

Abstract
The most recent papers describing the stereoselective synthesis of cyclic quaternary α-amino acids are collected in this review. The diverse synthetic approaches are classified according to the size of the ring and taking into account the bond that is formed to complete the quaternary skeleton.
1. Introduction
Linear peptides are highly flexible molecules that can adopt many conformations in solution and, of these, only a few are responsible for their biological activity. The construction of novel peptide sequences with tailor made enhanced properties is one of the most challenging areas in biomimetic research. The incorporation of rigid amino acid surrogates provides very useful information on the bioactive conformation and results in beneficial physiological effects. Between these rigid amino acids the use of quaternary compounds is one of the most interesting approaches, and for this reason during the last few years many procedures towards the stereoselective synthesis of these compounds have been described. In this context, we have previously reviewed (1998 and 2000) the stereoselective synthesis of these interesting compounds,1,2 and taking into account the great quantity of procedures reported, more recently we have published an update about the stereoselective synthesis of the acyclic α-amino acids3 that we would like to complete now with a corresponding update of the cyclic systems.
Before beginning the summary of the new procedures concerning the stereoselective synthesis of these cyclic derivatives it is worth mentioning that apart from our own contributions, during the last years some reviews focused on some particular aspects have been published in relation to the synthesis of some cyclic amino acid and derivatives,4,5 the synthesis of heterosubstituted carbocyclic α-amino acids,6 the synthesis of some fluorinated acyclic and cyclic amino acids,7 the synthesis of unnatural α-amino acids8 and the modelling and synthesis of some conformationally constrained amino acids.9 Much more recently, the synthesis of the family of enantiomerically pure 1-amino-2-phenylcycloalkanecarboxylic acids,10 an excellent review of 1-aminocyclopropane-carboxylic acids,11,12 the catalytic asymmetric synthesis of α-amino acids including some quaternary derivatives,13 the synthesis of cyclic α-amino acids and their use in the preparation of stable conformational short peptides,14 and also some recent approaches towards the asymmetric synthesis of quaternary amino acids15 have been reported.
Nevertheless, and in spite of all these reviews, some of which are from a general point of view and others focused on some particular aspects or families of compounds, we would like to review all methodologies in a manner that is useful to organic experimentalists.
Some data concerning the structural analysis of cyclic amino acids in small and medium size Acnc (1-aminocycloalkanecarboxylic acids) has been completed during these years and the synthesis and structural studies of model peptides containing these cyclic amino acids has been reported.16–24 Apart from these classical rings, the conformational tendencies of other cyclic amino acids such as Hms(Ipr) or O,O-isopropylidene-α-hydroxymethylserine,25 Afc or 9-amino-9-fluorenecarboxylic acid,26,27 Daf or 9-amino-4,5-diazafluorene-9-carboxylic acid,28,29 Adt 4-amino-1,2-dithiolane-4-carboxylic acid,30 the axially chiral α-amino acids Bip and Bin31–37 or the Bip system incorporating a crown ether receptor38 have been reported. More recently, the synthesis and properties of antAib, a novel tetrasubstituted α-amino acid of the Ac5c type possessing a fused anthracene fluorophore has also been reported.39,40
Theoretical calculations focused on the study of the conformational tendencies of 1-amino-cycloalkanecarboxylic acids (Acnc) have been reported.41–43 Of these compounds the cyclopropane derivatives have attracted the attention of many researchers, probably due to the particular characteristics that the cyclopropane ring confers to the amino acid. When additional substituents are incorporated into the ring, two chiral centres are formed and, as a consequence, new stereoisomers are possible. In the particular case of the incorporation of one phenyl ring as a substituent (named, c3Phe), the compound can be considered as a constrained phenylalanine and in this case several theoretical studies has been reported44 to explain the behaviour previously described by our group.45,46 The presence of an additional phenyl group in a different carbon atom (c3diPhe) confers peculiar characteristics to the molecule these have been reported both from an experimental47 and theoretical point of view.48 The case of the cyclopropane derivative in which both phenyl substituents are on the same carbon atom (c3Dip) seems particularly interesting since it has been reported that it confers important tendencies to give a γ-turn in some model peptides.49,50 The structural tendencies of other cyclopropane derivatives such as c3Val51,52 or other 2-phenyl-1-aminocycloalkanecarboxylic acids such as c5Phe53 and c6Phe54–56 have also been reported. Additionally, the theoretical study of 8-aminopentacycloundecane-8-carboxylic acid has been reported.57 Very recently, the helical screw sense exclusively governed by chiral centres in the side chain of some cyclic amino acids has been reported.58–60
Finally, some systematic structure-activity relationships between biological properties of peptides incorporating quaternary cyclic amino acids has also been reported.61–63
2. Synthesis of 1-aminocycloalkanecarboxylic acids
2.1. Using cyclic compounds as starting materials
One of the most useful methodologies to prepare 1-aminocycloalkanecarboxylic acids in a stereoselective manner involves the use of cyclic compounds (typically aldehydes but ketones for the synthesis of quaternary α-amino acids) as starting materials, although in this case the introduction of both functional groups (amino and carboxylic acid) is necessary. Of all reported methodologies, the Strecker reaction64,65 and related synthesis have been repeatedly used. The diastereoselective Strecker reaction involves the addition of cyanide or its equivalents to the previously formed C=N bond from the corresponding ketone and a chiral amine, and subsequent hydrolysis of the nitrile group. For the Strecker reaction several chiral auxiliaries such as (S)-α-methylbenzylamine (α-MBA),66 (R)-phenylglycinol,67 (R)-phenylglycine amide,68 (SS)-p-toluene- and (SS)-tert-butane-sulfinimides,69 (S)-1-amino-2-methoxymethylpyrrolidine (SAMP),70 (S)-1-amino-2-methoxymethyl-indoline (SAMI),71 and 2,3,4,6-tetra-O-pivaloyl-β-D-galactopyranosyl amine72 have been used.
The stereoselective synthesis of 1-aminocycloalkanecarboxylic acids using this methodology have been grouped depending on the size and type of the starting carbonyl compound.
The 1-aminocyclopropanecarboxylic acids probably are the most interesting carbocyclic α-amino acids and several methodologies have been described for their stereoselective synthesis. However, to the best of our knowledge the work of Fadel et al.66 is the only example reported in the literature in which, the Strecker reaction has been used for their stereoselective synthesis. In this context, reaction of cyclopropanone hemiacetal (2S)-1 with the chiral amine [(S)-α-MBA or (S)-α-methoxymethylbenzylamine ((S)-MOMBA)] afforded the imines 2a,b, which, by addition of NaCN gave the α-aminonitriles (1R,2S)-3a,b with moderate diastereoselective excess. Hydrolysis of diastereoisomerically pure (1R,2S)-3a,b with concentrated sulfuric acid at 0 °C, followed by hydrogenolysis over Pd(OH)2/C provided the amine amide (1R,2S)-4a, which, by treatment with 6 N HCl at reflux, gave the (1R,2S)-1-amino-2-methylcyclopropanecarboxylic acid 5a (allo-norcoronamic acid) in 85% yield (Scheme 1).
Scheme 1.

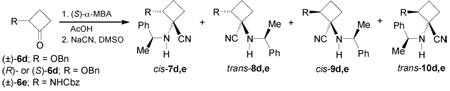
On the other hand, condensation of the cyclobutanones (±)-6a–c73 with (S)-α-MBA in the presence of a catalytic amount of acetic acid or p-toluenesulfonic acid (TsOH), followed by addition of sodium cyanide74 or trimethylsilylcyanide (TMSCN)75 in the presence of ZnCl2, afforded the α-amino nitriles cis-7a–c, trans-8a–c, cis-9a–c, and trans-10a–c in moderate yield and diastereoisomeric ratio. The results are summarized in Table 1.
Table 1.
Preparation of α-amino nitriles from the ketones (±)-6a–c.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Conditions | R | Yield (%) | Diastereoisomeric ratio | |||
| 7 | 8 | 9 | 10 | ||||
| 1 | NaCN, DMSO, 55–60 °C | a = Pha | 54 | 51 | <2.5 | 45 | <1.5 |
| 2 | NaCN, MeOH, 55–60 °C | a = Ph | 43 | 52 | 2.5 | 44 | <1.5 |
| 3 | NaCN, DMSO, 55–60 °C | b = i-Pr | 46 | 56 | 1.0 | 42 | <1 |
| 4 | NaCN, MeOH, 55–60 °C | b = i-Pr | 42 | 54 | <1.0 | 44 | <1 |
| 5 | TMSCN, MeOH, ZnCl2, 0 °C | c = Meb | -- | 6 | 46 | 11 | 37 |
| 6 | TMSCN, hexane, ZnCl2, −10 °C | c = Meb | -- | 11 | 15 | 33 | 41 |
Similar results were obtained using (R)-phenylglycinol and (R)-MOMBA.
The chiral amine was (R).
Hydrolysis of diastereoisomerically pure cis-7b (R = i-Pr) with concentrated sulfuric acid at 0 °C gave the amide 11b in 85% yield, which, by hydrogenolysis over Pd(OH)2/C, furnished the amine amide (1S,2S)-12b in 98% yield. Finally, hydrolysis of (1S,2S)-12b with 6 N HCl under reflux followed by treatment with propylene oxide in ethanol gave the α-amino acid (1S,2S)-13b in 90–94% yield. Under identical conditions, cis-9b was transformed into (1R,2R)-13b (Scheme 2).74
Scheme 2.

In a similar way, treatment of the mixture of α-amino nitriles 8–10c with concentrated sulfuric acid at 0 °C followed by separation and subsequent hydrogenolysis with HCO2NH4, Pd/C conditions, and hydrolysis with concentrate HCl led to (1R,2R)-13c, (1S,2S)-13c and (1R,2S)-14c as chlorohydrate salt (Scheme 3).75
Scheme 3.

Recently, Fadel et al.76 have reported the stereoselective synthesis of (1R,2R)-1-amino-2-hydroxy-cyclobutanecarboxylic acid 13d, a serine derivative from racemic or optically pure 2-benzyloxy-cyclobutanone 6d (R = OBn), and (1R,2R)- and (1S,2S)-1,2-diaminocyclobutanecarboxylic acid 13e an ornitine derivative, from racemic 2-aminocyclobutanone 6e. For this purpose, the condensation of either (±)-or enantiopure 6d with (S)-α-MBA, followed by addition of sodium cyanide gave the corresponding α-amino nitrile mixture 7–10d. The formation of the four diastereoisomers 7–10d using (R)- or (S)-6d was probably due to the partial racemization of enantiomerically pure starting ketone under Strecker conditions.76a On the other hand, one-pot reaction of 6e with (S)-α-MBA in the presence of AcOH and NaCN afforded, under thermodynamic control, only two major stereoisomers 8e and 10e in 55:45 ratio and excellent yield.76b The results are summarized in Table 2.
Table 2.
Asymmetric Strecker reaction of ketones 6d,e.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ketone | Conditions | Diastereoisomeric ratio | |||||
| Time | Temperature (°C) | Yield (%) | 7d,e | 8d,e | 9d,e | 10d,e | ||
| 1 | (±)-6d | 4 h | 20 | 52 | 35 | 15 | 35 | 15 |
| 2 | (±)-6d | 4 days | 50 | 54 | 10 | 40 | 10 | 40 |
| 3 | (R)-6d | 4 h | 20 | 55 | 75 | 13 | 6 | 6 |
| 4 | (R)-6d | 4 days | 50 | 54 | 28 | 60 | 2 | 10 |
| 5 | (S)-6d | 5 h | 20 | 55 | 5.8 | 2.5 | 72.5 | 19.2 |
| 6 | (±)-6e | 3 days | 50 | 90 | -- | 45 | -- | 55 |
Hydrolysis of diastereoisomerically pure 8d (R = OBn) with hydrogen peroxide and ethanolic potassium hydroxide solution, followed by hydrogenolysis over Pd(OH)2/C in the presence of di-tert-butylcarbonate [(Boc)2O], afforded the amide (1R,2R)-15d in 69% yield. Finally, hydrolysis of (1R,2R)-15d with 6 M HCl under reflux gave the quaternary α-amino acid (1R,2R)-13d in 74% yield as chlorohydrate salt. Under identical conditions trans-8e and trans-10e were transformed into quaternary 1,2-diamino acids (1R,2R)- and (1S,2S)-13e in good chemical yield (Scheme 4).76
Scheme 4.

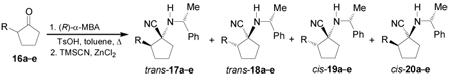
On the other hand, condensation of the 2-alkylpentanones 16a–e77 with (R)-α-MBA in the presence of a catalytic amount of TsOH, followed by addition of TMSCN and ZnCl2 in methanol or hexane under thermodynamically or kinetically controlled conditions, produced the four diastereoisomeric α-amino nitriles 17a–e to 20a–e. The results are summarized in Table 3.78
Table 3.
Asymmetric Strecker reaction of the ketones (±)-16a–e.
 | |||||||
|---|---|---|---|---|---|---|---|
| Diastereoisomeric ratio | |||||||
| Entry | R | Conditions | Yield (%) | 17 | 18 | 19 | 20 |
| 1 | a = OMe | MeOH, 20 °C, 12 h | 98 | 41 | 22 | 29 | 8 |
| 2 | a = OMe | Hexane, −10 °C, 18 h | 98 | 10 | 0 | 61 | 29 |
| 3 | b = Me | MeOH, 25 °C, 24 h | 100 | 55 | 24 | 16 | 5 |
| 4 | b = Me | MeOH, −10 °C, 3 h | 100 | 45 | 30 | 21 | 4 |
| 5 | b = Me | Hexane, −10 °C, 3 h | 100 | 43 | 5 | 47 | 5 |
| 6 | c = Et | MeOH, 25 °C, 24 h | 98 | 57 | 23 | 14 | 6 |
| 7 | c = Et | MeOH, −10 °C, 3 h | 98 | 45 | 21 | 28 | 6 |
| 8 | c = Et | Hexane, −10 °C, 3 h | 82 | 39 | 5 | 45 | 11 |
| 9 | d = i-Pr | MeOH, 25 °C, 24 h | 86 | 44 | 23 | 28 | 5 |
| 10 | d = i-Pr | MeOH, −10 °C, 3 h | 92 | 37 | 4 | 47 | 12 |
| 11 | d = i-Pr | Hexane, −10 °C, 3 h | 84 | 12 | 4 | 57 | 27 |
| 12 | e = t-Bu | MeOH, 25 °C, 24 h | 74 | 61 | 23 | 14 | 2 |
| 13 | e = t-Bu | MeOH, −10 °C, 3 h | 36 | 26 | 0 | 59 | 15 |
| 14 | e = t-Bu | Hexane, −10 °C, 3 h | 37 | 5 | 1 | 66 | 28 |
Hydrolysis of the mixture of α-amino nitriles 17a–e to 20a–e obtained using methanol as solvent with concentrate sulfuric acid, produced the diastereoisomerically pure α-amino carboxyamides 21a–e to 24a–e, after separation by flash chromatography and preparative HPLC. Hydrogenolysis of diastereoisomerically pure α-amino carboxyamides 21a–e, 22a–e, 23a–c and 24a–c with HCO2NH4 and Pd/C, followed by hydrolysis with concentrate HCl and subsequent treatment with cation exchange resin, gave the 2-alkylated 1-aminocyclopentanecarboxylic acids (1R,2R)- and (1S,2S)-25a–e, and (1R,2S)- and (1S,2R)-26a–c (Scheme 5).78
Scheme 5.

Condensation of the 5-bromo-1-indanone 27, which is easily obtained from 3-bromobenzaldehyde, with (R)-phenylglycinol followed by addition of TMSCN and subsequent treatment with HCl, afforded the mixture of α-amino esters 28 in 61% yield and 7:1 diastereoisomeric ratio, which, under reflux in toluene gave the spiro derivatives (S,R)-29 and (R,R)-30 in 59% yield. Palladium-catalyzed carbonylation of diastereoisomerically pure (S,R)-29 with Pb(OAc)2 and 1,3-bis(diphenyl-phosphino) propane (dppp) in ethanol produced the derivative (S,R)-31 in 67% yield, which, by cleavage of spiro ring with K2CO3 in methanol produced the diester (S,R)-32 in 70% yield. Finally, oxidative cleavage of benzyl fragment in (S,R)-32 with Pb(OAc)2 followed by acidic hydrolysis and subsequent treatment with propylene oxide furnished the 1-aminoindane-1,5-dicarboxylic acid 33 [(S)-AIDA] in 65% yield and this is an antagonist of metabotropic glutamate receptors (Scheme 6).79
Scheme 6.

On the other hand, palladium-catalyzed phosphonylation of diastereoisomerically pure (S,R)-29 whith diethyl phosphite produced the ethyl phosphonate (S,R)-34 in 83% yield, which, by cleavage of spiro ring with K2CO3 in methanol, led to diester (S,R)-35 in 70% yield. Finally, oxidative cleavage of benzyl fragment in (S,R)-35 with Pb(OAc)2, followed by acidic hydrolysis and subsequent treatment with propylene oxide furnished the 1-amino-5-phosphoindane-1-carboxylic acid 36 [(S)-APICA] in 65% yield (Scheme 7).79
Scheme 7.

Schann et al.80 reported the first stereoselective synthesis of aminopyrrolidinedicarboxylic acids 41 and 42, which have been used in the preparation of glutamate receptor compounds.81 Thus, the Bucherer-Bergs reaction82 of (S)-37, readily obtained from (2S,4R)-4-hydroxyproline, with (NH4)2CO3 and KCN in ethanol gave the spirohydantoin mixture 38 in 68–78% yield, which, by basic hydrolysis followed by treatment with SOCl2 and methanol under reflux afforded, after chromatographic separation, the amino esters (2S,4S)-39 and (2S,4R)-40. N-Boc protection of (2S,4S)-39 and (2S,4R)-40, followed by cleavage of benzyl protective group by hydrogenolysis under HCO2NH4 and Pd/C conditions, and subsequent saponification and cleavage of Boc protective group with HCl, gave the amino acids (2S,4S)-41 and (2S,4R)-42, respectively. Under identical conditions (R)-37 was transformed into (2R,4R)-41 and (2R,4S)-42 (Scheme 8).
Scheme 8.

On the other hand, reaction of the ulose 43, readily obtained by oxidation of diacetone-D-glucose, with ammonia in the presence of Ti(Oi-Pr)4, followed by addition of TMSCN, provided the glycol-α-amino nitrile 44 in 80% yield as the only detectable stereoisomer, which, by treatment with carbon dioxide in MeOH at 75 atm/85 °C or (NH4)2CO3 in MeOH-H2O at 70 °C, gave the spirohydantoin 45 in 80% yield. Selective hydrolysis of one of the acetonides of 45 with 1 N HCl, followed by hydantoin ring opening with barium hydroxide and subsequent ion-exchange chromatography, furnished the quaternary glycoamino acid 46 in 55% yield in three steps (Scheme 9).83
Scheme 9.

Recently De Micheli et al.84 reported the stereoselective synthesis of conformationally constrained α-amino acid 50, an analogue of aspartic acid, based on the Strecker methodology. Thus, TsOH-catalyzed condensation of the ketone 47 with 4-methoxybenzylamine (PMB-NH2), followed by addition of TMSCN in the presence of ZnCl2, afforded the cyano derivative 48 as a single detectable stereoisomer. Cleavage of PMB protective group in 48 with cerium(IV) ammonium nitrate (CAN) provided the α-amino nitrile 49, which, by hydrolysis and subsequent ion-exchange chromatography gave the conformationally constrained α-amino acid 50 in 27% overall yield (Scheme 10).
Scheme 10.

Conformationally constrained (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane 2,6-dicarboxylic acid, also known as (LY354740),85 is a highly potent and selective agonist for group II metabotropic glutamate (mGlu) receptors, specifically mGlu2 and mGlu3, that has been found to possess anxiolytic, antipsychotic, anticonvulsant, anti-Parkinsonian, analgesic, and neuroprotective properties in vivo.86 Additionally the peptides of type 51 are effective prodrugs of LY354740.87 For this reason several analogues of LY354740 have been prepared.

