scholar]
- 15.Sartori C, Allemann Y, Duplain H, et al. Salmeterol for the prevention of high-altitude pulmonary edema. N. Engl. J. Med. 2002;346:1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- •.Establishes the importance of active Na+ transport in pulmonary fluid reabsorption. Subjects prone to developing high altitude pulmonary edema had lower than normal nasal potential difference. Prophylactic inhalation of salmeterol decreased the incidence of PE presumably by upregulating Na+-dependent fluid reabsorption.
- 16.Sartori C, Duplain H, Lepori M, et al. High altitude impairs nasal transepithelial sodium transport in HAPE-prone subjects. Eur. Respir. J. 2004;23:916–920. doi: 10.1183/09031936.04.00115304. [DOI] [PubMed] [Google Scholar]
- 17.Sartori C, Allemann Y, Scherrer U. Pathogenesis of pulmonary edema: learning from high-altitude pulmonary edema. Respir. Physiol. Neurobiol. 2007;159:338–349. doi: 10.1016/j.resp.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Iles K, Dickinson D, Watanabe N, Iwamoto T, Forman H. AP-1 activation through endogenous H2O2 generation by alveolar macrophages. Free Radic. Biol. Med. 2002;32:1304–1313. doi: 10.1016/s0891-5849(02)00840-7. [DOI] [PubMed] [Google Scholar]
- 19.Iles K, Forman H. Macrophage signaling and respiratory burst. Immunol. Res. 2002;26:95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- 20.Parola M, Robino G, Marra F, et al. HNE interacts with JNK isoforms in human hepatice stellate cells. J. Clin. Invest. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Punjabi C, Laskin J, Pendino K, Goller N, Durham S, Laskin D. Production of nitric oxide by rat type II pneumocytes: increased expression of inducible nitric oxide synthase following inhalation of a pulmonary irritant. Am. J. Respir. Cell Mol. Biol. 1994;11:165–172. doi: 10.1165/ajrcmb.11.2.7519435. [DOI] [PubMed] [Google Scholar]
- 22.Haddad I, Hu P, Galliani C, Beckman J, Matalon S. Quantitation of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J. Clin. Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •.Determined that excessive nitration is present in frozen lung sections from pediatric patients who had died from acute lung injury, consistent with the presence of nitrating species generated either by peroxynitrite, or from the myloperoxidase-catalyzed reaction of nitrite and hydrogen peroxidase. Excessive nitration was also found in the lungs of rats exposed to hyperoxia.
- 23.Kobayashi A, Hashimoto S, Kooguchi K, et al. Expression of inducible nitric oxide synthase and inflammatory cytokines in alveolar macrophages of ARDS following sepsis. Chest. 1998;113:1632–1639. doi: 10.1378/chest.113.6.1632. [DOI] [PubMed] [Google Scholar]
- 24.Sittipunt C, Steinberg K, Ruzinski J, et al. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001;163:503–510. doi: 10.1164/ajrccm.163.2.2004187. [DOI] [PubMed] [Google Scholar]
- 25.Ignarro L. Haem-dependent activation of cytosolic guanylate cyclase by nitric oxide: a widespread signal transduction mechanism. Biochem. Soc. Trans. 1992;20:465–469. doi: 10.1042/bst0200465. [DOI] [PubMed] [Google Scholar]
- 26.Stamler J, Simon D, Jaraki O, et al. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc. Natl Acad. Sci. USA. 2009;89:8087–8091. doi: 10.1073/pnas.89.17.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo C, Atochina-Vasserman E, Abramova E, et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6:e266. doi: 10.1371/journal.pbio.0060266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall H, Potts E, Kelleher Z, Stamler J, Foster W, Auten R. Protection from LPS-induced lung injury by augmentation of airway S-nitrosothiols. Am. J. Respir. Crit. Care Med. 2009;180(1):11–18. doi: 10.1164/rccm.200807-1186OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 29.Beckman J, Beckman T, Chen J, Marshall P, Freeman B. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu P, Ischiropoulos H, Beckman J, Matalon S. Peroxynitrite inhibition of oxygen consumption and sodium transport in alveolar type II cells. Am. J. Physiol. 1994;266:L628–L634. doi: 10.1152/ajplung.1994.266.6.L628. [DOI] [PubMed] [Google Scholar]
