

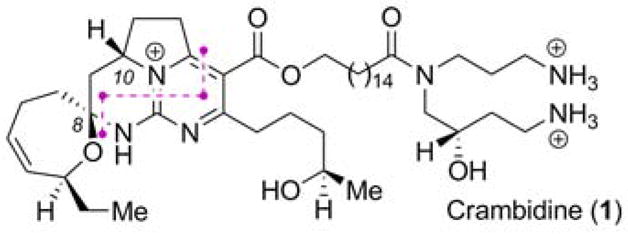
Certain members of the crambescidin natural products,1–5 derived from the marine sponge Crambe crambe, have exhibited remarkable biological properties, including anticancer, anti-HIV, antifungal, and Ca2+ ion channel blocking activities. Many compounds within this class are characterized by a polycyclic guanidine core linked to a hydroxyspermidine moiety by a linear ω-hydroxy fatty acid. Crambidine (1, Figure 1) is atypical within this family of alkaloids in that it possesses a fused pyrimidine heterocyclic core, while most of its other congeners exist in more highly reduced forms. Several elegant strategies have been reported for the synthesis of crambescidin alkaloids.6–9 However, only a single reported synthesis of crambidine (1) has appeared,10 involving dihydropyrimidine construction via Biginelli condensation, followed by oxidation. We report herein a synthesis of (−)-crambidine that capitalizes on two key processes, including a [4+2] annulation of thioimidates with vinyl carbodiimides and a hydroamination of alkynes with 2-aminopyrimidine nucleophiles.
Figure 1.
Vinylcarbodiimides derived from pyrimidine-diones have been shown to converge with O-methyl imidates to furnish 2-aminopyrimidines.11 However, when this annulation process was applied to vinylcarbodiimides and O-imidates more appropriate to the synthesis of 1, the reaction was inefficient. For example, heating a mixture of O-methyl imidate 2 (Scheme 1A) with benzylvinylcarbodiimide 4 at elevated temperature in C6D6 for 24 h provided only 24% conversion to the 2-aminopyrimidine 5. By contrast, when the corresponding thiomethyl imidate 3 was exposed to carbodiimide 4 under otherwise identical conditions, conversion to the pyrimidine 5 was accomplished with marked improvement (59%). Addition of the thiol scavenger AgOTf further enhanced the efficiency of the thioimidate annulation, allowing for the preparation of a variety of bicyclic 2-aminopyrimidines (Scheme 1B, 8–10).
Scheme 1.

Extension of this [4+2] annulation reaction to access a complex substrate more suited to the synthesis of 1 commenced with the synthesis of a fully elaborated vinyl carbodiimide 14 (Scheme 2). Silyl ether 11, obtained in enantiopure form according to the procedure of Campagne and coworkers,12 was acylated with allyl chloroformate to afford the propargylic ester 12. This intermediate was subjected to conjugate addition with azide to afford a separable E/Z mixture (2:1) of the corresponding β-azidoacrylate 13. Reduction of the azide in (E)-13 with PPh3, followed by condensation with TBSOCH2N=C=O, provided the vinyl carbodiimide 14. Importantly, advancement of (Z)-13 under analogous conditions also led to the formation of the (E)-isomer of 14 in similar yield, wherein stereochemical convergence occurs at the iminophosphorane stage.
Scheme 2a.
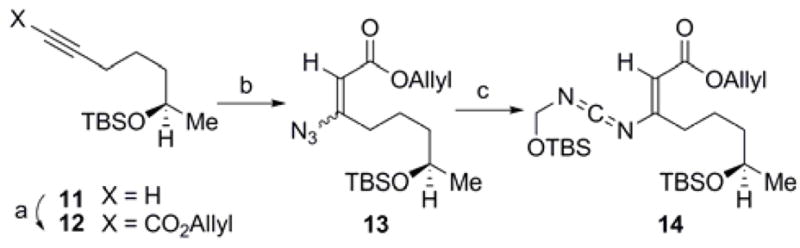
aReagents and conditions: (a) n-BuLi, THF, −78 °C; AllylOCOCl, 90%; (b) (Me2N)2C=NH2N3, CHCl3, 23 °C, 69% (2:1, E:Z); (c) PPh3, CH2Cl2, 23 °C; add TBSOCH2N=C=O, PhH, 80 °C, 49% from (E)-13, 43% from (Z)-13.