Monn et al.88 reported the synthesis of conformationally constrained α-amino acids (+)- and (+)-55 and (−)-56, which, were evaluated as mGlu receptors. In this context, reaction of the optically pure bicyclic ketone (+)-53, obtained from 52,89 with (NH4)2CO3 and KCN in ethanol gave the spirohydantoin (+)-54 in 28% yield after crystallization, which, by basic hydrolysis, gave the conformationally constrained α-amino acid (+)-55 in 55% yield. In a similar way, (−)-53 was transformed into (−)-56 (Scheme 11).
Scheme 11.

On the other hand, Lee and Miller90 reported the stereoselective synthesis of conformationally constrained α-diamino acid (−)-60 starting from the cyclic ketone (−)-57. In this context, the intermolecular cyclopropanation of the α,β-unsaturated ketone (−)-57 with the sulfonium ylide obtained from (ethoxycarbonylmethyl)dimethylsulfonium bromide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), afforded the bicyclic ketone (−)-58 in 60–73% yield,91 which, by a Bucherer-Bergs reaction with (NH4)2CO3 and KCN in ethanol, provided the spirohydantoin (−)-59 in 59% yield and 96% ee. Basic hydrolysis of (−)-59 and sequential treatment with copper(II) carbonate, benzoyl chloride and ion-exchange chromatography, furnished the α-diamino acid (−)-60 with >98% ee (Scheme 12).
Scheme 12.

Mann et al.92 reported the synthesis of constrained cycloalkyl analogue of glutamic acid 64 with a ω-phosphonic acid function, an analogue of AP4.93 Thus, reaction of the bicyclic ketone 61 with (NH4)2CO3 and KCN in H2O produced the spirohydantoins 62 and 63 in 68% yield and 4:1 ratio as an inseparable mixture of diastereoisomers, from which, by acidic hydrolysis and crystallization, the α-amino acid 64 could be obtained in 56% yield (Scheme 13).
Scheme 13.

On the other hand, reaction of the optically pure bicyclic ketone (−)-66, obtained in 9 steps from chiral methyl ester (1R,5R)-65, with ammonia in the presence of Ti(Oi-Pr)4 in methanol followed by addition of TMSCN, afforded the α-amino nitrile 67 in 80% yield and 13.1:1 diastereoisomeric ratio, which, by cristallyzation and subsequent hydrolysis with 8 N HCl and AcOH, furnished the optically pure conformationally constrained fluoro α-amino acid (+)-68 in 94% yield (Scheme 14).94
Scheme 14.

Nakazato et al.95,96 reported the synthesis of several conformationally constrained fluoro α-amino acids, which were evaluated as potent and selective group II metabotropic glutamate receptor antagonists. For example, reaction of the optically pure cyclic sulfates (+)-70a,b, obtained in 3 steps from the bicyclic ketone (−)-69, with sodium azide followed by treatment with sulfuric acid, gave the azide derivatives (1R,2R,3R,5R,6R)-71a,b in good yield. Catalytic hydrogenation of benzyl ester and azide functions in 71b followed by acidic hydrolysis provided the conformationally constrained α-amino acid (1R,2R,3R,5R,6R)-72 in 79% yield (Scheme 15).
Scheme 15.

On the other hand, reaction of (1R,2R,3R,5R,6R)-71a with trifluoromethanesulfonyl anhydride (Tf2O) in pyridine afforded the derivative (−)-73, which, by treatment with KNO2 in the presence of 18-crown-6 and subsequent addition of water, gave the compound (1R,2R,3S,5R,6R)-74 in 80% yield. Reduction of azide group in 74 under Staudinger conditions97 using PMe3, followed by basic hydrolysis, produced the α-amino acid (1R,2R,3S,5R,6R)-75 in 48% yield (Scheme 16).95
Scheme 16.

Additionally, optically pure (1R,2R,3R,5R,6R)-71a and (1R,2R,3S,5R,6R)-74 have been transformed into conformationally constrained α-amino acids (1R,2R,3R,5R,6R)-76 and (1R,2R,RS,5R,6R)-77a,b, respectively, which have been evaluated as potent and selective group II metabotropic glutamate receptor antagonists (Scheme 17).95,96
Scheme 17.

On the other hand, reaction of cyclic sulfate (1S,2S,3R,5R,6S)-78a with sodium azide, followed by treatment with sulfuric acid, gave the azide derivative (1S,2R,3R,5R,6S)-79a in 91% yield. Reaction of 79a with benzyl trichloroacetimidates (ArCH2OC(=NH)CCl3) in the presence of a catalytic amount of trifluoromethanesulfonic acid (TfOH) afforded the corresponding ether derivatives (1S,2R,3R,5R,6S)-80. Reduction of the azide function in 80 with PMe3 and subsequent basic hydrolysis produced the α-amino acids (1S,2R,3R,5R,6S)-81 (several aryl groups were used) (Scheme 18).96a
Scheme 18.

Very recently, Woltering et al.98 reported the stereoselective synthesis of (1S,2R,3R,5R,6S)-2-amino-3-hydroxybicyclo[3.1.0]hexane-2,6-dicarboxylic acid 82 [(+)-HYDIA], a group II mGlu receptor. Thus, the selective ring opening of cyclic sulfate (1S,2S,3R,5R,6S)-78b with sodium azide afforded the azide derivative (1S,2R,3R,5R,6S)-79b in 62% yield. Catalytic hydrogenation of the benzyl ester and azide functions of 79b, followed by acidic hydrolysis and subsequent treatment with propylene oxide, produced the (+)-HYDIA, 82 in 87% yield (Scheme 19).
Scheme 19.

Oxidation of the alcohol group in (1S,2R,3R,5R,6S)-79b with PCC gave the corresponding ketone (1S,2R,5R,6S)-83 in 67% yield, and subsequent reduction with NaBH4 afforded the alcohol (1S,2R,3S,5R,6S)-84 in 51% as a single diastereoisomer. Catalytic hydrogenation of the benzyl ester and azide functions in 84, followed by acidic hydrolysis and subsequent treatment with propylene oxide, produced the β-hydroxy-α-amino acid (1S,2R,3S,5R,6S)-85 in 86% yield. On the other hand, treatment of 79b with Tf2O in pyridine provided the triflate (1S,2R,3R,5R,6S)-86 in 86% yield and a subsequent SN2 reaction using sodium azide furnished (1S,2R,3S,5R,6S)-87 in 49% yield as a single diastereoisomer. Catalytic hydrogenation of the benzyl ester and azide functions of 87, followed by acidic hydrolysis and subsequent treatment with propylene oxide produced the α,β-diamino acid (1S,2R,3S,5R,6S)-88 in 79% yield (Scheme 20).98
Scheme 20.

Reaction of the (+)-pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-one 89 with (NH4)2CO3 and KCN in H2O produced the spirohydantoin 90 in 83% yield as the main product and hydrolysis of 90 with barium hydroxide gave the quaternary α-amino acid (−)-91 in 67% yield. Under identical conditions, the enone (−)-92 was transformed into the quaternary α-amino acid (+)-94 through the spirohydantoin (+)-93 (Scheme 21).99
Scheme 21.

Condensation of the 2-metoxycyclohexanone (±)-95a with (S)-α-MBA, followed by addition of TMSCN in the presence of ZnCl2 in methanol, gave the α-amino nitriles mixture 96a (cis/trans = 26:74 ratio) under thermodynamic control, and (cis/trans = 75:25) under kinetic control conditions. Hydrolysis of the mixture of α-amino nitriles cis/trans-96a with concentrate sulfuric acid produced, after chromatographic separation, the mixture of the α-amino carboxyamides cis/trans-97a, and the hydrogenolysed product (1S,2R)-98a. Low pressure liquid chromatography (LPLC) separation of the carboxyamides cis/trans-97a afforded the diastereoisomerically pure α-amino carboxyamide trans-97a [(1S,2S,1'S)-97a], which, by hydrogenolysis over Pd/C followed by hydrolysis with 12 M HCl and subsequent ion-exchange chromatography on a Dowex 50W column, led to (1S,2S)-1-amino-2-hydroxycyclohexanecarboxylic acid 99a. On the other hand, acidic hydrolysis of 98a and subsequent treatment with Dowex resin gave the quaternary α-amino acid (1S,2R)-100a (Scheme 22).100
Scheme 22.

Frahm et al.101 reported the stereoselective synthesis of 1,2-diaminocyclohexanecarboxylic acids (1R,2R)- and (1S,2S)-99b starting from the 2-benzoylaminocyclohexanone (±)-95b by applying the Strecker methodology. Thus, the condensation reaction of (±)-95b with (R)-α-MBA, followed by addition of TMSCN in the presence of ZnCl2 in methanol or hexane, under thermodynamic conditions, afforded the corresponding α-amino nitriles mixture cis/trans-96b in 99% yield, which, by hydrolysis with concentrate sulfuric acid at −20 °C produced, after LPLC separation, the α-amino carboxyamides (1R,2R,1'R)-97b and (1S,2S,1'R)-101b in 19 and 8% yield, respectively. Hydrogenolysis of diastereoisomerically pure 97b and 101b over Pd/C, followed by hydrolysis with 12 M HCl and subsequent ion-exchange chromatography gave the corresponding α,β-diamino acids (1R,2R)- and (1S,2S)-99b in 97% yield (Scheme 23).
Scheme 23.

On the other hand, condensation of ethyl 2-cyclohexanoneacetate (±)-95c with (R)-α-MBA, followed by addition of TMSCN in the presence of ZnCl2 in methanol under kinetic or thermodynamic control, gave the α-amino nitriles mixture cis/trans-96c in 96% yield. Hydrolysis of the mixture of α-amino nitriles 96c with concentrate sulfuric acid at −20 °C, followed by chromatographic separation and subsequent hydrogenolysis under HCO2NH4, Pd/C conditions produced the azabicyclo compounds (1R,2S)- and (1S,2R)-102 (Scheme 24).102
Scheme 24.

Reaction of the enantiopure ketone 103103 with (NH4)2CO3 and KCN, followed by treatment with (Boc)2O produced the spirohydantoins 104 and 105 in 49% yield and 5:2 dr. Basic hidrolysis of diastereoisomerically pure 104, obtained after chromatographic separation, afforded the quaternary α-amino acid (S,S)-106 in 91% yield (Scheme 25).104
Scheme 25.

Condensation of the ketones 107a,b with (S)-α-MBA, followed by addition of HCN in the presence of a catalytic amount of ZnI2 afforded the α-amino nitriles mixture 108a,b. Hydrolysis of 108a with concentrate sulfuric acid furnished the α-amino carboxyamides (1S,1'S)-109a and (1R,1'S)-110a in 86% yield and 10:1 diastereoisomeric ratio. Hydrolysis of 108b under identical conditions gave the α-amino carboxyamides (1S,1'S)-109b and (1R,1'S)-110b in 50% yield and 45:55 dr. Hydrogenolysis of diastereoisomerically pure (1S,1'S)-109a over Pd/C, followed by acidic hydrolysis provided the quaternary α-amino acid (S)-111a in quantitative yield. In a similar way, (1R,1'S)-110b was transformed into (R)-111b in quantitative yield (Scheme 26).105
Scheme 26.

In a similar way, condensation of the ketones 107a–c with (R)-phenylglycinol, followed by addition of TMSCN afforded the α-amino nitriles mixture 112a–c. Hydrolysis of 112a,b with concentrate sulfuric acid produced the corresponding α-amino carboxyamides (1S,1'R)-113a,b and (1R,1'R)-114a,b with a predominance of (1S,1'R)-113a,b, and small quantities of the lactones (1S,1'R)-115a,b. On the other hand, hydrolysis of 112c under identical conditions gave the lactone (1S,1'R)-115c as the principal product, which, by treatment with dry ammonia led t o the α-amino carboxyamide (1S,1'R)-113c. Oxidative cleavage of the chiral auxiliary fragment in diastereoisomerically pure (1S,1'R)-113a–c with Pb(OAc)2, followed by acidic hydrolysis and subsequent treatment with propylene oxide, provided the quaternary α-amino acids (S)-111a–c in 60–72% yield (Scheme 27).105
Scheme 27.

Warmuth et al.106 reported the stereoselective synthesis of conformationally constrained lysine derivatives (S,S)-122 and (R,S)-123. In this context, selective monoprotection of one of the carbonyl groups of the diketone 116 using 1,2-ethanedithiol in the presence of a catalytic amount of BF3.OEt2, followed by reaction with (NH4)2CO3 and KCN, gave the corresponding spirohydantoins mixture 117 in almost quantitative yield, which, by cleavage of the thiocetal group with AgNO3 and subsequent treatment with (Boc)2O in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP), produced the N,N'-bis-Boc-protected spirohydantoins mixture 118 in 79% yield. Condensation of 118 with (R)-phenylglycinol, followed by reduction of the imine formed with NaBH(OAc)3 in THF and subsequent chromatographic separation, afforded the diastereoisomerically pure (S,S,1'R)-119 and (R,S,1'R)-120 in 38 and 45% yield, respectively. Oxidative cleavage of chiral auxiliary fragment in (S,S,1'R)-119 with Pb(OAc)2, followed by hydrolysis with HCl and subsequent N-Boc-protection led to (S,S)-121 in 62% yield, which, by hydrolysis and esterification, gave the lysine analogue (S,S)-122 in 41% yield. In a similar way, (R,S,1'R)-120 was transformed into (R,S)-123 (Scheme 28).
Scheme 28.

Condensation of the ketone (±)-124 with (S)-α-MBA in the presence of TiCl4, followed by addition of TMSCN in the presence of AlCl3 afforded, after separation, the corresponding α-amino nitriles 125a and 125b in 31 and 39% yield, respectively. Reaction of the enantiomerically pure ketone (+)-124 under identical conditions, gave the α-amino nitrile 125a in 68% yield as a single stereoisomer. Hydrolysis of the benzyl ester, nitrile and N-debenzylation in the diastereoisomerically pure 125a with HCl and acetic acid at 160 °C, followed by addition of diazomethane gave the dimethyl ester (−)-126 in 49% yield and this was transformed into dibenzyl ester (−)-127 by ester exchange reaction with benzyl alcohol in the presence of Ti(Oi-Pr)4. Finally, cleavage of the benzyl groups in (−)-127 under hydrogenolysis over Pd(OH)2/C produced the conformationally constrained glutamic acid derivative (−)-128 in 68% yield. Under identical conditions, 125b was transformed into (+)-128 (Scheme 29).107
Scheme 29.

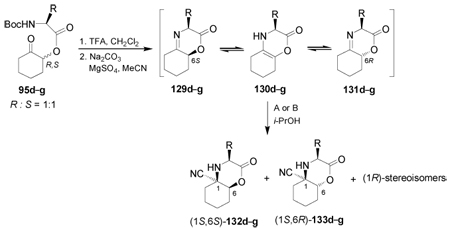
In some cases the intramolecular Strecker reaction has been used as an interesting methodology focused on the synthesis of quaternary α-amino acids. For example, intramolecular condensation of the ketones 95d–g in the presence of TFA afforded the ketimine mixture 129d–g and 131d–g, presumably under a rapid equilibrium through the enamines 130d–g. Addition of NaCN/TFA (condition A) or TMSCN/ZnCl2 (condition B) to the imine mixture 129d–g and 131d–g gave the α-amino nitriles (1S,6S)-132d–g and (1S,6R)-133d–g in moderate to excellent yield and with low to good diastereoselective ratio. The cyanide addition to the ketimines having an alkyl side chain gave a small amount of the (1R)-stereoisomers. The results are summarized in Table 4.108
Table 4.
Formation of α-amino nitriles (1S,6S)-132d–g and (1S,6R)-133d–g from 95d–g.
 | ||||
|---|---|---|---|---|
| Entry | R | Ratio of 129131 | Ratio of 132 : 133 : A: NaCN/TFA |
(1R)-isomers (Yield %) B: TMSCN/ZnCl2 |
| 1 | d = Bn | 50 : 50 | 66 : 34 : ND (96) | 41 : 59 : ND (89) |
| 2 | e = Indma | 50 : 50 | 77 : 23 : ND (56) | 33 : 67: ND (48) |
| 3 | f = i-Bu | 50 : 50 | 39 : 42 : 19 (86) | 18 : 71 : 11 (73) |
| 4 | g = t-Bu | 20 : 80 | 16 : 82 : 02 (88) | 05 : 90 : 05 (80) |
Indm = 2-indolylmethyl fragment. ND = (1R)-isomers were not detected by 1H NMR.
Oxidation of (1S,6S)-132d (R = Bn) with 1,4-diazabicyclo[2.2.2]octane (DABCO) and tert-butyl hypochlorite (t-BuOCl), followed by hydrolysis with concentrate HCl gave the (1R,2S)-1-amino-2-hydroxycyclohexanecarboxylic acid 100a in 92% yield. On the other hand, oxidation of (1S,6R)-133g (R = t-Bu) with ozone and subsequent hydrolysis with concentrate HCl afforded the (1R,2R)-99a in 90% yield (Scheme 30).108
Scheme 30.

This methodology has also been used in the stereoselective synthesis of several quaternary α-amino acids.109
The electrophilic α-amination of carbonyl compounds is a conceptually attractive method for the synthesis of nitrogenated compounds by C–N bond formation.110 In this context, apart from Strecker and related reactions, it has been reported that cyclic quaternary α-amino acids can be alternatively obtained through electrophilic amination reactions starting from molecules containing a carbonyl functionality. For example, Pellacani et al.111 reported that the α-amination of enamine 134 bearing (R)-α-MBA, with ethyl N-[(4-nitrobenzenesulphonyl)oxy]carbamate (NsONHCO2Et) as electrophilic aminating reagent, gave the quaternary α-amino derivative 136 in 95% yield and 60% ee, through the aziridine intermediate 135. The stereochemistry of 136 was not reported (Scheme 31).
Scheme 31.

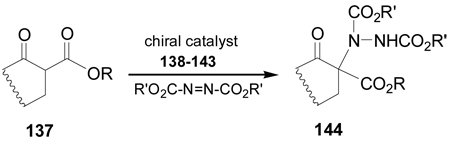
On the other hand, the α-amination of α-keto esters 137 using azodicarboxylates as the electrophilic aminating reagent, in the presence of 5–20 mol% of chiral catalyst such as β-isocupreidine 138112 (a constrained quinidine-derivative), the urea 139,113 cinchonine 140,114 chiral guanidine 141 with a seven-membered-ring structure,115 palladium complex 142,116 and (S,S)-ip-pybox 143,117 afforded the corresponding α-aminated derivatives 144 in good yield and excellent levels of enantioselectivity, which are important precursors of quaternary cyclic α-amino acids. The results are summarized in Table 5.
Table 5.
Enantioselective hydrazination of 137 in the presence of chiral catalysts 138–143.
 | ||||||
|---|---|---|---|---|---|---|
| Substrate | R | R’ | Catalyst | Yield (%) | ee (%), (Config) | Ref. |
|
t-Bu | t-Bu | 138 | 99 | 89 (S) | 112 |
| i-Pr | t-Bu | 139 | 98 | 91 (S) | 113 | |
| Et | Bn | 140 | 95 | 90 (R) | 114 | |
| Et | t-Bu | 141 | 99 | 97 (R) | 115 | |
| Et | Bn | 142 | 73 | 93 (R) | 116 | |
| t-Bu | t-Bu | 143 | 81 | 95 (R) | 117 | |
|
Et | t-Bu | 138 | 86 | 83 (S) | 112 |
| Me | t-Bu | 139 | 52 | 87 (S) | 113 | |
| Et | Bn | 140 | 92 | 84 (R) | 114 | |
| Et | t-Bu | 141 | 99 | 98 (R) | 115 | |
|
t-Bu | t-Bu | 139 | 90 | 90 (S) | 113 |
|
t-Bu | t-Bu | 139 | 93 | 90 (S) | 113 |
| Et | Bn | 142 | 94 | 99 (S) | 116 | |
|
Me | t-Bu | 139 | 99 | 87 (S) | 113 |
| Et | t-Bu | 141 | 99 | 97 (R) | 115 | |
| Me | t-Bu | 142 | 56 | 95 (S) | 116 | |

Asymmetric organocatalysis utilizes organic molecules to induce chirality in various C-C, C–N, and C–O bond-forming reactions.118 For example, the enantioselective catalytic α-amination of the carboxaldehydes 145a–c with dibenzyl azodicarboxylate in the presence of (R)-proline (20 mol%) produced the corresponding α-aminated products 146a–c in good yield and >99% ee. Oxidation of the aldehyde group in 146a,b with NaClO2, followed by esterification with (trimethylsilyl)-diazomethane (TMSCHN2) gave the esters 147a,b in 82% yield (Scheme 32).119
Scheme 32.