- 31.van der Vliet A, Hoen P, Wong P, Bast A, Cross C. Formation of S-nitrosothiols via direct nucleophilic nitrosation of thiols by peroxynitrite with elimination of hydrogen peroxide. J. Biol. Chem. 1998;273:30255–30262. doi: 10.1074/jbc.273.46.30255. [DOI] [PubMed] [Google Scholar]
- 32.Hickman-Davis J, McNicholas-Bevensee C, Davis I, et al. Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am. J. Respir. Crit. Care Med. 2006;173:334–344. doi: 10.1164/rccm.200501-155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazrak A, Iles K, Liu G, Noah D, Noah J, Matalon S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 2009 doi: 10.1096/fj.09-135590. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Zentner M, Deng H, et al. Oxidative stress disrupts glucocorticoid hormone-dependent transcription of the amiloride-sensitive epithelial sodium channel α-subunit in lung epithelial cells through ERK-dependent and thioredoxin-sensitive pathways. J. Biol. Chem. 2000;24:8600–8609. doi: 10.1074/jbc.275.12.8600. [DOI] [PubMed] [Google Scholar]
- 35.Matthay M, Wiener-Kronish J. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am. Rev. Respir. Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- •.Proposed that vectorial transport of Na+ ions across lung epithelial cells creates an osmotic force leading to the reabsorption of edema fluid. Consistent with this, patients with acute respiratory distress syndrome who could concentrate proteins in their edema fluid had a better outcome (mortality and oxygenation) than those who could not. This study provided the first evidence for the importance of active sodium transport in lung fluid balance across the lungs of patients with severe lung injury.
- 36.Ware L, Matthay M. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 37.Yue G, Matalon S. Mechanisms and sequelae of increased alveolar fluid clearance in hyperoxic rats. Am. J. Physiol. 1997;272:L407–L412. doi: 10.1152/ajplung.1997.272.3.L407. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki S, Zuege D, Berthiaume Y. Sodium-independent modulation of Na+-K+-ATPase activity by β-adrenergic agonist in alveolar type II cells. Am. J. Physiol. 1995;268:L983–L990. doi: 10.1152/ajplung.1995.268.6.L983. [DOI] [PubMed] [Google Scholar]
- 39.Sakuma T, Okaniwa G, Nakada T, Nishimura T, Fujimura S, Matthay M. Alveolar fluid clearance in the resected human lung. Am. J. Respir. Crit. Care Med. 1994;150:305–310. doi: 10.1164/ajrccm.150.2.8049807. [DOI] [PubMed] [Google Scholar]
- 40.Pittet J, Lu L, Morris D, et al. Reactive nitrogen species inhibit alveolar epithelial fluid transport after hemorrhagic shock in rats. J. Immunol. 2001;166:6301–6310. doi: 10.4049/jimmunol.166.10.6301. [DOI] [PubMed] [Google Scholar]
- 41.Zhu S, Ware L, Geiser T, Matthay M, Matalon S. Increased levels of nitrate and surfactant protein a nitration in the pulmonary edema fluid of patients with acute lung injury. Am. J. Respir. Crit. Care Med. 2001;163:166–172. doi: 10.1164/ajrccm.163.1.2005068. [DOI] [PubMed] [Google Scholar]
- 42.DuVall M, Zhu S, Fuller C, Matalon S. Peroxynitrite inhibits amiloride-sensitive Na+ currents in Xenopus oocytes expressing αβγ-rENaC. Am. J. Physiol. 1998;274:C1417–C1423. doi: 10.1152/ajpcell.1998.274.5.C1417. [DOI] [PubMed] [Google Scholar]
- 43.Matalon S, Beckman J, Duffey M, Freeman B. Oxidant inhibition of epithelial active sodium transport. Free Radic. Biol. Med. 1989;6:557–564. doi: 10.1016/0891-5849(89)90061-0. [DOI] [PubMed] [Google Scholar]