Synthesis of an initial crambidine-relevant thioimidate employed methyl thioimidate 1513 (Scheme 3). This served as a suitable acylation agent for the alkyl lithium species derived from the homoallylic iodide 16, prepared in a manner similar to that described by Overman.14 The resulting keto-thioimidate 17 was then heated with vinyl carbodiimide 14 to effect [4+2] annulation. Unfortunately, the bicyclic pyrimidine 18 was not observed; instead, the sole isolable product was pyrimidine 19, presumably a result of C9–C10 β-elimination of the heterocycle in 18.7
Scheme 3a.

aReagents and conditions: (a) 16, t-BuLi, Et2O, Hexanes, −78 °C; add 15, −78 → 23 °C, 67%; (b) 14 (2 equiv), (CH2Cl)2, 60 °C, 70%.
Given that the fragmentation (18→19) and concomitant destruction of the C10 stereoconfiguration could not be avoided, attention turned to thioimidate 26 (Scheme 4) as an alternate annulation substrate in which the internal alkyne would serve as a less acidic ketone surrogate. Thus, Z-selective Wittig olefination of (S)-2-(t-butylsilyloxy)butyraldehyde (20) with the phosphonium ylide derived from 4-(triphenylphosphonium)but-1-yne bromide (21) afforded enyne 22. Following iodination of the terminal alkyne, the alkynyl iodide 23 was subjected to a sequence involving: (1) Cu-mediated coupling with pyrrolidinone 24 to afford internal alkyne 25; (2) a two step conversion of the lactam 25 to the thioimidate 26 via carbonyl thionation and S-alkylation; (3) [4+2] annulation with vinylcarbodiimide 14 to furnish bicyclic pyrimidine 27; and (4) chemoselective N-deprotection to provide the free 2-aminopyrimidine 28, a substrate poised for intramolecular alkyne hydroamination.
Scheme 4a.

aReagents and conditions: (a) 21, n-BuLi, THF, −78 → 0 °C; add 20, −78 → 23 °C, 85%; (b) NIS, AgNO3, Me2CO, 23 °C, 91%; (c) 24, Zn, DMF, 0 °C; CuCN, LiCl, THF, DMF, −40 → 23 °C; add 23, −40 → 23 °C, 54% (d) Lawesson’s Rgt, THF, 0 °C, 94%; (e) MeI, K2CO3, THF, 23 °C, 95%; (f) 14 (2 equiv), (CH2Cl)2, 23 °C, 65%; (g) NH4F, MeOH, 23 °C, 79%; (h) AuCl3, MeCN, 40 °C, 78%; (i) p-TsOH·H2O, MeCN, 23 °C, 77%; (j) Pd(PPh3)4, pyrrolidine, MeCN, 23 °C, 81%; (k) Cs2CO3, DMF, 23 °C, 88%; (l) HCl, Et2O, 0 °C, 77%.
Transition metal catalyzed hydroamination of alkynes15, 16 is a powerful reaction in synthesis;17–19 however, the paucity of guanidine or 2-aminopyrimidine nucleophiles engaging in this reaction is notable. After extensive experimentation, this transformation was validated by treatment of alkyne 28 (Scheme 4) with 10 mol% AuCl320 at 40 °C, leading to efficient production of the tricyclic pyrimidine 29 as a single isomer (78%).