Hydrolysis of 147b with pyridine and trifluoroacetic anhydride (TFAA), followed by N-N bond cleavage with SmI2 and subsequent treatment with propylene oxide gave the (S)-AIDA 33 in 70% yield. On the other hand, palladium-catalyzed phosphonylation of 147a furnished the ethyl phosphonate 148 in 77% yield, which, by hydrolysis followed by N-N bond cleavage and subsequent treatment with propylene oxide led to (S)-APICA 36 in 80% yield. (Scheme 33).119
Scheme 33.

Very recently, Shibasaki et al.120 reported the catalytic asymmetric α-amination of the succinimide 149. Thus, reaction of 149 with di-tert-butyl azodicarboxylate in the presence of a catalytic amount of (R)-150 derived from D-valine, La(Oi-Pr)3 and N,N-dimethylacetamide (DMA) in chloroform at 0 °C, afforded the corresponding α-aminated product (R)-151 in quantitative yield and 92% ee (condition A). Identical results were obtained using a catalytic amount of (R)-150, and readily available and much less expensive La(NO3)3 and H-D-Val-Ot-Bu in ethyl acetate at 0 °C.121 Treatment of 151 with HCl(g) in toluene, followed by cleavage of the N-N bond by hydrogenation over Raney-Ni and recrystallization led to the enantiomerically pure (R)-3-amino-3-ethoxycarbonyl-pyrrolidin-2,5-dione 152 in 66% yield (Scheme 34). The quaternary α-amino derivative 152 is a key intermediate in the synthesis of AS-3201 (Ranirestat), a highly potent aldose reductase inhibitor.122
Scheme 34.

Other methodology focused on the stereoselective synthesis of cyclic α-amino acids starting from cyclic carbonyl compounds is the amidation reaction, which, is carried out using nitrogen as the nucleophilic reagent. For example, Satoh et al.123 reported the synthesis of the cyclic quaternary α-amino acids (R)- and (S)-157 through the selective ring-opening of diastereoisomerically pure sulfinyloxiranes (2S,3R,RS)-154 and (2R,3R,RS)-155. In this context, reaction of the β-tetralone with the lithium α-sulfinyl carbanion generated from enantiomerically pure (R)-chloromethyl p-tolyl sulfoxide and lithium diisopropylamide (LDA), afforded the adduct 153 as a mixture of two diastereoisomers in 82% yield, which, by treatment with t-BuOK gave the sulfinyloxiranes (2S,3R,RS)-154 and (2R,3R,RS)-155 in 93% yield and 3:1 dr. These compounds were separated by column chromatography. Treatment of diastereoisomerically pure (2S,3R,RS)-154 with sodium azide, followed by oxidation of the resulting aldehyde intermediate with a methanolic solution of iodine and KOH, produced the azido methyl ester (R)-156 in 82% yield, which, by catalytic hydrogenation, led to enantiomerically pure (R)-157 in 98% yield. In a similar way, (2R,3R,RS)-155 was transformed into (S)-157 (Scheme 35).
Scheme 35.

Recently, Honda et al.124 in order to obtain the (R)-deoxydysibetaine and 4-epi-dysibetaine, they carried out the addition of the lithium salt of chloroform to the ketone 158, which is readily obtained from (R)-4-hydroxyproline, to give the alcohol 159 in 74% yield and high diastereoselectivity, which, by treatment with DBU and sodium azide in the presence of 18-crown-6 under modified Corey-Link reaction,125 gave the dimethyl ester 161 in 56% yield through the intermediate epoxide 160. Reduction of azide group in 161 with H2 over Raney-Ni, followed by protection of resulting primary amine with (Boc)2O, furnished the protected quaternary α-amino acid 162 in 75% yield, and subsequent treatment with SmI2 in THF-HMPA or THF-DMEA afforded the δ-lactam126 (R)-163 in >90% yield (Scheme 36).
Scheme 36.

2.2. Construction of the ring by cyclization reactions
Due to the wide range of methodologies reported to the construction of the cyclic by C-C bond formation, we have decided to organize this section according to the size of the ring to be prepared. Since the Grubbs reaction is common to different cycles it can be considered independently.
Enantiomerically pure epichlorohydrins have been used as bifunctional electrophiles for the asymmetric synthesis of aminocyclopropanecarboxylic acids. For example, treatment of chiral glycine equivalent 165 obtained from enantiopure 164, with 2.1 equiv. of sodium bis(trimethylsilyl)amide (NaHMDS), followed by addition of (R)-epichlorohydrin gave the cyclopropane as derivative 166 in 69% yield. Hydrolysis of 166 afforded the (1R,2R)-1-amino-2-(hydroxymethyl)cyclopropanecarboxylic acid 167 in 59% yield. On the other hand, Swern-oxidation of 166, followed by reductive amination – performed with aniline and NaBH3CN – produced the compound 168 in good yield. Subsequent hydrolysis furnished the diamino acid (1R,2S)-169 in 62% yield. Under identical conditions, the alkylation of 165 with (S)-epichlorohydrin and subsequent reactions produced the quaternary α-amino acids (1R,2S)-170 and (1R,2R)-171 (Scheme 37).127
Scheme 37.

In a similar way, alkylation of chiral glycine equivalent (S)-173, obtained in four steps from carboxylic acid (S)-172, with (R)-epichlorohydrin gave the cyclopropane derivative 174, which under identical conditions to those described in Scheme 37 was transformed into quaternary α-amino acids (1S,2R)-170 (X = OH) and (1S,2S)-171 (X = NHPh). Alkylation of (S)-173 with (S)-epichloro-hydrin afforded the cyclopropane derivative 175, which was transformed into quaternary α-amino acids (1R,2S)-170 and (1R,2R)-171 (Scheme 38).128
Scheme 38.

On the other hand, treatment of 174 with 1-phenyl-3-(trifluoroacetyl)urea 176 and diisopropyl azodicarboxylate (DIAD) under Mitsunobu129 conditions afforded the urea 177 in 65% yield, which, by cleavage of the trifluoroacetyl group with aqueous K2CO3, led to compound 178 in 76% yield. Finally, hydrolysis of 178 produced the quaternary diamino acid (1S,2S)-179 in 24% yield (Scheme 39).128
Scheme 39.

Recently, Acher et al.130 reported the utility of (1S,2R)- and (1R,2R)-1-amino-2-(hydroxy-methyl) cyclopropanecarboxylic acid derivatives 180 and 185131 in the synthesis of (1S,2R)- and (1R,2R)-1-amino-2-phosphonomethylcyclopropanecarboxylic acids 184 and 186 (APCPr),132 which were evaluated at the recombinant group III metabotropic glutamate receptor. Thus, the bromination of (1S,2R)-180 with CBr4 and polymer bond PPh3 in the presence of triethylamine led to bromo derivative (1S,2R)-181 in 56% yield. In order to prevent the cyclopropane cleavage in the next Arbuzov reaction,133 the Boc protective group was replaced by a more electron-withdrawing trifluoroacetyl group, obtaining (1S,2R)-182 in 95% yield. Arbuzov reaction of 182 with trimethyl phosphite gave the corresponding phosphonate (1S,2R)-183 in 51% yield, which, by hydrolysis, followed by ion exchange chromatography, produced the optically pure (1S,2R)-184, APCPr in 96% yield. Under identical conditions, (1R,2R)-185 was transformed into (1R,2R)-186, APCPr (Scheme 40).
Scheme 40.

Carboni et al.134 reported the application of Belokon's Ni(II) complex (S)-187 (a glycine equivalent) in the diastereoselective synthesis of (1S,2R)- and (1R,2S)-allonorocoronamic acid 5a through a double alkylation. In this context, treatment of Ni(II) complex (S)-187 with potassium tert-butoxide followed by addition of sulfate (S)-188 gave the corresponding enolate 189, which, by intramolecular alkylation, afforded the cyclopropane derivative (S,1S,2R)-190 in 70% yield. Acidic hydrolysis of 190 followed by ion exchange chromatography produced the (1S,2R)-allonorcoronamic acid 5a in 96% yield. Alkylation of Ni(II) complex (S)-187 with the sulfate (R)-188, followed by hydrolysis, gave the (1R,2S)-allonorcoronamic acid 5a (Scheme 41).
Scheme 41.

Recently, Fox et al.135 reported the catalytic stereoselective synthesis of (1R,2S)-dehydrocoronamic acid methyl ester 196, through a double alkylation of glycine anion equivalents 191a,b. Thus, asymmetric allylic alkylation of 191a,b with 3,4-epoxy-1-butene in the presence of a catalytic amount of (S,S)-192-(allylPdCl)2 complex, afforded the allyl derivatives mixture 193a,b in quantitative yield and 3:2 dr, which, by mesylation followed by treatment with NaH or potassium tert-butoxide in THF, gave the cyclopropanes 194a,b and dihydroazepines 195a,b. Hydrolysis of the mixture of 194a and 195a followed by separation led to (1R,2S)-196 in 14% yield and 88% ee (Scheme 42).136
Scheme 42.

Reaction of 2,3-epoxy-1,1,1-trifluoropropane 197 with the sodium salt of 198 gave the γ-hydroxy nitrile derivative 199 in 73% yield and 30% de, which, by reaction with tosyl chloride (TsCl) followed by treatment with NaH and subsequent recrystallization, afforded the diastereoisomerically pure cyclopropyl cyanide (1S,2S)-200 in 70% yield. Oxidative degradation of the pyrrole ring of 200 with NaIO4 in the presence of a catalytic amount of RuCl3 produced the α-amino nitrile (1S,2S)-201 in 71% yield. Subsequent hydrolysis with HCl furnished the optically pure trifluoronorcoronamic acid (1S,2S)-202 in 67% yield (Scheme 43).137
Scheme 43.

In a similar way, reaction of 197 with the sodium salt of 203 gave the γ-hydroxy nitrile 204, which, by reaction with TsCl followed by treatment with sodium hydride and subsequent recrystallization, furnished the diastereoisomerically pure cyclopropyl cyanide (1R,2S)-205 in 70% yield. Hydrolysis of nitrile function of 205 with hydrogen peroxide under basic conditions produced the amide (1R,2S)-206 in 79% yield, which, by Hoffman rearrangement138 followed by oxidative degradation of aromatic ring of 207 with NaIO4 in the presence of a catalytic amount of RuCl3, produced the optically pure N-Boc-trifluoronorcoronamic acid (1R,2S)-208 in 30% yield (Scheme 44).137
Scheme 44.

Synthesis of optically pure 1-aminocycloalkanecarboxylic acids starting from α-amino acids is another methodology that has been used. For example, Donkor et al.139 reported the synthesis of all four diastereoisomers of N-Cbz-2,3-methanoleucine from L- and D-valine. Deamination of L-valine with NaNO2/H2SO4 followed by reduction of the carboxylic acid with LiAlH4 produced the corresponding diol (S)-209 in 42% yield. This compound was transformed into cyclic sulfate (S)-210 in 91% yield. Reaction of (S)-210 with the sodium dimethyl malonate afforded the cyclopropane derivative (R)-211 in 84% yield and selective hydrolysis with KOH and subsequent Curtius rearrangement140 with diphenylphosphorazide (DPPA) in the presence of triethylamine (TEA), followed by addition of benzyl alcohol, gave the diprotected α-amino acid (1S,2R)-212 in good yield. Finally, hydrolysis of (1S,2R)-212 with KOH gave (1S,2R)-213 in 91% yield (Scheme 45).
Scheme 45.

On the other hand, selective hydrolysis of (R)-211 with KOH followed by treatment with hydrazine gave the compound 214, which, by reaction with NaNO2/H2SO4 and subsequent esterification with diazomethane provided the azide derivative 215. Curtius rearrangement of 215 followed by hydrolysis with KOH furnished (1R,2R)-216 (Scheme 46). Under indentical conditions the diastereoisomers (1R,2S)-213 and (1S,2S)-216 were obtained from D-valine.139
Scheme 46.

Frick et al.141 reported the stereoselective synthesis of protected 2,3-methano amino acids (1S,2S)-224 and (1R,2R)-225, which are analogues of ornithine and glutamic acid, respectively. Initially, treatment of 218, obtained from epoxide 217,142 with 3,5-dinitrobenzoic acid under Mitsunobu conditions gave the corresponding 3,5-dinitrobenzoate 219 in 91% yield. This compound was reacted with NaH to give the cyclopropane derivative 220 in 81% yield. Cleavage of the benzyl protective group with H2 over Pd/C followed by treatment with TFA furnished the lactone (1R,6R)-221 in 97% yield. The synthesis of the lactone (1S,6S)-221 was reported by Frick et al.142 (Scheme 47).
Scheme 47.

Reaction of (1S,6S)-221142 with ethyl chloroformate followed by treatment with sodium azide and subsequent Curtius rearrangement of the corresponding azide under heating and the addition of benzyl alcohol, produced the N-Cbz-amino derivative (1S,6S)-222 in 90% yield, which, by hydrolysis with LiOH and subsequent esterification with MeI, afforded the protected amino acid (1S,2S)-223 in 93% yield. Reaction of (1S,2S)-223 with methanesulfonyl chloride (MsCl), followed by reaction with sodium azide and subsequent reduction of the azido group with H2 over Pd-BaSO4 in the presence of (Boc)2O, gave the protected 2,3-methanoornithine analogue (1S,2S)-224 in 79% yield (Scheme 48).141
Scheme 48.

Under identical conditions to those described in the scheme 48, (1R,6R)-221 was transformed into (1R,2R)-223 in good yield and subsequent oxidation with pyridine-SO3 followed by treatment with sodium chlorite gave, the methyl 2,3-methanoglutamate derivative (1R,2R)-225 in 80% (Scheme 49).141
Scheme 49.

Chiral didehydroamino acid derivatives from a cyclic glycine template have been used in the stereoselective synthesis of cyclopropane amino acid derivatives through diastereoselective cyclopropanation reactions by using Corey's ylide. For example, reaction of (S)-oxazinone 227, obtained in four steps from (S)-2-hydroxyisovaleric acid 226, with acetaldehyde and propanaldehyde in the presence of K2CO3 and tetrabutylammonium bromide (TBAB), produced the didehydroamino acid derivatives (S)-228a,b with high selectivity in 50–55% yield. Treatment of these compounds with Corey's dimethylsulfoxonium methylide gave the cyclopropanation products 229a,b in moderate yield and 9:1 diastereoisomeric ratio. Hydrolysis of diastereoisomerically pure 229a,b with HCl afforded the (1S,2R)-allo-norcoronamic acid 5a in 60% yield and (1S,2R)-allo-coronamic acid 5b in 67% yield (Scheme 50).143
Scheme 50.

On the other hand, condensation of the protected (S)-pyrazine-2-one 231, obtained in three steps from (S)-α-aminoketone 230, with acetaldehyde and propanaldehyde furnished the Z-α,β-unsaturated compounds (S)-232a,b in 88 and 86% yield, respectively. Treatment of these compounds with Corey's dimethylsulfoxonium methylide gave the cyclopropanation products 233a,b in moderate yield and 23:1 dr. Hydrolysis of diastereoisomerically pure 233a with HCl afforded the enantiomerically pure (1S,2R)-allo-norcoronamic acid 5a in 24% yield (Scheme 51).144
Scheme 51.

Didehydroamino acid derivatives from cyclic glycine templates have also been used in the stereoselective synthesis of cyclopropane amino acid derivatives, through diastereoselective cyclopropanation reactions with phosphorus or sulfur ylides. For example, addition of Me2C(Li)PPh3 and (CD3)2CD2(Li)SO to the dehydroalanine (S)-234 afforded the spiro derivatives (3S,6S)-235a,b in excellent yield and diastereoselectivity (>98:2), which, by treatment with TFA, gave the diketopiperazines (3S,6S)-236a,b in good yield. Finally, hydrolysis of 236a,b with 6 M HCl followed by esterification with SOCl2/MeOH produced the corresponding methyl ester hydrochloride salts (S)-237a,b in excellent yield (Scheme 52).145
Scheme 52.

Enantioselective organocatalytic intermolecular cyclopropanation of protected dehydroalanine 238 with the ammonium ylide generated from reaction of tert-butyl bromoacetate with catalytic amounts of quinine derivatives 239 or 240 and Cs2CO3 as a base, afforded the cyclopropane compound (+)-241 in 97% ee using 239 as a catalyst, and (−)-241 in 90% ee using 240 as a catalyst (Scheme 53).146
Scheme 53.

On the other hand, condensation of (R)-242 with benzylamine, followed by addition of TMSCN afforded a mixture of α-amino nitriles (1R,2S)-243 and (1S,2S)-244 in 75% yield and 85:15 diastereoisomeric ratio. Protection of the amino function of diastereoisomerically pure (1R,2S)-243 with methyl chloroformate (MocCl) gave (1R,2S)-245 in 98% yield, which, by selective cleavage of the tert-butyldimethylsilyl (TBS) protective group with acetic acid and subsequent reaction with PPh3 and chloroform or bromoform, led to the derivatives (1R,2S)-246a,b in excellent yield. Intramolecular alkylation of (1R,2S)-246a with KOH-DMF or potassium tert-butoxide in THF gave the cyclopropylaminonitrile (1S,2R)-247 in 82% yield and >98:2 dr, which, by treatment with hydrogen peroxide under basic conditions, furnished the amide (1S,2R)-248 in 87% yield (Scheme 54).147
Scheme 54.

Wanner et al.148 reported the synthesis of all four stereoisomers of 1-amino-2-(hydroxymethyl)-cyclobutanecarboxylic acid (1S,2S)- and (1R,2R)-13f, (1S,2R)- and (1R,2S)-14f through a double alkylation of the chiral glycine equivalent (R)-173. In this context, reaction of (R)-173 with s-BuLi in THF at −78 °C, followed by addition of but-3-enyl triflate, afforded the alkylated products 249a and 249b in 69% yield and 95.5:4.5 dr. The use of other bases and 4-bromobut-1-ene as the alkylating reagent gave both low yield and diastereoselectivity. Oxidation of the terminal double bond of the butenyl side chain in 249a,b with a catalytic amount of OsO4 in combination with Me3NO as a co-oxidant, followed by selective protection of primary hydroxy group with tert-butyldimethylsilyl chloride (TBSCl) and subsequent selective replacement of secondary hydroxy group with PPh3 and I2, produced the iodohydrins 250a–d in good overall yield and 4:4:1:1 dr. Reaction of 250a–d with phosphazenic base (t-BuP4) gave the corresponding cyclobutane derivatives, which, by treatment with tetrabutylammonium fluoride (TBAF) and subsequent separation by preparative HPLC, furnished the hydroxyl derivatives 251a–d in good yield and 48:31:18:3 dr. Finally, hydrolysis of diastereoisomerically pure 251a led to (1S,2S)-13f in 71% yield. In a similar way, 251b afforded (1S,2R)-14f in 76% yield. The diastereoisomers (1R,2R)-13f and (1R,2S)-14f were obtained from (S)-173 (Scheme 55).
Scheme 55.

Dialkylation of N-(diphenylmethylene)glycine ethyl ester 191a with 1,4-diiodo derivative (S)-253, obtained in 4 steps from (S)-malic acid dimethyl ester 252, afforded the cyclopentane derivative mixture 254 in 2:1 dr. Hydrolysis of 254 with 2 M HCl followed by treatment with (Boc)2O and subsequent chromatographic separation, gave the diprotected quaternary α-amino acids (1S,3S)-255 and (1R,3S)-256 in 36 and 19% yield, respectively, and these were used in the preparation of the thymine derivatives 257 and 258 (Scheme 56).149
Scheme 56.