- 44.Vadász I, Morty R, Kohstall M, et al. Oleic acid inhibits alveolar fluid reabsorption: a role in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2005;171:469–479. doi: 10.1164/rccm.200407-954OC. [DOI] [PubMed] [Google Scholar]
- 45.Vadász I, Morty R, Olschewski A, et al. Thrombin impairs alveolar fluid clearance by promoting endocytosis of Na+, K+- ATPase. Am. J. Respir. Cell Mol. Biol. 2005;33:343–354. doi: 10.1165/rcmb.2004-0407OC. [DOI] [PubMed] [Google Scholar]
- 46.Yucel O, Kunak Z, Macit E, et al. Protective efficiacy of taurine against pulmonary edema progression: experimental study. J. Cardiothorac. Surg. 2008;3:57. doi: 10.1186/1749-8090-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sivrikoz M, Tunçözgür B, Cekmen M, et al. The role of tissue reperfusion in the reexpansion injury of the lungs. Eur. J. Cardiothorac. Surg. 2002;22:721–727. doi: 10.1016/s1010-7940(02)00447-5. [DOI] [PubMed] [Google Scholar]
- 48.Tibor B, Zsolt R. High altitude and free radicals. J. Sports Sci. Med. 2009;3:64–69. [PMC free article] [PubMed] [Google Scholar]
- 49.Sarada S, Himadri P, Mishra C, Geetali P, Ram M, Ilavazhagan G. Role of oxidative stress and NFκB in hypoxia-induced pulmonary edema. Exp. Biol. Med. (Maywood) 2008;233:1088–1098. doi: 10.3181/0712-RM-337. [DOI] [PubMed] [Google Scholar]
- 50.Dickinson DA, Liu RM, Iles KE, Forman HF. Signaling for the synthesis of glutathione. In: Yoshikawa T, editor. Free Radicals in Chemistry, Biology and Medicine. OICA International; London, UK: 2000. [Google Scholar]
- 51.Iles K, Liu R. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. J. Free Radic. Biol. Med. 2005;38:547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 52.Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann. NY Acad. Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 53.Wild AC, Gipp JJ, Mulcahy T. Overlapping antioxidant response element and PMA response element sequences mediate basal and β-naphthoflavone-induced expression of the human γ-glutamylcysteine synthetase catalytic subunit gene. Biochem. J. 1998;332:373–381. doi: 10.1042/bj3320373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fu Z, Jiang P, Ren Y, et al. Changes of antioxidative capacity and endothelial function before and after treatment among patients with high altitude pulmonary edema. Zhonghua Jie He He Hu Xi Za Zhi. 2002;25:33–35. [PubMed] [Google Scholar]
- 55.Vats P, Singh V, Singh S, Singh S. Glutathione metabolism under high-altitude stress and effect of antioxidant supplementation. Aviat. Space Environ. Med. 2008;79:1106–1111. doi: 10.3357/asem.2305.2008. [DOI] [PubMed] [Google Scholar]
- 56.Sciuto A, Cascio M, Moran T, Forster J. The fate of antioxidant enzymes in bronchoalveolar lavage fluid over 7 days in mice with acute lung injury. Inhal. Toxicol. 2003;15:675–685. doi: 10.1080/08958370390197245. [DOI] [PubMed] [Google Scholar]
- 57.Dickinson D, Iles K, Watanabe N, et al. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic. Biol. Med. 2002;33:974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 58.Dickinson D, Iles K, Zhang H, Blank V, Forman H. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17:473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- •.Definitively showed that curcumin could simultaneously modulate key cell signaling pathways to robustly enhance the de novo production of the antioxidant glutathione. It was demonstrated that curcumin could remodel transcription factor complexes and alter activation and translocation to signal for the upregulation of protective genes. This established a molecular rationale for using diet to increase the production of GSH in lung epithelia, which may have applications in reducing the severity of NPE, or delaying or preventing its onset.
- 59.Guidot D, Modelska K, Lois M, et al. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:L127–L135. doi: 10.1152/ajplung.2000.279.1.L127. [DOI] [PubMed] [Google Scholar]