Subsequent spiroaminal formation at C8 in enamine 29 was conducted under carefully controlled acidic conditions, being mindful of the possibility of undesired C10-N bond rupture via potential C8-iminium reactivity. This liability was precluded by treatment with TsOH, effecting TBS removal and spirocyclization to provide the tetracyclic pyrimidinium 30 (77%).21 The final stages of the synthesis involved conversion of the allyl ester 30 to its Cs-carboxylate. This nucleophile, obtained from carboxylic acid 31, was amenable to selective alkylation with iodide 32.13 The resulting ester 33 was then subjected to t-butylcarbamate removal to afford (−)-crambidine (1).
A convergent synthesis of crambidine has been described, showcasing a [4+2] thioimidate-vinyl carbodiimide annulation and an intramolecular alkyne-guanidine hydroamination. This strategy should not only prove useful for preparing other members of the crambescidins, but also provide an attractive means with which to access complex N-heterocycles in general.
Supplementary Material
Acknowledgments
This research was supported by the NIH (GM57859) and Merck, Inc. We thank Prof. Larry Overman for providing a copy of a 13C NMR spectrum of synthetic 1.
Footnotes
Supporting Information Available: Experimental details (PDF). This information is available at http://pubs.acs.org.
References
- 1.Heys L, Moore CG, Murphy PJ. Chem Soc Rev. 2000;29:57–67. [Google Scholar]
- 2.Jareserijman EA, Sakai R, Rinehart KL. J Org Chem. 1991;56:5712–5715. [Google Scholar]
- 3.Ohtnai I, Kusumi T, Kakisawa H, Kashman Y, Hirsh S. J Am Chem Soc. 1992;114:8472–8479. [Google Scholar]
- 4.Berlinck RGS, Braekman JC, Daloze D, Bruno I, Riccio R, Ferri S, Spampinato S, Speroni E. J Nat Prod. 1993;56:1007–1015. doi: 10.1021/np50097a004. [DOI] [PubMed] [Google Scholar]
- 5.Jareserijman EA, Ingrum AL, Carney JR, Rinehart KL, Sakai R. J Org Chem. 1993;58:4805–4808. [Google Scholar]
- 6.Snider BB, Shi ZP. J Am Chem Soc. 1994;116:549–557. [Google Scholar]
- 7.Coffey DS, McDonald AI, Overman LE, Rabinowitz MH, Renhowe PA. J Am Chem Soc. 2000;122:4893–4903. [Google Scholar]
- 8.Nagasawa K, Georgieva A, Koshino H, Nakata T, Kita T, Hashimoto Y. Org Lett. 2002;4:177–180. doi: 10.1021/ol0168263. [DOI] [PubMed] [Google Scholar]
- 9.Moore CG, Murphy PJ, Williams HL, McGown AT, Smith NK. Tetrahedron Lett. 2003;44:251–254. [Google Scholar]
- 10.Overman LE, Rhee YH. J Am Chem Soc. 2005;127:15652–15658. doi: 10.1021/ja055464h. [DOI] [PubMed] [Google Scholar]
- 11.Wamhoff H, Schmidt A. Heterocycles. 1993;35:1055–1066. [Google Scholar]
- 12.Petry N, Parenty A, Campagne JM. Tetrahedron-Asymmetry. 2004;15:1199–1201. [Google Scholar]
- 13.See supporting information.
- 14.Coffey DS, McDonald AI, Overman LE, Stappenbeck F. J Am Chem Soc. 1999;121:6944–6945. [Google Scholar]
- 15.Pohlki F, Doye S. Chem Soc Rev. 2003;32:104–114. doi: 10.1039/b200386b. [DOI] [PubMed] [Google Scholar]
- 16.Muller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 17.McGrane PL, Livinghouse T. J Am Chem Soc. 1993;115:11485–11489. [Google Scholar]
- 18.Crawley SL, Funk RL. Org Lett. 2003;5:3169–3171. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]
- 19.Kadzimirsz D, Hildebrandt D, Merz K, Dyker G. Chem Commun. 2006:661–662. doi: 10.1039/b516017k. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda Y, Utimoto K. Synthesis. 1991:975–978. [Google Scholar]
- 21.This and subsequent aminopyrimidine intermediates were purified by RP-HPLC (water/MeCN/TFA).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.