On the other hand, reduction of dicarboxylic acid (S,S)-259 with LiAlH4, followed by treatment with I2 and PPh3, afforded the diiodide 260 in 83% yield. Dialkylation of ethyl isocianoacetate with 260, followed by hydrolysis and subsequent treatment with (Boc)2O, gave the ethyl 1-N-Boc-aminocyclopentanecarboxylate 261 in 59% yield. Ozonolysis of 261 followed by treatment with NaBH4 and subsequent oxidation of the resulting diol with oxone gave the dicarboxylic acid 262 in 25% overall yield. On the other hand, hydrogenation of 261 over Pd/C produced the diprotected quaternary α-amino acid 263 in 99% yield. Finally, ozonolysis of 261 followed by treatment with benzylamine and subsequent reduction with NaBH3CN produced the compound 264 in 53% yield (Scheme 57).150
Scheme 57.

Ma et al.151 reported the stereoselective synthesis of (S)-1-aminoindane-1,6-dicarboxylic acid 269 and related analogues, through the intramolecular acylation of enantiopure α,α-disubstituted amino acid (S)-266. In this context, the protection of (R)-phenylglycine with methyl chloroformate followed by condensation with benzaldehyde dimethyl acetal, afforded the cis-oxazolidinone (2R,4S)-265, which, by alkylation with tert-butyl bromoacetate followed by hydrolysis, produced the carboxylic acid (S)-266. Reaction of (S)-266 with PCl5 followed by treatment with AlCl3 gave the acylated product (S)-267 in 92% yield, and this was hydrogenated over Pd/C to provide the cyclic α-amino acid (S)-268 in 94% yield. Sequential iodination with I2/Hg(OTf)2, palladium-catalyzed carbonylation under Pd(OAc)2/CO/MeOH conditions, and hydrolysis led to (S)-269 in 40% overall yield (Scheme 58).
Scheme 58.

On the other hand, treatment of (S)-268 with acetyl chloride catalyzed with AlCl3, followed by Baeyer-oxidation using m-chloroperbenzoic acid (m-CPBA) and subsequent hydrolysis, produced the phenol derivative (S)-270 in good yield. Iodination of (S)-270 with I2/pyridine followed by palladium-catalyzed carbonylation using Pd(OAc)2/CO/EtOH afforded the diester (S)-271 in 55% overall yield, and this compound was hydrolysed with TMSI to give the conformationally constrained (S)-272 in 75% yield. Additionally, iodination of (S)-270 followed by palladium-catalyzed phosphonylation with Pd(PPh3)4/HP(O)(OEt)2 afforded the phosphonate (S)-273 in 58% yield, which, by hydrolysis with TMSI, gave the phosphonic acid (S)-274 in 82% yield (Scheme 59).151
Scheme 59.

Asymmetric Strecker reaction of 4-methylbenzaldehyde with (R)-phenylglycinol and NaCN, followed by hydrolysis and subsequent intramolecular esterification with TsOH afforded the corresponding lactone mixture 275 in 57% yield. Alkylation of 275 with tert-butyl bromoacetate and subsequent opening of the lactone ring with Et3N/MeOH, produced the alkylated products (S,R)-276 and (R,R)-277 in 65% yield and 4:1 dr. Cleavage of the chiral auxiliary of diastereo-isomerically pure (S,R)-276 with Pd(OAc)4/NaOAc, followed by acidic hydrolysis and subsequent protection of the resulting amino group with methyl chloroformate led to (S)-278 in 75% yield. Subsequent intramolecular acylation with oxalyl chloride and AlCl3 gave (S)-279 in 93% yield. Benzylic bromination of (S)-279 with N-bromosuccinimide (NBS) catalyzed with 2,2’-azoiso-butyronitrile (AIBN) produced the bromo derivative (S)-280 in 87% yield. This compound was oxidised with Ag2O and AgNO3 and subsequent esterification with MeI/K2CO3 furnished the diester (S)-281 in 64% yield. Reduction of the ketone function of (S)-281 by Pd/C-catalyzed hydrogenation gave (S)-282 in 97% yield, which, by hydrolysis with 6 N HCl and subsequent treatment with propylene oxide afforded the (S)-AIDA 33 in 78% yield (Scheme 60).151
Scheme 60.

The alkylidene carbene C–H insertion is another strategy for the enantioselective synthesis of conformationnaly constrained α-amino acids. For example, reaction of (R)-283, obtained in six steps from L-serine, with lithium (trimethylsilyl)diazomethane generated in situ by treatment of (trimethylsilyl)diazomethane with n-BuLi, gave the alkylidene carbene 284, which, through a 1,5-C–H insertion reaction, produced the spiro compound (S)-285 in 62% yield. Catalytic hydrogenation of (S)-285 over Pd/C afforded the hydrogenated product (1S,3R)-286 in 79% yield as a single diastereoisomer, which, by cleavage of the acetonide function with BF3.2AcOH furnished the alcohol (1S,3R)-287 in 60–70% yield. Finally, Dess-Martin periodinane oxidation of (1S,3R)-287 followed by treatment with sodium chlorite afforded the (1S,3R)-N-Boc-2,5-methanoleucine 288 in 70% yield and >95% ee (scheme 61).152
Scheme 61.

In a similar way, treatment of (R)-289, obtained from L-serine, with lithium (trimethylsilyl)-diazomethane led to a 1,5-C–H insertion reaction that gave the spiro compound (S)-290 in 69% yield. Catalytic hydrogenation of this compound over Pd/C produced (1S,3R)-291 in 79% yield and >10:1 dr. Selective cleavage of TBS protective group in (1S,3R)-291 with HF/MeCN led to diol (1S,3R)-292 in 81% yield, which, by oxidation of hydroxy groups with RuCl3/NaIO4 followed by treatment with HCl and subsequent ion exchange chromatography, afforded the quaternary α-amino acid (1S,3R)-ACPD, 293 in 49% yield (scheme 62).153
Scheme 62.

Treatment of 295a,b, obtained from (R)-glyceraldehyde dimethyl acetal 294, with NBS in MeCN afforded the bicyclic lactone 297 in 42% yield. This product is probably obtained through the oxidation of 295 to the imine intermediate 296 followed by an intramolecular attack of the free OH group on the carbon-nitrogen double bond. Hydrolysis of bicyclic lactone 297 with HCl and subsequent cleavage of the TBS protective group gave the quaternary α-amino acid methyl ester 298 (Scheme 63).154,155
Scheme 63.

Alkylation of commercially available (R)-bislactim ether 299 with the dibromide 300 and n-BuLi in THF at −78 °C afforded the alkylated product (2R,5S)-301 in 95% yield and 93:7 dr, which, by treatment with diluted n-BuLi, furnished the corresponding spiro derivative (2R,5R)-302 in 99% yield as a single diastereoisomer. Hydrolysis of (2R,5R)-302 gave the methyl ester of 2-amino-tetraline-2-carboxylic acid (R)-157 in 98% yield (Scheme 64).156
Scheme 64.

Jørgensen et al.157 reported the first highly enantioselective catalytic alkylation of ketimines, a methodology used for the synthesis of quaternary α-amino acids. In this context, addition of ketene acetal 304 to the ketimine 303 in the presence of a catalytic amount of (R,R)-Ph-pybox-Zn(OTf)2 305 afforded the Mannich base 306 in 98% yield and 93% ee. Selective N-protection of 306 with (Boc)2O gave the compound 307 in 78% yield, which, by treatment with Cs2CO3, produced the δ-lactone 308 in 79% yield by spontaneous cyclization of the resulting phenol function (Scheme 65).
Scheme 65.

On the other hand, the first direct organocatalytic enantioselective Mannich reaction of the ketimine 309 with several aldehydes in the presence of a catalytic amount of the chiral amine 310, afforded the corresponding Mannich products 311a–h and 312a–h in good yield and with moderate to excellent diastereoisomeric ratio (4:1 to >20:1), with a predominance of 311a–h. These compounds can be used as intermediates in the synthesis of quaternary α-amino acids (Scheme 66).158
Scheme 66.

Olefin metathesis is a fundamental chemical reaction involving the rearrangement of carbon–carbon double bonds and can be used to couple, cleave, ring-close, ring-open, or polymerize olefinic molecules. The widely accepted view that olefin metathesis revolutionized the different fields of synthetic chemistry led to the award of the 2005 Nobel Prize in Chemistry to Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock “for the development of the metathesis method in organic synthesis”.159 The ring closing metathesis (RCM) synthetic methodology has also been used in the stereoselective synthesis of different size of cyclic α-amino acids,160 and in this review we present this methodology as an independient section. For example, Ru(II)-catalyzed ring-closing metathesis reaction of dialkylated compounds 313a,b, obtained from bislactim ether (R)-299, in 1,2-dichloroethane (DCE) gave the spiro derivatives 314a,b, which, by dihydroxylation of the five and six-membered-rings with a catalytic amount of OsO4 in combination of morpholine N-oxide (NMO) afforded the diols 315a,b and 316a,b. Treatment of the diols 315a,b with methyl iodide and sodium hydride, followed by hydrolysis with TFA, produced the conformationally constrained cyclic α-amino acids methyl esters 317a,b. Hydrolysis of diol 315a with TFA, followed by acetylation, gave the acetylated α-amino acid methyl ester 318a (Scheme 67).161
Scheme 67.

Ring closing metathesis of dialkylated derivatives 313a–d in the presence of a catalytic amount of Grubbs second generation catalyst PhCH=RuCl2(IMes)(PCy3) under microwave assisted heating, gave the corresponding spiro compounds with five-, six- and seven-membered rings containing a double bond 314a–d in 63–99% yield. Hydrolysis of the bis-lactim ether of 314a–d with TFA at room temperature or under microwave conditions, followed by treatment with (Boc)2O afforded the N-Boc protected quaternary amino acid ethyl esters 319a–d in good yield, which, by basic hydrolysis under microwave assisted heating, produced the amino acids 320a–d in 76–93% yield (Scheme 68).162
Scheme 68.

Cascade Ru(II)-catalyzed ring closing metathesis reaction of 321a,b produced the RCM products 322a,b in excellent yield. In a similar way, reaction of 323a–c, obtained from 321a,b, under identical conditions furnished the conformationally constrained amino acids 324a–c in good yield (Scheme 69).163,164
Scheme 69.

Diels-Alder reaction of 324a,b,d with diethyl acetylenedicarboxylate (DEAD) followed by aromatization with MnO2 gave the conformationally constrained α-amino acids 325a,b,d in good yield (Scheme 70).164
Scheme 70.

On the other hand, ring closing metathesis reaction of triyne 327, readily obtained from 326, in the presence of a catalytic amount of PhCH=RuCl2(PCy3)2 in toluene at 85 °C gave the product (2R,7R)-328 in 58% yield.165 Ring closing metathesis reaction of triyne 326 in the presence of a catalytic amount of PhCH=RuCl2(PCy3)2 in toluene at 85 °C gave 329 in 90% yield,165,166 and a quantitative yield was obtained when the reaction of 326 was carried out under microwave assisted heating. Identical results were obtained using PhCH=RuCl2(IMes)(PCy3)2 as a catalyst.166 Hydrolysis of 329 with 0.1 M TFA gave the constrained α-amino acid methyl ester (2R,7R)-330 in 35% yield (Scheme 71).167
Scheme 71.

Ring closing metathesis reaction of tetraene 331 in the presence of 10 mol% of PhCH=RuCl2(PCy3)2 in toluene at 85 °C in order to obtain the spirane 332 was unsatisfactory, probably due to a sterically congested substrate. The more reactive PhCH=RuCl2(IMes)(PCy3) catalyst also failed to effect the spiroannulation of 331. However, ring closing metathesis reaction of less bulky tetraene 333, obtained by hydrolysis of 331, under identical conditions gave the spiranes 334 and 335 in 73% yield and 3:2 isomeric ratio (Scheme 72).168
Scheme 72.

On the other hand, Ru(II)-catalyzed ring closing metathesis reaction of diastereoisomerically pure 336 gave the cyclic α-amino acid methyl ester 337 in 96% yield (Scheme 73).169
Scheme 73.

Chemoselective allylation of imino ester 338 with allylzinc bromide afforded the diene 339 in 95% yield as a single diastereoisomer, which, by ring closing metathesis reaction in the presence of Grubbs first generation catalyst PhCH=RuCl2(PCy3)2, gave the cyclic amino ester 340 in 92% yield and >98% de. Cleavage of the benzyl group in 340 under H2/Pd(OH)2 conditions provided the amino ester 341 in almost quantitative yield, which, by treatment with TBAF and subsequent ion-exchange chromatography, furnished the quaternary α-amino acid 342 in 70% yield (Scheme 74).170
Scheme 74.

Undheim et al.171 reported the stereoselective synthesis of rigidified homoserine analogues 350 and 351 through the ring closing metathesis reaction. In this context, reaction of hydroxy derivative 343 in the presence of a catalytic amount of PhCH=RuCl2(PCy3)2 in DCE at 65 °C gave the spiro compound 344 in 72% yield. Swern oxidation of 344 produced the α,β-unsaturated ketone 346 in 74% yield. On the other hand, oxidation of 343 under Swern conditions furnished the ketone 345 in 79% yield, which, by ring closing metathesis reaction in the presence of a catalytic amount of PhCH=RuCl2(PCy3)2 in benzene at 70 °C, afforded the spiro derivative 346 in 37% yield. Conjugate addition of lithium dimethylcuprate to 346 furnished 347 in 91% yield and high diastereoselectivity (Scheme 75).
Scheme 75.

Reduction of the carbonyl group of 347 with NaBH4 in methanol afforded the alcohols 348 and 349 in 37 and 57% yield, respectively. Hydrolysis of diastereoisomerically pure 348 with 0.1 M TFA gave the amino ester 350 in 38% yield, whereas the hydrolysis of 349 under identical conditions afforded the dipeptide 351 in 77% yield (Scheme 76).171
Scheme 76.

Møller and Undheim172 reported the synthesis of spiro derivatives 354 and 357 by palladium-mediated 5-exo-trig-spiroannulation and these compounds are precursors of functionalized cyclic quaternary α-amino acids. Thus, the lithiation of 352 followed by addition of 2,3-dibromopropene gave the diene 353 in 60% yield and >98% de and treatment of 353 with a catalytic amount of Pb(OAc)2 in the presence of PPh3/Ag2CO3 afforded the spiro compound 354 in 60% yield. In a similar way, reaction of 355, obtained from 352,172b produced the spiro derivative 356 in 60% yield and treatment with a catalytic amount of NiCl2(dppp) and MeMgBr led to compound 357 in 64% yield (Scheme 77).
Scheme 77.

On the other hand, aldol reaction of 358a,b using Cs2CO3 as a base in acetonitrile, afforded the spiroannulated compounds 359a,b in 49 and 63% yield, respectively, and subsequent hydrolysis with 0.1 M TFA gave the amino esters 360a,b in 56 and 59% yield, respectively (Scheme 78). Using this methodology the α-amino acid methyl esters 360c–f were obtained from the appropriate substrates.173
Scheme 78.

2.3. Cicloadditions and related reactions
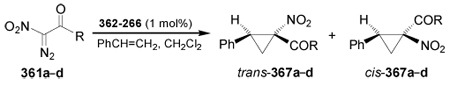
Direct incorporation of an “amino acid synthetic equivalent” into an alkene by transition metal catalyzed diazo decomposition has also been used for the synthesis of quaternary 1-amino-cyclopropanecarboxylic acids.174,175 For example, the asymmetric catalytic cyclopropanation of styrene with α-nitro-α-diazocarbonyl compounds 361a–d in the presence of a catalytic amount of 362–366 as a chiral catalyst, afforded the cyclopropane derivatives trans- and cis-367a–d in good selectivity trans:cis, but with low enantioselectivity.176 The results are summarized in Table 6.
Table 6.
Asymmetric cyclopropanation of styrene with α-nitro-α-diazocarbonyl compounds 361a–d.
 | ||||||
|---|---|---|---|---|---|---|
| Substrate | Catalyst | Aditive | Yield (%) |
Ratio (trans:cis) |
% ee (trans) |
% ee (cis) |
| 361a; R = OMe | 362 | -- | 75 | 86 : 14 | 28 | 13 |
| 361b; R = OEt | 362 | -- | 72 | 83 : 17 | 30 | 0 |
| 361c; R = Ot-Bu | 362 | -- | 68 | 68 : 32 | 41 | 6 |
| 361d; R = Ph | 362 | -- | 64 | 39 : 61 | 31 | 13 |
| 361b; R = OEt | 363 | -- | 71 | 75 : 25 | 13 | 16 |
| 361b; R = OEt | 364 | -- | 76 | 86 : 14 | 33 | 0 |
| 361b; R = OEt | 365a | -- | 89 | 89 : 11 | 2 | 17 |
| 361b; R = OEt | 365b | -- | 74 | 79 : 21 | 8 | 10 |
| 361a; R = OMea | 366a | (BzO)2 | 27 | 90 : 10 | nd | nd |
| 361a; R = OMea | 366a | EDA (20%) | 55 | 90 : 10 | 72 | 51 |
| 361a; R = OMea | 366a | EDA (10%) | 52 | 90 : 10 | 66 | 49 |
| 361a; R = OMeb | 366a | PhNHNH2 | 39 | 90 : 10 | 70 | 49 |
| 361a; R = OMea | 366b | EDA (10%) | 16 | 95 : 05 | 68 | nd |
| 361a; R = OMea | 366c | EDA (10%) | 7 | 95 : 05 | 63 | nd |
in the presence of 5 mol% of Cu(MeCN)4PF6.
Cu(II)OTf2 was used as the copper source

Moreau and Charette177 reported the catalytic asymmetric cyclopropanation of styrene with iodonium ylides derived from nitroacetates. For example, reaction of phenyliodonium with methyl nitroacetate gave the corresponding phenyliodonium ylide 368, which, by cyclopropanation reaction with styrene in the presence of a catalytic amount of isopropilidene bis(4-phenyl-2-oxazoline) 366a and AgSbF6, afforded the methyl 1-nitrocyclopropyl carboxylate 367a in 79% yield and excellent diastereo- and enantioselectivity (similar results were obtained using others alkyl and aryl alkenes). Reduction of the nitro group of 367a with Zn/HCl in 2-propanol furnished the aminoester 369 in 89% yield (Scheme 79).
Scheme 79.

On the other hand, reaction of chiral carbenes 370a,b with terminal olefins in toluene under reflux produced the corresponding cyclopropanes 371a,b and 372a,b as a mixture of both cis diastereoisomers in low yield and 2:1 and 1.5:1 diastereoisomeric ratio, respectively, with a predominance of 371a,b (Scheme 80).178
Scheme 80.

Reaction of (Z)-373 with (−)-menthol in the presence of bis-(dibutylchlorotin)oxide gave the aminoacrylate (Z)-374 in 78% yield and subsequent treatment with diazomethane in dichloromethane produced the Δ1-pyrazolines 375a and 375b in 93% yield and 1.8:1 dr, (reversal of diastereoselectivity was observed when (+)-menthol was used as chiral auxiliary). Heating of diastereoisomerically pure 375a and 375b at 150 °C afforded the constrained cysteines derivatives (1S,2S)- and (1R,2R)-376, respectively, in good yield and diastereoselectivity. Saponification of (1S,2S)-376 with NaOH in methanol gave the carboxylic acid (1S,2S)-377 in 44% yield (Scheme 81).179
Scheme 81.

1,3-Dipolar cycloaddition of α,β-unsaturated compound 379, obtained from Horner-Wadsworth-Emmons reaction of (S)-294 and the phosphonate 378, with diazomethane followed by photolysis of the resultant pyrazoline gave the cyclopropane derivative 380 in 87% yield.180 Hydrolysis of the acetonide in 380 with HCl afforded the corresponding diol, which was oxidised with NaIO4 produced the aldehyde (1R,2S)-381 in good yield. Reduction of 381 with NaBH4 and subsequent saponification of methyl ester gave (1R,2S)-382 in good yield. Finally, cleavage of the N-Boc protective group in (1R,2S)-382 followed by treatment with propylene oxide provided the α-aminocyclopropanecarboxylic acid (1R,2S)-170 in excellent yield (Scheme 82).181
Scheme 82.

Reaction of aldehyde (1S,2R)-381 with N-methylglycine and [60]fullerene afforded the fulleropyrrolidine 384 in 25% yield, through the 1,3-dipolar cycloaddition of the in situ produced azomethine ylide 383. Cleavage of the N-Boc protective group of 384 with TMSI in chloroform led to compound 385 in 89% yield (Scheme 83).182
Scheme 83.

On the other hand, 1,3-dipolar cycloaddition of diazomethane to α,β-unsaturated compound 386 followed by photolysis of the resultan pyrazoline gave the cyclopropane derivative 387 in 48% yield, which, by saponification of the methyl ester and cleavage of the acetonide using pyridinium p-toluenesulfonic acid (PPTS), gave the keto amino acid (1R,2R,1'R,3'R)-388 in 35% yield. In a similar way, reaction of 389 with diazomethane followed by photolysis produced the diprotected quaternary α-amino acid (1S,2S,1'S,3'R)-390 in 45% yield (Scheme 84).183
Scheme 84.

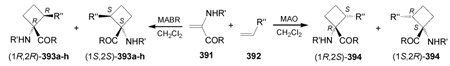
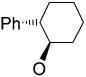
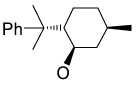
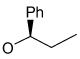
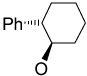
Recently, Avenoza et al.184 reported the asymmetric [2+2] cycloaddition of 2-acylaminoacrylates 391 with donor olefins 392 in the presence of a catalytic amount of sterically hindered aluminum aryloxides, such as methylaluminum bis(4-bromo-2,6-di-tert-butyl phenoxide) (MABR) and methylaluminoxane (MAO) as a Lewis acid. These reactions gave the constrained protected serine analogues c4Ser(OBn) 393a–h and 394.185 The results are summarized in Table 7. The best diastereoselectivity was obtained in the reaction of vinyl ether bearing (1R,2S)-2-phenylcyclohexyl gragment as a chiral auxiliary (entries 3, 6 and 9).
Table 7.
[2+2] Cycloaddition of dehydroamino esters 391 with vinyl ethers 392.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Product | R | R’ | R’’ | Yield (%) | (1R,2R) : (1S,2S) |
| 1 | 393a |
|
COMe | OEt | 13 | 15 : 85 |
| 2 | 393b |
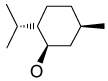
|
COMe | OEt | 30 | 40 : 60 |
| 3 | 393c | OMe | COMe |
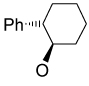
|
45 | >98 : 02 |
| 4 | 393d | OMe | COMe |
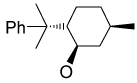
|
70 | 76 : 24 |
| 5 | 393e | OMe | COMe |
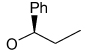
|
55 | 60 : 40 |
| 6 | 393f | OMe | COCF3 |
|
18 | >98 : 02 |
| 7 | 393g | OMe | COCF3 |
|
56 | 80 : 20 |
| 8 | 393h | OMe | COCF3 |
|
30 | 60 : 40 |
| 9 | 394 | OMe | COMe |
|
62 | 02 : 98a |
The configuration of the products was (1R,2S) and (1S,2R) with the predominance of (1S,2R).
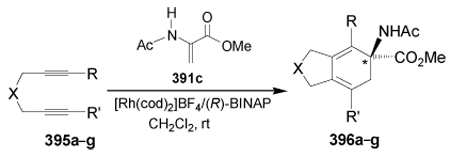
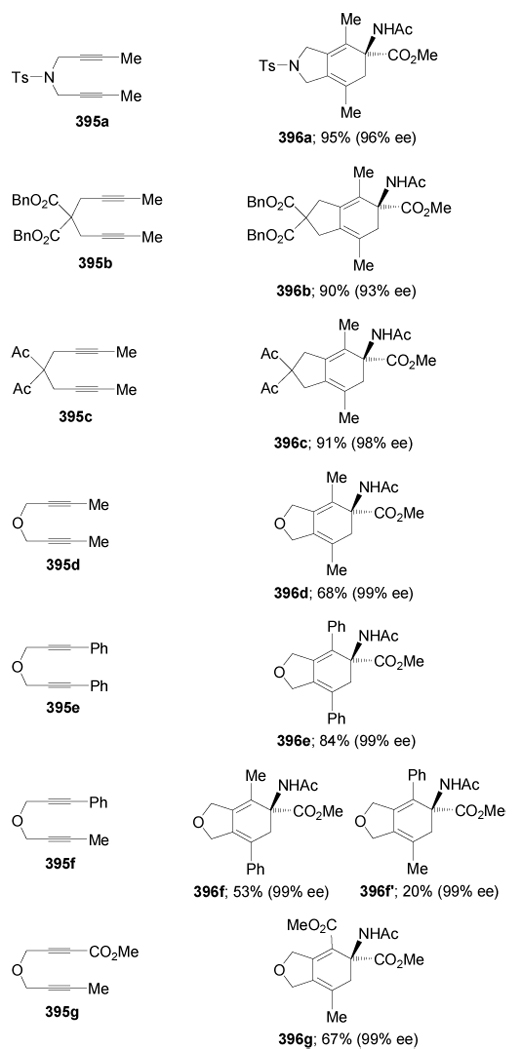
Recently, Tanaka et al.186 reported the synthesis of α,α-disubstituted α-amino esters 396a–g by Rh-catalyzed [2+2+2] cycloaddition of 1,6-diynes 395a–g with protected dehydroamino ester 391c. In all cases the compounds 396a–g were obtained in good yield and with good enantioselectivity and, in the case of unsymmetrical 1,6-diynes, moderate regioselectivity was observed. The results are summarized in Table 8.
Table 8.
Rh(I)/(R)-BINAP-catalyzed enantioselective [2+2+2] cycloaddtion of 395a–g with 391c.
 | |
|---|---|
| Sustrate | Product, % yield (% ee) |
 | |
Pyne et al.187 reported the synthesis of conformationally constrained cyclopentenylglutamate analogues in a regioselective and diastereoselective manner using a formal [3+2] cycloaddition reaction of chiral dehydroamino esters. For example, [3+2] cycloaddition of ylide 398a generated in situ from ethyl 2,3-dienoate 397a, with the chiral dehydroamino ester (R)-399 gave the mixture of the two regioisomers 400a and 401a in 17 and 49% yield, respectively, after column chromatographic separation. In a similar way, cycloaddition of 398b,c, obtained from 397a,b, with (R)-399 afforded the spiro compounds 400b,c in 38 and 78% yield, respectively, as a single diastereoisomers. Hydrolysis of optically pure 400a–c with HCl followed by ion-exchange chromatography and subsequent treatment with HCl produced the conformationally constrained amino acids 402a–c in good yield as chlorohydrate salt. In a similar way, 401a was transformed into the quaternary α-amino acid (S)-403 (Scheme 85).
Scheme 85.

Reaction of dehydroamino ester 404 with ethyl butynoate 405 in the presence of PPh3 gave the cycloadducts 406 and 407 in 87% yield and 60:40 dr, which, by successive selective hydrolysis of the N=CPh2 group, N-Cbz protection, preparative HPLC separation and hydrolysis of the esters, afforded the cyclic glutamic acid analogues (R)- and (S)-402a in good yield (Scheme 86).187
Scheme 86.

Diels-Alder reaction of (S)-228c with cyclopentadiene at room temperature gave, after flash chromatography, the cycloadduct endo-408 in 85% yield and 15% of other diastereoisomers, and with cyclohexa-1,3-diene at 90 °C afforded the cycloadduct endo-409 in 88% yield and 12% of other diastereoisomers. Hydrolysis of the imine moiety of the cycloadducts endo-408 and endo-409, followed by catalytic hydrogenation of double bound C=C and subsequent hydrolysis of the ester function with 6 N HCl, produced the constrained α-amino acids (S)-410 and (S)-411, respectively, in good yield (Scheme 87).144,188
Scheme 87.

In a similar way, Diels-Alder reaction of (S)-232c with cyclopentadiene and cyclohexa-1,3-diene gave, after flash chromatography, the cycloadducts endo-412 and endo-413 as the main diastereoisomers, respectively. Catalytic hydrogenation of these compounds over Pd/C, followed by hydrolysis with 6 N HCl and subsequent ion-exchange chromatography, furnished the α-amino acids (S)-410 and (S)-411 in moderate yield (Scheme 88).144
Scheme 88.

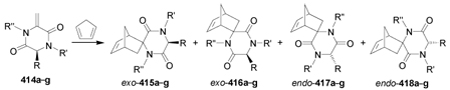
Diels-Alder cycloaddition of chiral methylene piperazine-2,5-diones 414a–g with cyclopentadiene gave the four diastereoisomers exo-415a–g, exo-416a–g, endo-417a–g and endo-418a–g in low to moderate yield and with good exo/endo selectivity. The results are summarized in Table 9.189
Table 9.
Diels-Alder cycloaddition of 414a–g with cyclopentadiene.
 | |||||
|---|---|---|---|---|---|
| dienophile | R | R' | R" | Yield (%) | 415 : 416 : 417 : 418 |
| 414a | Me | Ac | H | 60 | 10 : 17 : 1 : 1 |
| 414b | Me | Ac | Ac | 50 | trace : 1.2 : trace : 1 |
| 414c | Me | Me | H | <10 | not determined |
| 414d | i-Pr | Ac | H | 60 | 12 : 1.4 : 1 : 0 |
| 414e | i-Pr | Ac | Ac | trace | not determined |
| 414f | 4-AcOC6H4CH2 | Ac | H | 60 | 7 : 1 : 1 : 0 |
| 414g | 4-AcOC6H4CH2 | Ac | Ac | trace | not determined |
Diels-Alder cycloaddition of chiral acrylates 419a,b, bearing (+)- or (−)-menthyl as a chiral auxiliary, with cyclopentadiene in the presence of EtAlCl2 or Mg(ClO4)2 under thermal or ultrasound conditions, gave the four diastereoisomers exo-420a,b, exo-421a,b, endo-422a,b and endo-423a,b in moderate to good yield, good exo/endo selectivity, and good enantioselectivity. The results are summarised in Table 10.190
Table 10.
Asymmetric Diels-Alder cycloaddition of 419a,b with cyclopentadiene.
 | ||||||
|---|---|---|---|---|---|---|
| R | Conditions | Yield (%) | exo : endo | 420 : 421 (de) | 422 : 423 (de) | |
| 419a | (−)-menthyl | EtAlCl2, ))) | 36 | 70 : 30 | 23 : 77 (54) | 87 : 13 (74) |
| 419b | (+)-menthyl | Mg(ClO4)2, Δ | 50 | 77 : 23 | 90 : 10 (80) | 07 : 93 (86) |
| 419a | (−)-menthyl | Mg(ClO4)2, Δ | 50 | 77 : 23 | 10 : 90 (80) | 93 : 07 (86) |
| 419b | (+)-menthyl | Mg(ClO4)2, ))) | 90 | 78 : 22 | 89 : 11 (78) | 07 : 93 (86) |
| 419a | (−)-menthyl | Mg(ClO4)2, ))) | 90 | 77 : 23 | 10 : 90 (80) | 92 : 08 (84) |
On the other hand, enantioselective Diels-Alder cycloaddition of achiral acrylate 424 with cyclopentadiene in the presence of a catalytic amount of chiral ligands 366a, 425, 426, and 427, and Mg(ClO4)2 or Ce(OTf)4.H2O, gave the spiro compounds mixture of two endo-428a,b and two exo-429a,b, both with poor enantioselectivity.190,191 The results are summarised in Table 9.

Recently, Pellegrino et al.192 reported that the Diels-Alder cycloaddition of acylaminoacrylate 430, bearing the (−)-8-phenylmenthyl group as a chiral auxiliary, with cyclopentadiene in the presence of a catalytic amount of Mg(ClO4)2 under ultrasound conditions gave the adducts exo-431a,b and endo-432a,b in 87% yield and 7:1 dr, with a predominance of exo-431a (only trace amount of the second exo-431b (0.3%) and endo432b (0.9%) isomers were detected). Hydrogenation of the C=C double bond of diastereoisomerically pure exo-431 followed by selective hydrolysis with Na2CO3 and subsequent oxidation of alcohol group, led to the β-keto ester exo-433 in excellent yield. This compound was used in the synthesis of cis-3-carboxycyclopentylglycine (1S,3R,1'S)-434a and its epimer (1S,3R,1'R)-434b (Scheme 89).
Scheme 89.

The same authors193 reported that Diels-Alder cycloaddition of chiral aminoacrylate 391a, bearing the (−)-8-phenylmenthyl group as a chiral auxiliary, with cyclopentadiene in the presence of a catalytic amount of Mg(ClO4)2 under ultrasound conditions. This reaction gave the norbornenes exo-435 and endo-436 in 84% yield, a ratio of 83:17 and with high diastereoselectivity (exo 97% and endo 96%). Selective hydrolysis of major exo-435 produced the enantiopure constrained α-amino acid exo-437 in 79% yield (Scheme 90).
Scheme 90.

On the other hand, oxidative cleavage of the C=C double bond of norbornene exo-435 with potassium permanganate furnished (1S,2R,4S)-438 in 81% yield and selective hydrolysis of the ester function under basic conditions provided the tricarboxylic acid (1S,2R,4S)-439 in 80% yield. Finally, hydrolysis of amide function of (1S,2R,4S)-439 with 6 M HCl gave the quaternary α-amino acid (1S,2R,4S)-440 in 83% yield. In a similar way, (1R,2S,4R)-endo-436 was transformed into quaternary α-amino acid (1S,2S,4R)-441 (Scheme 91).193,194
Scheme 91.

Dihydroxylation of C=C double bond in the norbornene exo-435 with NMO in the presence of a catalytic amount of osmium tetroxide afforded the diol exo-442,195 which, by cleavage of C5−C6 bond with sodium periodate, gave the bisaldehyde (1S,2R,4S)-443. Reductive amination of bisaldehyde 443 with p-methoxybenzylamine (PMBNH2) and sodium triacetoxyborohydride as a reducing agent, provided the derivative (1S,5S,6S)-exo-444, which, by treatment with sodium in methanol produced the constrained α-amino acid (1S,5S,6S)-exo-445 in 57% overall yield. In a similar way, (1R,2S,4R)-endo-436 was transformed into quaternary α-amino acid (1R,5R,6S)-endo-446 (Scheme 92).196
Scheme 92.

On the other hand, reductive amination of bisaldehydes (±)-447a,b using (R)-α-MBA and sodium triacetoxyborohydride afforded, after chromatographic separation, the azabiciclo derivatives (1R,5R,6R,1'R)-exo-448a, (1S,5S,6S,1'R)-exo-448b, (1R,5R,6S,1'R)-endo-449a and (1S,5S,6R,1'R)-endo-449b in 25%, 28%, 12% and 10% yield, respectively. Cleavage of the benzyl group by hydrogenolysis over Pd/C on diastereoisomerically pure exo-488a,b and endo-489a,b, followed by hydrolysis with 6 N HCl gave the constrained α-amino acids (1R,5R,6R)-exo-450, (1S,5S,6S)-exo-450, (1R,5R,6S)-exo-451 and (1S,5S,6R)-exo-451 in good yield (Scheme 93).197
Scheme 93.

2.4. Resolution procedures
2.4.1. Chemical resolution
Mash et al.198 reported the synthesis of 2-amino-4-bromo-7-methoxyindane-2-carboxylic acid (S)-461 by chemical resolution. In this context, a double alkylation of ethyl glycinate 452a with 2,3-bis(bromomethyl)-4-bromoanisole 453 afforded the racemic compound (±)-454, which, by hydrolysis of imine function furnished the corresponding α-amino ester (±)-455 in 33% yield. Coupling of (±)-455 with the N-Boc-phenylalanine (S)-456 in the presence of benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP) gave a 50:50 mixture of the dipeptides (R,S)-457 and (S,S)-458 in 93% yield and these were separated by column chromatography. Cleavage of the N-Boc protective group of diastereoisomerically pure (S,S)-458 with TFA produced the dipeptide (S,S)-459, which was treated with phenylisothiocyanate and triethylamine to furnish the corresponding thiourea (S,S)-460 in 71% yield. Finally, hydrolysis of (S,S)-460 with HCl provided the quaternary α-amino acid (S)-461 in 83% yield (Scheme 94). The α-amino acid (S)-461 has been used in the synthesis of piperazine-2,5-diones.
Scheme 94.

Monn et al.199 reported the synthesis of heterobicyclic α-amino acids (−)- and (+)-465a,b by resolution and these compounds were evaluated as agonist for group II mGlu receptors. In this context, reaction of furan or thiophene with ethyl diazoacetate in the presence of Rh2(OAc)4 produced the bicyclic adducts (±)-462a,b in (20–40%) yield. Reaction of (±)-462a,b with (NH4)2CO3 and KCN, followed by saponification, gave the (±)-carboxylic acids 463a,b in 72% yield. These compounds were resolved by selective crystallization of either the (R)- or (S)-phenylglycinol salts (−)-464 or (+)-464, respectively. Hydrolysis of diastereoisomerically pure salts (−)-464a,b and (+)-464a,b followed by ion-exchange chromatography furnished the optically pure (−)- and (+)-465a,b in good yield (Scheme 95).
Scheme 95.

The diastereoisomer (−)-465b has been transformed into sulfoxides 469 and 470 as well as sulfone 472, and these compounds were evaluated as potent, selective, and orally bioavailable agonist for mGlu2/3 receptors. Thus, esterification of (−)-465b with thionyl chloride in methanol followed by treatment with (Boc)2O gave the methyl ester 466 in 79% yield, which, by oxidation with m-CPBA afforded the sulfoxides mixture 467 and 468 in 5% and 85% yield, respectively. Hydrolysis of diastereoisomerically pure 467 and 468 produced the quaternary α-amino acids 469 and 470 in 65% and 69% yield, respectively. On the other hand, oxidation of sulfoxide function in 467 with m-CPBA provided the corresponding sulfone 471 in 84%, which, by hydrolysis afforded the quaternary α-amino acid 472 in 71% yield (Scheme 96).200
Scheme 96.

On the other hand, Bucherer-Bergs reaction of 6-bromo-2-tetralone 473 with (NH4)2CO3 and KCN gave the spirohydantoin 474 in 81% yield, which, by cleavage of the hydantoin ring and esterification, afforded the methyl (±)-2-amino-6-bromotetraline-2-carboxylate 475 in 54% yield. Resolution of (±)-475 as the L-mandelic acid salt produced the ammonium salt (S,S)-476 in 25% yield and treatment with (Boc)2O/Et3N, followed by basic hydrolysis, produced the constrained N-Boc α-amino acid (S)-477 in 77% yield. This compound was converted in four steps into (2S)-N-Boc-amino-6-(diethylphosphono)tetraline-2-carboxylic acid 478 (Scheme 97).201
Scheme 97.

Treatment of (±)-480, which is readily obtained from (±)-479, with (S)-phenylalanine cyclohexylamide 481 in N-methylpyrrolidin-2-one (NMP) at 90 °C, followed by column chromatography separation, produced the diastereoisomerically pure (1S,2S,1'S)-482a and (1R,2R,1'S)-482b in 36 and 35% yield, respectively. Subsequent hydrolysis and treatment with propylene oxide furnished the 1-amino-2-hydroxycyclohexanecarboxylic acid (1S,2S)- and (1R,2R)-99a in good yield (Scheme 98).202
Scheme 98.

In a similar way, treatment of (±)-483 with (S)-2-acetoxypropanoyl chloride (S)-484 in the presence of triethylamine, followed by column chromatography separation, gave the diastereoisomerically pure (1S,2R,1'S)-485 and (1R,2S,1'S)-486 in 40 and 50% yield, respectively. Hydrogenation of (1S,2R,1'S)-485 and (1R,2S,1'S)-486 over Pt/C, followed by hydrolysis and subsequent treatment with propylene oxide, led to (1S,2R)- and (1R,2S)-100a in good yield (Scheme 99).202
Scheme 99.

Recently, Gelmi et al.194 reported the synthesis of the four diastereoisomers of constrained α-amino acids exo-437 and exo-487 by resolution. In this context, treatment of (±)-exo-437 with (R)-α-MBA gave the corresponding diastereoisomeric salts, hydrolysis of which afforded, after crystallization, the enantiomerically pure (1R,2R,4R)-437 and (1S,2S,4S)-437 in 31 and 37% yield, respectively. In a similar way, the resolution of (±)-endo-487 gave the enantiomerically pure (1R,2S,4R)-487 and (1S,2R,4S)-487 in 42 and 40% yield, respectively (Scheme 100).
Scheme 100.

Reaction of N-protected amino acids RCO-Bin-OH (±)-488a,b with (S)-481 in the presence of N-hydroxybenzotriazole (BtOH) and N-ethyl-N'-dimethylaminopropylcarbodiimide hydrochloride (EDC) in CH2Cl2, followed by column chromatography separation, afforded the diastereoisomerically pure (R,S)-489a,b and (S,S)-490a,b in good yield. Hydrolysis of diastereoisomerically pure (R,S)-489a,b and (S,S)-490a,b, followed by esterification with MeOH/HCl, furnished the H-Bin-OMe (R)- and (S)-491 in good yield (Scheme 101).203
Scheme 101.

On the other hand, reaction of racemic diesters (±)-cis-492 with commercially available (1S,2S,5S)-2-hydroxy-3-pinanone 493 in the presence of BF3.OEt2 afforded the corresponding Schiff bases (S)-cis and (R)-cis-494. Crystallization of the diastereoisomeric mixture gave (S)-cis-494 as a single diastereoisomer. The remaining diastereoisomer (R)-cis-494 could not be isolated from mother liquor either by crystallization or by chromatography on silica gel. However, (R)-cis-494 could be converted into the (S)-cis-494 diastereoisomer by thermal equilibration. Hydrolysis of diastereoisomerically pure (S)-cis-494 followed by treatment with (Boc)2O furnished the enantiomerically pure N-Boc-protected methyl ester (S)-cis-495. Under identical conditions, N-Boc-protected methyl ester (S)-trans-497 was obtained from (±)-trans-496 (Scheme 102).204
Scheme 102.

Treatment of racemic Boc-[OH]2-Bip-OMe (±)-498 with the ditosylate (R)-499 in the presence of Cs2CO3 in DMF at 60 °C gave the methyl esters N-Boc-[20-C-6]-(R)-Bip-OMe (R,R)-500 and N-Boc-[20-C-6]-(S)-Bip-OMe (R,S)-501 in 25 and 26% yield, respectively. Cleavage of ether function of (R,R)-500 with large excess of BBr3 followed by esterification with thionyl chloride and methanol, produced the (R)-binaphthol and H-[OH]2-Bip-OMe (R)-502 (Scheme 103).205,206
Scheme 103.

On the other hand, epoxidation of 319a with m-CPBA afforded the epoxides 503 and 504 in 59% yield and 85:15 dr. Desymetrization of the major epoxide isomer 503 with s-BuLi in the presence of (−)-sparteine gave the allyl alcohol (1S,4R)-505 in 14% yield and 33% ee, which, by hydrogenation over Pd/C provided the alcohol (1S,3R)-256 in 71% yield. Treatment of (1S,3R)-256 with acetic acid under Mitsunobu conditions produced the alcohol (1S,3S)-255 in 58% yield and subsequent mesylation followed by reaction with NaCN and hydrolysis with 6 M HCl led to (1S,3R)-ACPD 293 in 45% yield. In a similar way, (1S,3R)-256 was transformed into (1S,3S)-ACPD 506 in 34% overall yield (Scheme 104).207
Scheme 104.

Recently, Varie et al.208 reported a pilot-plant desymetrization of the cyclic meso-epoxide 507a using a chiral lithium amide prepared from (R,R)-diamine 508 and n-BuLi to give the allyl alcohol (1S,4R)-509 in 72% yield and 99.3% ee. However, treatment of meso-epoxide 507b under identical conditions gave the allyl alcohol (1S,4S)-510 in only 3% yield and 48% ee (Scheme 105).
Scheme 105.

2.4.2. Enzymatic resolution
Enzymatic hydrolysis of prochiral bis(2,2,2-trifluoroethyl)-2,2-dimethylcyclopropane-1,1-dicarboxylate 511 with pig liver esterase (PLE) gave the (R)-2,2-dimethyl-1-(2,2,2-trifluoroethoxycarbonyl)-cyclopropane-1-carboxylic acid (R)-512 in 62% yield and >95% ee, which, by Curtius rearrangement with diphenylphosphoryl azide (DPPA), followed by work-up with ethanol gave the diprotected α-amino acid (S)-513 in 34% yield. Finally, basic hydrolysis of (S)-513 produced the (S)-1-amino-2,2-dimethylcyclopropane-1-carboxylic acid (S)-514 in 75% yield and >84% ee (Scheme 106).209
Scheme 106.

Recently, Beaulieu et al.210 reported the pilot plant large-scale synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid methyl ester (1R,2S)-196 from (±)-trans-515 using inexpensive esterase enzyme (Alcalase) as a resolution agent. In this context, treatment of (±)-trans-515, obtained in three steps from 452b, with a large excess of Alcalase under Na2HPO4 buffer conditions at pH 8.1–8.2 produced (1R,2S)-515 in 49% yield and 97% ee, and (1S,2R)-516 in with 99% ee, after separation.211 Hydrolysis of (1R,2S)-515 with HCl gave the (1R,2S)-516 in 64% yield as hydrochloride salt with >97% ee (Scheme 107). The vinyl-ACCA derivative (1R,2S)-196 is an important building block for the preparation of HCV protease inhibitors.212
Scheme 107.

Kirihara et al.213 reported an efficient synthesis of (R)- and (S)-1-amino-2,2-difluorocyclo-propanecarboxylic acid 521 by lipase-catalyzed desymetrization of diol 517 or diacetate 522. Thus, lipase-catalyzed transesterification of prochiral diol 517 with vinyl acetate as the acyl donor in the presence of lipase PS from Pseudomonas cepacia in benzene and diisopropyl ether, afforded the corresponding mono-acetylated product (R)-518 in 97% yield and 91.3% ee. Oxidation of (R)-518 followed by treatment with DPPA and subsequent work-up with tert-butyl alcohol and Et3N under reflux gave the carbamate (R)-519 in 51% yield. Cleavage of the acetyl group of (R)-519 produced the N-protected aminoalcohol (R)-520 in 66% yield and >99% ee, which, by oxidation followed by hydrolysis led to (R)-521 in 99% yield (Scheme 108).
Scheme 108.

On the other hand, lipase-catalyzed deacetylation of the prochiral diacetate 522 with lipase PS in a mixed solvent of acetone and phosphate buffer gave the corresponding mono-acetylated product (S)-518 in 86% yield and 91.7% ee, which, under identical conditions to those described in the Scheme 108, was transformed into (S)-1-amino-2,2-difluorocyclopropanecarboxylic acid 521 as hydrochloride salt (Scheme 109).213
Scheme 109.

Catalytic hydrolysis of (±)-523 with lipase CALB from Pseudomonas cepacia in a mixed solvent of acetone and phosphate buffer gave the corresponding mono-acid (3aS,5S,6aS)-524 and the residual diester (3aR,5R,6aR)-523, both with >99% ee. Hydrolysis of (3aS,5S,6aS)-524 and (3aR,5R,6aR)-523 furnished the constrained α-amino acids (3aS,5S,6aS)-525 and (3aR,5R,6aR)-525 in 60 and 74% yield, respectively (Scheme 110).214
Scheme 110.

In a similar way, hydrolysis of (±)-526 using lipase proleather (Subtilysin Carlsberg) in acetone and phosphate buffer gave the monoacid (3aR,5S,6aR)-527, the product derived from the hydrolysis of methyl ester linked to position 5, and the residual diester (3aS,5R,6aS)-526, both with >99% ee. Similar results were obtained with papain-catalyzed hydrolysys of (±)-526, but with reversal of the stereochemistry. Hydrolysis of (3aR,5S,6aR)-527 and (3aS,5R,6aS)-526 led to the constrained α-amino acids (3aR,5S,6aR)-528 and (3aS,5R,6aS)-528 in 78 and 64% yield, respectively (Scheme 111).214,215
Scheme 111.

On the other hand, the pig liver esterase (PLE) enzymatic desymetrization of diacetate 529 afforded the monoacetate (S)-530 with 80% ee along with diol 531. The monoacetate (S)-530 was transformed in three steps into diprotected alkyne (S)- and (R)-532. Addition of the carbanion derived from (S)-532 to the lactone 533 gave the compound 534 in 68% as a 1:1 mixture of anomers. Partial reduction of the alkyne followed by the spiroketalization and subsequent cleavage of the silyl protecting groups with TBAF, acetylation and HPLC separation, produced the spiro derivative 535 in 53% overall yield. Reduction of the azide and olefin fuctional groups and simultaneous removal of (DMB) protective group in 535 in the presence of acetic anhydride furnished the acetamide 536 in 59% yield. Oxidation of 536 with Dess-Martin periodinane and NaClO2 gave the α-N-acetylgalactosaminylserine derivative 537 in 89% yield. In a similar way, (R)-532 was transformed into derivative 538 (Scheme 112). 216
Scheme 112.

Metathesis reaction of 539, which is readily obtained by dialkylation of dimethyl malonate with 4-bromo-1-butene, in the presence of Grubbs catalyst gave the cycloheptene 540 in 98% yield. Epoxidation of this compound with m-CPBA followed by hydrolysis with sulfuric acid afforded the cycloheptane-trans-1,2-diol (±)-541 in 80% yield. Kinetic resolution of (±)-541 with Amano AK in vinyl acetate produced the diol (4R,5R)-541 and the monoacetate (4S,5S)-542 in 43% and 33% yield, respectively, both with >99% ee. Methylation of (4R,5R)-541 with MeI and Ag2O produced the dimethoxy compound (4R,5R)-543 in quantitative yield and selective hydrolysis with NaOH, followed by Curtius rearrangement and subsequent workup with benzyl alcohol, furnished the cyclic amino acid (4R,5R)-544 in 92% yield (Scheme 113).217
Scheme 113.

2.4.3. HPLC resolution
Semipreparartive chiral HPLC resolution of (±)-546, obtained in four steps from α,β-dehydroamino acid derivative 545, using a mixture of 10-undecenoate/3,5-dimethylphenylcarbamate of amylose covalently attached to allylsilica gel (CSP-2) as a chiral stationary phase, afforded the enantio-merically pure (2R,3R)- and (2S,3S)-546. Hydrolysis of each enantiomer gave the constrained cyclopropane analogues of phenylalanine c3diPhe (2R,3R)- and (2S,3S)-547 in excellent yield as hydrochloride salt (Scheme 114).218
Scheme 114.

In a similar way, chiral HPLC resolution of (±)-549, obtained in three steps from α,β-dehydroamino acid derivative 548, using a mixture of 10-undecenoate/3,5-dimethylphenylcarbamate of cellulose linked to allylsilica gel (CSP-1) as a chiral stationary phase, afforded the enantiomerically pure (R)- and (S)-549, which, by cleavage of the benzoyl group with hydrazine followed by hydrolysis gave the constrained cyclopropane analogues of valine c3Val (R)- and (S)-514 in excellent yield as hydrochloride salt (Scheme 115).219
Scheme 115.

Chiral HPLC resolution of trans-c4Phe (±)-550, using CSP-1 as a chiral stationary phase and a mixture of hexane/2-propanol/chloroform (95/3/2) as eluent afforded the enantiomerically pure (1S,2R)- and (1R,2S)-550. Hydrolysis of each enantiomer gave the constrained cyclobutane analogues of phenylalanine (1S,2R)-N-Boc-c4Phe-OH (1S,2R)-551 and (1R,2S)-N-Boc-c4Phe-OH (1R,2S)-551 in excellent yield. Under identical conditions, (±)-552 gave (1R,2R)-N-Cbz-c4Phe-OH (1R,2R)-553 and (1S,2S)-N-Cbz-c4Phe-OH (1S,2S)-553 in excellent yield (Scheme 116).220
Scheme 116.

Chiral HPLC resolution of cis-N-(1-cyano-2-phenylcyclopentyl)benzamide (±)-554 using CSP-1 as a chiral stationary phase and a mixture of hexane/2-propanol/acetone (95/3/2) as eluent afforded the enantiomerically pure (1R,2R)- and (1S,2S)-554, hydrolysis of which produced the constrained cyclopentane analogues of phenylalanine (1R,2R)-c5Phe and (1S,2S)-c5Phe 555 in excellent yield as hydrochloride salt. Finally, reaction of (1R,2R)- and (1S,2S)-555 with TMSCl, followed by addition of benzyl chloroformate (CbzCl) furnished the (1R,2R)-N-Cbz-c5Phe-OH and (1S,2S)-N-Cbz-c5Phe-OH 556 in 60 and 65% yield, respectively. In a similar way, (±)-557 afforded the (1R,2S)-c5Phe and (1S,2R)-c5Phe 558 in 95 and 92% yield, respectively, and these compounds were transformed into (1R,2S)-N-Boc-c5Phe-OH and (1S,2R)-N-Boc-c5Phe-OH 559 (Scheme 117).221
Scheme 117.

Natalini et al.222 reported the preparative resolution of 1-aminoindane-1,5-dicarboxylic acid (±)-AIDA 33 by chiral ligand-exchange chromatography (CLEC), using (S)-N,N-dimethylphenylalanine as the chiral selector in the movile phase, obtaining the enantiomerically pure (S)- and (R)-AIDA 33 with high ee (Scheme 118).
Scheme 118.

On the other hand, chiral HPLC resolution of methyl cis-1-benzamido-2-phenylcyclohexane-carboxylate (±)-560 using CSP-2 as a chiral stationary phase and a mixture of hexane/2-propanol/chloroform (96/1/3) as eluent to afford enantiomerically pure (1R,2R)- and (1S,2S)-560, which, by hydrolysis with HCl under reflux, produced the constrained cyclohexane analogues of phenylalanine (1R,2R)-c6Phe and (1S,2S)-c6Phe 561 in quantitative yield as chlorhydrate salt. Finally, reaction of (1R,2R)- and (1S,2S)-561 with TMSCl, followed by addition of CbzCl, provided the (1R,2R)-N-Cbz-c6Phe-OH and (1S,2S)-N-Cbz-c6Phe-OH 562 in 70 and 80% yield, respectively. (Scheme 119).223
Scheme 119.

Esterification of (1R,2R)- and (1S,2S)-561 with thionyl chloride and methanol, followed by coupling with protected aspartic acid (S)-N-Cbz-Asp(Ot-Bu)-OH using i-BuOCOCl in the presence of NMM, produced the protected dipeptides (S,1R,2R)-563 and (S,1S,2S)-564 in 90% yield. Subsequent deprotection with TFA, followed by hydrogenolysis over Pd/C, led to the optically pure aspartame analogues H-(S)-Asp-(1R,2R)-c6Phe-OMe, (S,1R,2R)-565 (sweet) and H-(S)-Asp-(1S,2S)-c6Phe-OMe, (S,1S,2S)-566 (bitter), in 96 and 98% yield, respectively (Scheme 120).224
Scheme 120.

On the other hand, chiral HPLC resolution of trans-N-(1-cyano-2-phenylcyclohexyl)acetamide trans-(±)-567 using CSP-1 as a chiral stationary phase and a mixture of hexane and 2-propanol (93:7) as eluent, gave the enantiomerically pure (1R,2S)- and (1S,2R)-567, which, by hydrolysis with HCl under reflux, produced the constrained cyclohexane analogues of phenylalanine (1R,2S)-c6Phe and (1S,2R)-c6Phe 568 as chlorhydrate salts in 92 and 98% yield, respectively. Finally, reaction of (1R,2S)- and (1S,2R)-568 with (Boc)2O in tetramethylammonium hydroxide (TMAH) furnished the (1R,2S)-N-Boc-c6Phe-OH and (1S,2R)-N-Boc-c6Phe-OH 569 in 50 and 46% yield, respectively (Scheme 121).225
Scheme 121.

Enantiomerically pure (1R,2S)- and (1S,2R)-568 were transformed into optically pure aspartame analogues H-(S)-Asp-(1R,2S)-c6Phe-OMe (S,1R,2S)-570 (sweet) and H-(S)-Asp-(1S,2R)-c6Phe-OMe (S,1S,2R)-571 (bitter) under identical conditions to those described above (Scheme 122).226
Scheme 122.

3. Synthesis of azacycloalcane-2-carboxylic acids
2.1. Using cyclic compounds as starting materials
One of the most useful procedures to the stereoselective synthesis of these compounds involves alkylation reactions using the non-quaternary cyclic amino acids as starting materials whenever stereochemical control can be achieved. For example, Wulff et al.227 reported the highly diastereoselective alkylation of enantiopure aziridine-2-carboxylic acid ethyl esters 572 and 573228 with complete retention of the stereochemistry. In this context, treatment of 572 with LDA at −78 °C in 1,2-dimethoxyethane (DME) and diethyl ether, followed by addition of several alkylating agents, afforded the alkylated compounds 574a–j as single stereoisomers and in good yields with complete retention of the stereochemistry. Similar results were obtained in the methylation of 573 (R = Ph), with the methylated product 575 obtained with high diastereoselectivity and in 91% yield.229 Treatment of 575 with triflic acid and anisole produced the trisubstituted aziridine 576 in 84% yield (Scheme 123).
Scheme 123.

This methodology has been extensively used for the stereoselective synthesis of α-alkylprolines. Since most of the examples have been collected in a recent review, 230 we only report here the most recents papers. Sommer and Williams231 reported the stereoselective synthesis of 13C-labeled α-alkyl-β-methylproline ethyl ester (2R,3S)-579, a key intermediate in the elaboration of paraherquamides E, F and related derivatives, through the stereocontrolled allylation of β-methylproline ethyl ester (2S,3S)-577. In this context, treatment of β-methylproline ethyl ester (2S,3S)-577, obtained from expensive 1-13C-(S)-isoleucine, with KHMDS at −78 °C followed by addition of allyl iodide 578 afforded the α-allylated product (2R,3S)-579 in 88% yield as a single diastereoisomer. Identical results were obtained in the allylation of (2R,3S)-580, obtained in several steps from (R)-α-MBA and the cheap 1-13C-ethyl bromoacetate. The stereochemistry obtained in the allylation of (2S,3S)-577 and (2R,3S)-580 is influenced strongly by the methyl group in the β-position on the proline ring (Scheme 124).
Scheme 124.

Very recently, Chandan and Moloney232 reported the synthesis of 2,2,5-trisubstituted pirrolidines 585a–c from allylic pyroglutamates 581a–c by Ireland-Claisen ester rearrangement. Thus, treatment of 581a–c with LiHMDS and Al(i-OPr)3 in the presence of quinine under Kazmaier's conditions,233 gave the rearrangement products 582a–c in good yield as single diastereo-isomers through the transition state A. Interestingly, the Claisen rearrangement only occurred in the presence of quinine. Esterification of 582a–c with MeOH and TsOH, followed by treatment with Lawesson's reagent afforded the thiolactams 583a–c, which, by an Eschenmoser sulfide contraction234 with diethyl bromomalonate and sodium bicarbonate, produced the enamines 584a–c in good yield. Finally, reduction of enamine function in 584a–c with sodium cyanoborohydride provided the corresponding 2,2,5-trisubstituted pirrolidines 585a–c in good yield as single diastereoisomers (Scheme 125).
Scheme 125.

In 1981, Seebach et al.235 reported a methodology that formally allows the direct α-alkylation of L-proline without loss of the optical purity and with retention of the configuration, thus constituying a showcase of their concept of self-reproduction of chirality.236 In recent years this methodology has been used for the stereoselective synthesis of quaternary proline analogues. For example, treatment of the oxazolidinone (3S,7aR)-586, readily obtained from L-proline,237 with LDA in THF at −78 °C followed by addition of 3-bromoprop-1-yne gave the α-alkylated product (3S,7aR)-587 in 24% yield as a single diastereoisomer. Subsequent cleavage of the oxazolidinone moiety with TMSCl in methanol under microwave conditions, followed by treatment with CbzCl, furnished the N-Cbz protected α-propargyl proline (R)-588 in 73% yield. Cycloaddition of (R)-588 with the appropriate azide derivative, followed by treatment with CuSO4 and Cu(0) under microwave conditions, afforded the corresponding triazoles (R)-589a–d in 62–79% yield (Scheme 126).238
Scheme 126.

On the other hand, aldol reaction of the oxazolidinone (3S,7aR)-590, readily obtained from L-proline,235b,c with the Garner's aldehyde239 (R)-591 afforded the aldol products 592 and 593 in 58% yield and 4:1 dr. Dess-Martin oxidation of 592 followed by reduction with NaBH4 gave 593 in 43% yield and this was used in the synthesis of (2S,3S,4R,7R,9S)-kaitocephalin.240 Under identical conditions, aldol reaction of (3S,7aR)-590 with the aldehyde (S)-591 gave 594 and 595 in 51% yield and 2:3 dr. Oxidation of 594 followed by reduction with NaBH4 gave 595 in 31% yield and this was used in the synthesis of (2R,3S,4R,7R,9S)-kaitocephalin241 (Scheme 127).
Scheme 127.

Aldol reaction of enantiopure trans-596 with Garner's aldehyde (S)-597242 afforded the aldol products in 60% yield as a complex mixture, indicating a mis-matched double stereodifferentiation, whereas the same reaction using the aldehyde (R)-597 gave the aldol product 598 in 40–50% yield as a single diastereoisomer, which, confirm a matched double stereodifferentiation. On the other hand, treatment of trans-596 with LDA in THF at −78 °C, followed by addition of N-acylimidazole (S)-599, gave the corresponding β-keto ester 600 in 40–50% yield as a single diastereoisomer, which, by reduction of the keto function with DIBAL at −78 to 25 °C, gave the β-hydroxy ester 601 in 86–93% yield and >30:1 dr. The latter compound is an epimer of 598 and is a key compound for the synthesis of (2R,3S,4R,7R,9S)-kaitocephalin (Scheme 128).243
Scheme 128.

Recently, we reported244 a versatile methodology for the synthesis of (2R,3aS,7aS)-2-methyl-octahydroindole-2-carboxylic acid 605. Treatment of (S,S,S,R)-603, obtained in three steps from (S)-indoline-2-carboxylic acid 602, with LDA in THF at −78 °C followed by addition of several alkyl electrophiles produced the alkylated products 604a–c in good yield. In these compounds both the trichloromethyl group and the newly introduced substituent are cis to each other on the exo side of the bicyclic constituted by the two five-membererd rings, as in the pioneering investigations by Seebach.235b Hydrolysis of (S,S,S,R)-604a with 6 N HCl in acetic acid gave the α-methylated indoline (2R,3aS,7aS)-605 in 92% yield as the hydrochloride salt (Scheme 129).
Scheme 129.

Treatment of enantiopure 606 with LDA followed by addition of several alkyl halides produced the 3,3-disubstituted bicyclic derivatives 608a–f in good yield and with >95% diastereoselectivity, through the exocyclic lithium enolate 607 (Scheme 130).245
Scheme 130.

Symmetry-breaking enolization reaction of meso-diester 610, obtained in three steps from dipicolinic acid 609, with the chiral bis-lithium amide base 611 followed by addition of several alkylating reagents afforded the alkylated compounds 612a–f in good yield and with >98% ee (Scheme 131).246
Scheme 131.

Hou et al.247 have reported the synthesis of (R)- and (S)-2-alkyl pipecolic acids 617a–e by diastereoselective alkylation of (R)-5-phenylmorpholin-2-one 613. In this context, commercially available (R)-phenylglycinol was transformed in three steps into (R)-613 in 34% overall yield, and treatment of this compound with NaHMDS followed by the addition of 1,4-diiodobutane gave the iodide derivative (R,R)-614 in 65% yield as a single diastereoisomer. Cleavage of Boc protective group of (R,R)-614 with TFA and subsequent cyclization under basic conditions produced the (4R,9aR)-oxazin-2-one 615 in 65% yield. Treatment of 615 with KHMDS followed by addition of several alkyl halides afforded the corresponding alkylated compounds (4R,9aS)-616a–d and (4R,9aR)-616e in good yield and diastereoselectivity. Hydrogenation of (4R,9aS)-616a–d and (4R,9aR)-616e in the presence of Pearlman's catalyst gave the 2-substituted pipecolic acids (S)-617a–c and (R)-617e in quantitative yield (Scheme 132).
Scheme 132.

On the other hand, Porzi and Sandri248 reported the synthesis of unnatural dipeptides (2S,2'S)-622 and (3S,2'S)-623 through an alkylation-cyclization reaction using the mono-lactim ether (S)-618 as starting material. In this context, treatment of (S)-618 with LiHMDS, followed by addition of 1-chloro-4-iodobutane and α, α'-dibromo-o-xylene afforded the monoalkylated products 619a,b in moderate yield and 98:2 dr, which, by heating in DMF, gave the bicyclic derivatives 620a,b in good yield. Reaction of 620a,b with LiHMDS and subsequent addition of methyl iodide produced the methylated compounds 621a,b in 80–85% yield and with 1,4-trans induction. Successive cleavage of the benzyl group with Li/NH3, treatment with Et3OBF4 and acidic hydrolysis furnished the dipeptides (2S,2'S)-622 and (3S,2'S)-623a in 65% yield (Scheme 133).
Scheme 133.

3.2. Construction of the ring by cyclization reactions
The following paragraphs cover all current methodologies for cyclcization reactions and these are arranged into several according to the strategy involved. The first want involves the N-C bond formation starting from quaternary acyclic compounds in which the stereocentre has been previously formed. For example, nosylation reaction of methyl α-alkylserinates 624a–c with o-NsCl and excess of KHCO3 in acetonitrile under reflux provided the N-nosyl aziridines 625a–c in good yield (Scheme 134).249
Scheme 134.

On the other hand, treatment of (R)-5-phenylmorpholin-2-one 613 with NaHMDS, followed by addition of several alkylating reagents, gave the corresponding alkylated compounds (R,R)-626a–f in 49–79% yield as single diastereoisomers. Treatment of these compound with KHMDS and subsequent addition of 1,4-diiodobutane (the alkylation of 626e–f did not proceed), followed by cleavage of Boc protective group with TFA and subsequent cyclization under basic conditions produced the (4R,9aR)-oxazin-2-one 627a–d in 56–67% yield. Hydrogenation of 627a–d in the presence of Pearlman's catalyst gave the 2-substituted pipecolic acids (R)-617a–c in quantitative yield (Scheme 135).247
Scheme 135.

In a similar way, treatment of (S)-618 with LiHMDS followed by the addition of allyl bromide provided 628 in 85% yield and 85:15 dr,250 which, by alkylation using LiHMDS as a base and α,α'-dibromo-o-xylene as an alkylating reagent, afforded the dialkylated derivative (3R,6S)-629 in 80% yield. Heating of (3R,6S)-629 in DMF produced the bicyclic derivative 630 in 85% yield and treatment of this compound under identical conditions to those described for 621a,b, gave (3R,2'S)-631 in 67% yield (Scheme 136).248
Scheme 136.

Maruoka et al.251 reported the catalytic enantioselective synthesis of tetrahydroisoquinoline- and dihydroisoquinoline-3-carboxylic acid derivatives 634a–c and 636a,c by a phase-transfer alkylation-cyclization process. Thus, treatment of Shiff bases 632a–c, obtained from p-chlorobenzaldehyde and the appropriate α-amino acid tert-butyl esters, with α,α'-dibromo-o-xylene and 50% KOH in the presence of C2-symmetric chiral quaternary ammonium salt (S,S)-3,4,5-F3-Ph-NAS-Br 633, followed by hydrolysis with citric acid and subsequent treatment with excess of NaHCO3, produced the (3R)-3-alkyl-1,2,3,4-tetrahydroisoquinolines derivatives 634a–c in moderate yield and good enantioselectivity. In a similar way, alkylation of 632a,c with 635 in the presence of 633, followed by hydrolysis with HCl and subsequent treatment with excess of NaHCO3, produced the (3R)-3-alkyl-3,4-dihydroisoquinoline derivatives 636a,c in good yield and enantioselectivity (Scheme 137).
Scheme 137.

Formation of the C–N bond can be achieved by cyclization of carbenoid intermediates. For example, intramolecular cyclization of enantiopure carbenoids 637a,b, obtained from (R)-299, in the presence of a catalytic amount of Rh2(OAc)4 in CH2Cl2 gave the bicyclic compounds 638a,b with complete chemoselectivity at the adjacent annular nitrogen and a preference for carbon-carbon double bond additions or C–H insertions. Hydrolysis of 638a,b with 3 M HCl, followed by treatment with (Boc)2O and triethylamine, produced the protected dipeptides 639a,b (Scheme 138).252
Scheme 138.

In a similar way, intramolecular cyclization of enantiopure carbenoids (2S,5R)-640a,b in the presence of a catalytic amount of Rh2(OAc)4, afforded the bicyclic compounds 643a,b as the main products and 644a,b, probably due to isomerization at C-5 through the intermediates 641 and 642. Chemoselective opening ring of the iminoether function in the diastereoisomerically pure 643a,b with 3 M HCl, followed by treatment with (Boc)2O and triethylamine, produced the corresponding dipeptides (2S,2'R)-645a,b in moderated yield. Under identical conditions the enantiopure carbenoids (2R,5R)-646a,b were transformed into the dipeptides (2R,2'S)-645a,b (Scheme 139).253
Scheme 139.

Other protocol reported by our group involved the large scale reduction of enantiopure ketone 648,254 obtained in several steps from 647 by a Diels-Alder reaction. In this context, reduction of 648 with K-selectride in THF at −78 °C afforded the mixture of alcohols (axial 649 and equatorial 650) in 98% yield and an 85:15 ratio. Treatment of these compounds with MsCl and triethylamine followed by base-promoted internal nucleophilic displacement with sodium hydride and DMF, gave the 7-azabicyclo[2.2.1]heptane derivative 651 in 75% yield (Scheme 140).255
Scheme 140.

Cleavage of the acetonide function of 651 with PPTS in acetone-water provided the corresponding diol 652 in 67% yield, which, by oxidation with NaIO4 and RuCl3 followed by hydrolysis with 6 N HCl gave the (1S,2R,4R)-7-azabicyclo[2.2.1]heptane-1,2-dicarboxylic acid as hydrochloride salt 653 in 75% yield. On the other hand, oxidation of 652 with NaIO4 furnished the aldehyde (1S,2R,4R)-654 in 90% yield, which, by Wittig reaction with RCH=PPh3, provided the vinyl derivatives (1S,2R,4R)-655a–e in 75–99% yield. The vinyl derivative 655a was also obtained through the two-step Corey-Winter256 procedure. In this context, reaction of diol 652 with N,N'-thiocarbonyldiimidazole (TDCI) provided the thiocarbonate 656 in 83% yield. Treatment of 656 with 1,3-dimethyl-2-phenyl-1,3,2-diazaphospholidine (DMPDAP) led to 655a in 86% yield. Finally, hydrogenation of C=C double bond of 655a–e over Pd(OH)2 followed by hydrolysis with 6 N HCl afforded the (1S,2R,4R)-proline derivatives 657a–d in good yield (Scheme 141).257
Scheme 141.

Additionally, the aldehyde (1S,2R,4R)-654 has been used in the synthesis of 7-azanorbornane β-susbtituted prolines.258 For example, reduction of aldehyde function in (1S,2R,4R)-654 with NaBH4 followed by treatment with MsCl and triethylamine produced the corresponding mesylate (1S,2R,4R)-658, which, by nucleophilic substitution, afforded the compounds (1S,2R,4R)-659a–e in 60–100% yield. Hydrolysis of (1S,2R,4R)-659a,b,d with 6 M HCl provided the amino acids (1S,2R,4R)-660a,b,d in quantitative yield and these can be considered as (2S,3R)-3-methylproline, (2S,3R)-3-methylthio-methylproline and (2S,3R)-3-carboxymethylproline analogues. Additionally, (1S,2R,4R)-659e was transformed into amino compounds (1S,2R,4R)-661a–d (Scheme 142).
Scheme 142.

The second reported methodology involves the formation of a Cα-Cβ bond. In this case the stereoselectivity of the cyclization reaction depends on the chirality of the non-quaternary α-amino acid used as starting material, wherever the chirality of the stereocentre can be remembered to some extent (memory of chirality). This methodology has been repeatedly used for the stereoselective synthesis of four-, five-, six- and seven-membered rings. These results have been collected in recent reviews,259 and we therefore only describe here the most recents papers. For example, Kawabata et al.260,261 reported the four-membered cyclization for the straightforward synthesis of cyclic amino acids with tetrasubstituted stereocentres from chiral α-amino acids through memory of chirality. In this context, treatment of 662a,b with KHMDS in DMF at −60 °C furnished the four-membered compounds 663a,b with good enantioselectivity and retention of configuration, while that the use of lithium 2,2,6,6-tetramethylpiperidine (LTMP) as a base in THF at −20 °C led to 663a,b with good enantioselectivity but with inversion of the configuration. Treatment of (R)-663a with methanolic NaOMe followed by cleavage of the N-Boc protective group with 4 N HCl gave the azetidine derivative (R)-664 in 56% yield (Scheme 143).
Scheme 143.

In a similar way, treatment of 665 and 666 with KHMDS in DMF at −60 °C provided the piperidine derivative 667 in 84% yield and 97% ee, and the azepane 668 in 31% yield and 83% ee (666 was recovered). The stereochemical course of the cyclization was with retention of the configuration (Scheme 144).260
Scheme 144.

Recently, Kawabata et al.262 reported that the asymmetric cyclization of 662a–c (n = 2), 669a–c (n = 3), and 665a–c (n = 4) using powdered KOH as an efficient base in DMSO at 20 °C afforded the four- five- and six-membered compounds 663a–c, 670a–c and 667a–c, respectively, in good yield and excellent enantioselectivity. The results are summarised in Table 12.
Table 12.
Asymmetric cyclization of α-amino acids derivatives with KOH/DMSO at 20 °C.
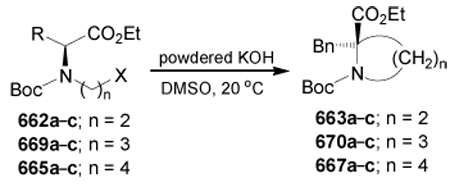 | |||||
|---|---|---|---|---|---|
| entry | R | X | product | Yield (%) | ee (%) |
| 1 | a = Bn | Br | 663a; n = 2 | 82 | 99 (R) |
| 2 | b = MeSCH2CH2 | Br | 663b; n = 2 | 85 | 99 (S) |
| 3 | c = i-Pr | Br | 663c; n = 2 | 79 | 99 a |
| 4 | a = Bn | Br | 670a; n = 3 | 91 | 99 (S) |
| 5 | b = MeSCH2CH2 | Br | 670b; n = 3 | 91 | 98 (S) |
| 6 | c = i-Pr | Br | 670c; n = 3 | 94 | 98 a |
| 7 | a = Bn | Br | 667a; n = 4 | 73 | 90 a |
| 8 | b = MeSCH2CH2 | Br | 667b; n = 4 | 86 | 88 a |
| 9 | c = i-Pr | Br | 667c; n = 4 | 74 | 94 a |
| 10 | a = Bn | I | 667a; n = 4 | 97 | 97 a |
| 11 | b = MeSCH2CH2 | I | 667b; n = 4 | 89 | 97 a |
| 12 | c = i-Pr | I | 667c; n = 4 | 90 | 98 a |
The configuration was not reported.
This protocol has been used in the synthesis of Fmoc-cyclic amino acid 671, which is expected to be an useful building block for conformationally constrained peptides of biological interest. In this context, treatment of isoleucine derivative 669d with powdered KOH in DMSO at 20 °C afforded the cyclic product 670d in 94% yield as a single diastereoisomer, which, by hydrolysis with HCl and subsequent N-Fmoc protection, led to the proline derivative 671 in 53% yield (Scheme 145).262
Scheme 145.

Very recently, Kawabata et al.263 reported the asymmetric intramolecular alkylation of β-alcoxy-α-amino esters through memory of chirality methodology. In this context, treatment of serine derivatives 672a, 673a–f and 674a with CsOH as an efficient base in DMSO at 20 °C afforded the cyclization products 675a, 676a–f and 677a in 13–89% yield and enantioselectivities in the range 82 to 94%. The results are summarized in Table 13.
Table 13.
Asymmetric cyclization of serine derivatives.
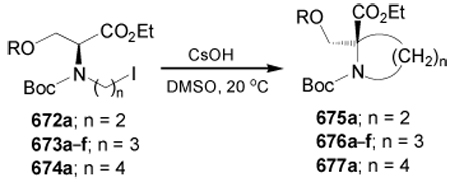 | ||||
|---|---|---|---|---|
| entry | substrate | product | Yield (%) | ee (%) |
| 1 | 672a; R = t-Bu | 675a; n = 2 | 74 | 92 a |
| 2 | 673a; R = Bn | 676a; n = 3 | 84 | 86 (S) |
| 3 | 673b; R = Me | 676b; n = 3 | 75 | 82 (S) |
| 4 | 673c; R = MOM | 676c; n = 3 | 72 | 82 (S) |
| 5 | 673d; R = TBDPS | 676d; n = 3 | 13 | 88 (S) |
| 6 | 673e; R = PMB | 676e; n = 3 | 88 | 92 (S) |
| 7 | 673f; R = t-Bu | 676f; n = 3 | 89 | 93 (S) |
| 8 | 674a; R = t-Bu | 677a; n = 4 | 77 | 94 a |
The configuration was not reported.
Memory of chirality in intramolecular conjugate addition of enolates is another metholodogy used for the asymmetric synthesis of nitrogen heterocycles with contiguous quaternary stereocentres. For example, treatment of α,β-unsaturated derivatives 678a,b with KHMDS in DMF-THF at −78 °C gave the piperidine derivatives 679a,b as a single detectable diastereoisomers in moderate yield. Seven-membered ring cyclization of α,β-unsaturated compound 678c proceeded to give 679c in 91% ee, albeit in only 19% yield (Scheme 146).264 Compounds 679a–c are precursors of conformationally constrained L-glutamate analogues.
Scheme 146.

On the other hand, treatment of α,β-unsaturated compound 680 with KHMDS in DMF-THF at −78 °C gave the tetrahydroisoquinoline derivative 681 as a single diastereoisomer in 95% ee and 94% yield.261,264 Treatment of 680 with LTMP in THF at 0 °C led to ent-681 as a single diastereoisomer in 91% ee and 62% yield (Scheme 147).261
Scheme 147.

The third methodology reported involves the cyclization by C-C bond formation, starting from the corresponding quaternary α-amino acids previously obtained with both chain appropriately functionalized. For example, ring closing metathesis of dialkylated derivative (R)-683, obtained in three steps from 682,265 in the presence of a catalytic amount of PhCH=RuCl2(PCy3)2 in benzene under reflux gave the corresponding six-membered derivative (R)-684 in 94% yield. Subsequent hydrogenation of the C=C double bond over Pd/C afforded the α-quaternary pipecolic acid derivative (R)-685 in 95% yield (Scheme 148).266
Scheme 148.

Finally, the one-pot aza-Darzens reaction has been reported as a competitive alternative for the synthesis of aziridine carboxylic acids. For example, aza-Darzens reaction267 of (S)-sulfinimine 686a with the lithium α-bromoenolate generated from methyl α-bromopropianate 687 and LiHMDS in THF at −78 °C, afforded the corresponding aziridines (SS,2R,3S)-688 and (SS,2S,3S)-689 in 55 and 21% yield, respectively. Oxidation of diastereoisomerically pure (SS,2R,3S)-688 with m-CPBA gave the N-tosyl aziridine (2R,3S)-690 in excellent yield. On the other hand, reaction of diastere-oisomerically pure (SS,2R,3S)-688 with MeMgBr provided the aziridine (2R,3S)-691 in 92% yield (Scheme 149).268
Scheme 149.

In a similar way, the one-pot reaction of (S)-sulfinimines 686b,c with the lithium α-bromoenolate generated from methyl 3-benzyloxy-2-bromopropionate 692269 and LiHMDS in THF at −78 °C, produced the aziridines (SS,2S,3S)-693b and (SS,2R,3S)-694b (R = Ph) in 70% yield and 95:5 dr, and the aziridines (SS,2S,3S)-693c and (SS,2R,3S)-694c (R = E-MeCH=CH) in 79% yield and 15:85 dr. Selective cleavage of N-sulfinyl bond in (SS,2S,3S)-693b with TFA gave the aziridine (2S,3S)-695b in 76% yield, whereas the treatment of (SS,2R,3S)-694c with excess of MeMgBr provided the aziridine (2R,3S)-696c in 86% yield (Scheme 150).270
Scheme 150.

3.3. Cycloadditions and related reactions
This strategy has been elegantly used to the synthesis of different types of prolines and derivatives and it is especially useful in the synthesis of polysubstituted (polyfunctional) prolines. Nevertheless, most of the published papers have been gathered in our recent review.230 As a result we only include here the reports that have appeared very recently. For example, Xie et al.271 reported a practical asymmetric synthesis of highly substituted proline derivatives 698 and 700 on a multi-kilogram scale. In this context, [3+2] cycloaddition reaction of methyl acrylate with the enantiopure imine (S)-697a, readily obtained by condensation of L-leucine tert-butyl ester with 2-thiazole-carboxaldeyde, in the presence of a catalytic amount of hydroquinine, AgOAc, and molecular sieves, produced the proline derivative 698 in 85:15 enantiomeric ratio (er). Treatment of the resulting compound with (R)-1,1'-binaphthyl-2,2'-dehydrogenphosphate in 2-propanol and subsequent crystallization gave 698 in 99.9:0.1 er and 57% overall yield. In asimilar way, [3+2] cycloaddition reaction of (S)-697a with methyl vinyl ketone in the presence of a catalytic amount of cinchonidine and AgOAc gave the proline derivative 699 as a mixture of α/β epimers in 98:2 ratio, which, by treatment with 10 mol% of DBU, gave the β epimer 700 as the main product with 73:27 er (Scheme 151).
Scheme 151.

Very recently, Kobayashi et al.272 reported the [3+2] cycloaddition reaction of different imines (±)-701 with several acrylates 702 in the presence of a catalytic amount of the bisoxazolines 703a–e and Ca(Oi-Pr)2 in THF and molecular sieves. The substituted pyrrolidine derivatives 704 were obtained in high yields and with high diastereoselectivities and enantioselectivities (Scheme 152)
Scheme 152.

The [3+2] cycloaddition reaction was used by Kobayashi el at.272 in the synthesis of optically pure pyrrolidine cores of hepatitis C virus RNA-dependent polymerase inhibitors and potentially effective antiviral agents. In this context, [3+2] cycloaddition reaction of tert-butyl acrylate with the enantiopure imines (R)-697a,b in the presence of a catalytic amount of the bisoxazolines 703a,b and Ca(Oi-Pr)2 in THF and molecular sieves, produced the pyrrolidine derivatives 705a,b in high yield with perfect diastereoselectivities and high enantioselectivities (Scheme 153).
Scheme 153.

Another approach to generate enantiomerically enriched polysubstitutted prolines or pyrrolidine derivatives is the 1,3-dipolar cicloaddition between electrophilic alkenes and stabilized or nonstabilized dipolarophiles, respectively. This strategy allows the creation of up four stereogenic centres in only one step and gives high regioselectivity and endo/exo-diastereoselectivities. For example, Nájera et al.273 reported the stereoselective synthesis of polysubstituted prolines (2R,4R,5S)- and (2S,4S,5R)-707 by a 1,3-dipolar cicloaddition, In this context, the cycloaddition reaction between the racemic imino ester (±)-697b with acrylate bonded to methy (S)-lactate 706 in the presence of a catalytic amount of AgOAc and KOH in toluene, afforded the polysubstituted proline (2R,4R,5S)-707 in 77% yield and 96% de. In similar way, reaction of (±)-697b with acrylate bonded to methy (R)-lactate 706 gave the polysubstituted proline (2S,4S,5R)-707 in 88% yield and 96% de. (2R,4R,5S)-707 And (2S,4S,5R)-707 were transformed into (2R,4R,5S)- and (2S,4S,5R)-708, two promising potential drugs, particularly for the hepatitis C virus RNA-dependent polymerase inhibitors and potential effective antiviral agents (Scheme 154).
Scheme 154.

Recently, Nájera et al.274 reported that the catalytic enantioselective 1,3-dipolar cycloaddition reaction of racemic benzylideneiminoglycinates 709a–d with tert-butyl acrylate in the presence of a catalytic amount of (Sa,R,R)-710, AgClO4 and triethylamine or 1,4-diazabicyclo[2.2.2]octane (DABCO) as a base, afforded the corresponding prolines 711a,b and 712c,d with high enantiomeric ratio (Scheme 155).275
Scheme 155.

1,3-Dipolar cycloaddition reaction of racemic methyl N-benzylidenealaninate 713 with the vinyl sulfone in the presence of copper(I)/click ferrophos complex 714 and CuOAc, produced the quaternary methyl prolinate derivative 715 in 83% yield and 93% ee (Scheme 156).276
Scheme 156.

Very recently, Carretero et al.277 reported the stereoselective synthesis of 3-pyrrolines 719a,b by asymmetric 1,3-dipolar cycloaddition reaction. Thus, the reaction of racemic methyl N-benzylidene-alaninates 713a,b with trans-1,2-bisphenylsulfonyl ethylene 716 in the presence of a catalytic amount of Cu(MeCN)4PF6, Fesulphos (R)-717 and Et3N, afforded the quaternary methyl prolinate derivatives 718a,b in good yield and with good enantioselectivity. Subsequent treatment of these compounds with Na(Hg) gave the quaternary derivatives 719a,b in 85 and 77% yield, respectively. (Scheme 157).
Scheme 157.

Cyclopropanation reaction of a cyclic dehydroaminoacid derivative allowed the synthesis of new constrained quaternary pipecolic derivatives. Thus, the reaction of (S)-2,3-didehydropipecolate 721, obtained in five steps from N,N-diprotected L-lysine methyl ester 720, with dimethylsulfoxonium methylide afforded the 2,3-methano-6-methoxypipecolate (2S,3R)-722 in 73% yield and treatment with NaBH4 in formic acid gave the 2,3-methanopipecolate (2S,3R)-723 in 75% yield and 85% ee. Finally, hydrolysis of (2S,3R)-723 with TMSI produced the (2S,3R)-methanopipecolic acid 724 in 50% yield (Scheme 158).278
Scheme 158.

3.4. Resolution procedures
3.4.1. Chemical resolution
Reaction of alcohol (±)-726, obtained from (±)-725,279 with (R)-methoxytrifluorophenylacetic acid [(R)-MTPA] in the presence of N,N'-dicyclohexylcarbodiimide (DCC) and DMAP, followed by crystallization, gave the diastereoisomeric esters (1S,2S,4R,2'R)-727 and (1R,2R,4S,2'R)-728 in 95% yield and >95% optical purity. Hydrolysis of 727 and 728 with methanolic NaOMe followed by hydrolysis with 6 N HCl at 60 °C furnished the enantiomerically pure (1S,2S,4R)-729 and (1R,2R,4S)-729, respectively, and these are analogues of 3-hydroxyproline (Scheme 159).280
Scheme 159.

3.4.2. HPLC Resolution
Preparative HPLC resolution of β-lactam (±)-730 on CSP-1 as a chiral stationary phase gave the β-lactams (S)- and (R)-730 with 85 and 92% enantiomeric purity. Subsequent saponification of these compounds provided the conformationally constrained amino acids (S)- and (R)-731. On the other hand, reduction of the amide function of (S)- and (R)-730 with Ph2SiH2 and RhH(CO)(PPh3)3 followed by cleavage of PMB protective group under H2 and Pd(OH)2, provided the optically pure Phe-derived conformationally constrained amino esters (S)- and (R)-732 (Scheme 160).281
Scheme 160.

Preparative HPLC resolution of (±)-733, obtained from 545, on CSP-1 as a chiral stationary phase gave the enantiomerically pure (1S,2S,4R)-733 and (1R,2R,4S)-733, which, were separately treated with 6 N HCl to give the enantiomerically pure proline-phenylalanine chimeras (1S,2S,4R)- and (1R,2R,4S)-734 in 95% yield. Oxidative cleavage of the phenyl substituent on the azabicyclic ring of (1S,2S,4R)- and (1R,2R,4S)-733 produced the corresponding carboxylic acids (1S,2R,4R)- and (1R,2S,4S)-735 in 45% yield. Hydrolysis of these compounds provided the enantiomerically pure (1S,2R,4R)- and (1R,2S,4S)-3-carboxyproline analogues 736 in 95% yield. On the other hand, the conversion of the carboxylic acid function of (1S,2R,4R)- and (1R,2S,4S)-735 into methyl alcohols (1S,2R,4R)- and (1R,2S,4S)-737 was carried out by treatment with isobutylchloroformate (IBCF) and triethylamine followed by reduction with NaBH4. Finally, Dess-Martin oxidation of the alchohol function of (1S,2R,4R)- and (1R,2S,4S)-737 gave the corresponding aldehyde derivatives (1S,2R,4R)- and (1R,2S,4S)-738 in 80% yield, and these proved to be a versatile synthetic intermediate in the preparation of a wide variety of β-substituted azabicyclic prolines (Scheme 161).282
Scheme 161.

3.5. Miscellaneus and notes added in proofs
Several other special cyclization procedures useful for very particular cases have been reported. For example, the Pictet-Spengler cyclization of N-sulfonyl-β-phenylethylamines 739a,b with menthyl α-chloro-α-phenylseleno propionate 740 in the presence of SnCl4 gave the corresponding 1,2,3,4-tetrahydrosioquinoline-1-carboxylates derivatives 741a,b in moderate yield and good diastereo-selectivity after crystallization (Scheme 162).283
Scheme 162.

Pictet-Spengler cyclization of quaternary oxazolidines 742a–d in the presence of TiCl4 and Et3N provided the corresponding tetrahydroisoquinolines 743a–d with good regioselectivity, which, by cleavage of the benzyl functionality under H2 and Pd(OH)2, gave the quaternary amino acids 744a–d in 30–96% yield (Scheme 163).284
Scheme 163.

On the other hand, Pictet-Spengler cyclization of quaternary N-Boc-N-MOM-α-methyl- and α-allyltrytophan derivatives 746a,b, obtained from alkylation of 745, with HCl in ethyl acetate afforded the corresponding tryptoline derivatives 747a,b in good yield, where the MOM protective group serves as a formaldehyde equivalent (Scheme 164).285
Scheme 164.

Reaction of enantiopure quaternary α-amino acid derivative 748 with paraformaldehyde in formic acid followed by treatment with H2O and TsOH provided the lactone cis-752 and the hydroxy ester trans-753 in 50 and 32% yield, respectively. The transformation from 748 to cis-752 and trans-753 should occur through the chairlike N-tosyliminium intermediate 749 followed by cyclization to give the secondary cation 750, which, is stabilized by the ester carbonyl group to produce the dioxycarbenium ion 751. Finally, the hydrolysis of 751 followed by treatment with TsOH gave the lactone cis-752 and the hydroxy ester trans-753 (Scheme 165).286
Scheme 166.

Clayden et al.287 reported the synthesis of α-methyl kainic acid (an α-methylproline 3,4-disubstituted system) by the stereospecific lithiation-dearomatizing cyclization of the chiral benzamide (R,R)-754. Thus, reaction of the benzamide (R,R)-754 with tert-BuLi at −78 °C followed by treatment with 0.5 M HCl produced the corresponding bicyclic compound 755 as a single stereo- and regioisomer in 70% yield. Conjugated addition of Me2CuLi to 755 followed by cleavage of the benzyl fragment with CAN and subsequent treatment with (Boc)2O gave 756 in 66% yield, which, in turn was transformed into α-methyl kainic acid after 12 steps (Scheme 166).
Finally, during the corrections of this review, Makosza et al.288 reported the stereoselective synthesis of (2R)-4-nitroarylprolines 759a–c through oxidative nucleophilic substitution of hydrogen in nitroarenes using the chiral carbanion of L-proline derivative (3S,7aR)-590, applying the self-reproduction of chirality methodology. Thus, treatment of (3S,7aR)-590 with KHMDS in THF-DMF at −78 °C followed by the addition of corresponding nitroarene, afforded the σH adduct 757, which, by oxidation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) gave the 4-nitroaryl derivatives 758a–c as a single detectable diastereoisomeres with 29–72% yield (reaction with 2-fluor, 2-chloro and 2-methylnitrobenzene failed). Hydrolysis of (3S,7aR)-758a–c with HBr and subsequent treatment with propylene oxide led to the (2R)-4-nitroarylprolines 759a–c with 55–85% yield (Scheme 167).
Scheme 167.

Concluding remarks
In this review, we have covered recent progress in the development of new synthetic methodologies for the preparation of cyclic α,α-dialkylamino acids and we have also discussed extensions to well established synthetic routes. The use of cyclic compounds as starting materials is one of the most convenient procedures reported.
The construction of the cycle using cyclization or cycloaddition reactions both in a diastereo-selective or enantioselective manner is one excellent alternative. All of these strategies can be completed with the use of resolution procedures (chemical, enzymatic or chromatographic) that have emerged as another good alternative.
All of these methodologies give the synthetic organic chemist the opportunity to select the most appropriate way to obtain the desired cyclic α,α-dialkylamino acid in enantiomerically pure form on both a laboratory scale and a multigram scale.
Scheme 165.

Table 11.
Enantioselective Diels-Alder cycloaddition of 424 with cyclopentadiene.
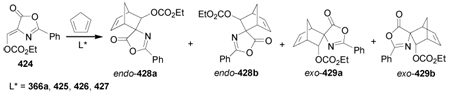 | ||||
|---|---|---|---|---|
| Conditions | t (h) | exo: endo | endo ratio | exo ratio |
| 366a-Mg(ClO4)2 | 48 | 75 : 25 | 43 : 57 | 50 : 50 |
| 366a-Ce(OTf)4.H2Oa | 75 | 70 : 30 | 50 : 50 | 50 : 50 |
| 425-Mg(ClO4)2 | 100 | 70 : 30 | 50 : 50 | 50 : 50 |
| 425-Ce(OTf)4.H2Oa | 150 | 75 : 25 | 50 : 50 | 50 : 50 |
| 426-Mg(ClO4)2 | 24 | 80 : 20 | 47 : 53 | 43 : 57 |
| 427-Mg(ClO4)2 | 24 | 75 : 25 | 56 : 44 | 66 : 34 |
| 427-Ce(OTf)4.H2Oa | 200 | 75 : 25 | 50 : 50 | 50 : 50 |
In the presence of molecular sieves.
Acknowledgements
This work was carried out with the financial support of CONACYT-MEXICO (Projects 62271 and 44126) and financial support from the Ministerio de Educación y Ciencia–FEDER (project CTQ2007-62245) and Gobierno de Aragón (group E40 and project MI041/2007) is gratefully acknowledged. This project has been funded as whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the view or the policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- Ac
acetyl
- AcOH
acetic acid
- ACCA
1-aminocyclopropanecarboxylic acid
- acac
acetylacetone
- ACPD
1-amino-1, 3-cyclopentane dicarboxic acid
- Adt
4-amino-1,2-dithiolane-4-carboxylic acid
- Afc
O,O-isopropylidene-α-hydroxymethylserine
- AIBN
2,2'-azoisobutyronitrile
- AIDA
1-aminoindane-1,5-dicarboxylic acid
- APCPr
1-amino-2-phosphonomethylcyclopropanecarboxylic acid
- APICA
1-amino-5-phosphoindane-1-carboxylic acid
- AP4
L-2-amino-4-phosphonobutanoic acid
- BINAP
2,20-bis(diphenylphosphanyl)-1,10-binaphthyl
- BINOL
1,10-bi-2-naphthol
- Bn
benzyl
- Boc
tert-butoxycarbonyl
- BOP
benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate
- BtH
benzotriazole
- BuLi
butyl lithium
- Bz
benzoyl
- CALB
Candida antarctica lipase B
- CAN
ceric ammonium nitrate
- Cbz
benzyloxycarbonyl
- CLEC
chiral ligand-exchange chromatography
- Daf
9-amino-9-fluorenecarboxylic acid
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DAM
di-p-anisylmethyl
- DBDA
dibenzyl azodicarboxylate
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCC
N,N'-dicyclohexylcarbodiimide
- DCE
1,2-dichloroethane
- DDQ
2,3-dichloro-5,6-dicyano-p-benzoquinone
- DEAD
diethyl acetylenedicarboxylate
- DEAD
diethyl azodicarboxylate
- DIAD
diisopropyl azodicarboxylate
- DMA
N,N'-dimethylacetamide
- DMB
3,4-dimethoxybenzyl
- DMAP
4-dimethylaminopyridine
- DMEA
dimethylethanolamine
- DME
1,2-dimethoxyethane
- DMF
N,N'-dimethylformamide
- DMPDAP
1,3-dimethyl-2-phenyl-1,3,2-diazaphospholidine
- DMSO
dimethylsulfoxide
- DPPA
diphenylphosphorazide
- dppp
1,3-bis(diphenylphosphino)propane
- dr
diastereoisomeric ratio
- EDA
ethylenediamine
- EDC
N-ethyl-N'-dimethylaminopropylcarbodiimide hydrochloride
- ee
enantiomeric excess
- er
enantiomeric ratio
- HMPA
hexamethylphosphoramide
- HOBt
N-hydroxybenzotriazole
- HPLC
High Performance Liquid Chromatography
- HYDIA
amino-3-hydroxybicyclo[3.1.0]hexane-2,6-dicarboxylic acid
- IBCF
isobutylchloroformate
- LDA
lithium diisopropylamide
- LiHMDS
lithium bis(trimethylsilyl)amide
- LPLC
Low pressure liquid chromatography
- LTMP
lithium 2,2,6,6-tetramethylpiperidide
- KHMDS
potasium bis(trimethylsilyl)amide
- MABR
methylaluminum bis(4-bromo-2,6-di-tert-butyl phenoxide)
- MAO
methylaluminoxane
- MBA
methylbenzylamine
- m-CPBA
m-chloroperbenzoic acid
- MOM
methoxymethyl
- MOMBA
methoxymethylbenzylamine
- MS
molecular sieves
- Ms
methanesulfonyl (mesyl)
- MTPA
methoxytrifluorophenylacetic acid
- NaHMDS
sodium bis(trimethylsilyl)amide
- NBS
N-bromosuccinimide
- NMM
N-methylmorpholine
- NMO
morpholine N-oxide
- NMP
N-methylpyrrolidin-2-one
- Ns
nitrobenzenesulphonyl
- PCC
pyridinium chlorochromate
- PLE
pig liver esterase
- PMB
p-methoxybenzyl
- mGlu
metabotropic glutamate
- PPTS
pyridinium p-toluenesulfonic acid
- RCM
ring closing metathesis
- rt
room temperature
- SAMI
(S)-1-amino-2-methoxymethylindoline
- SAMP
(S)-1-amino-2-methoxymethylpyrrolidine
- TBAB
tetrabutylammonium bromide
- TBAF
tetra-n-butylammonium fluoride
- TBS
tert-butyldimethylsilyl
- TDCI
N,N'-thiocarbonyldiimidazole
- TEA
triethylamine
- Tf
trifluoromethanesulfonyl
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- TfOH
trifluoromethanesulfonic acid
- THF
tetrahydrofuran
- TMAH
tetramethylammonium hydroxide
- TMSCl
trimethylsilyl chloride
- TMSI
trimethylsilyl ioide
- TMSCN
trimethylsilylcyanide
- TMSE
trimethylsilylethyl
- Tol
tolyl
- TsOH
p-toluenesulfonic acid
- Ts
p-toluenesulfonyl (tosyl)
References
- 1.Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 1998;9:3517–3599. [Google Scholar]
- 2.Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 2000;11:645–732. [Google Scholar]
- 3.Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 2007;18:569–623. [Google Scholar]
- 4.Undheim K. Amino Acids. 2008;34:357–402. doi: 10.1007/s00726-007-0512-5. [DOI] [PubMed] [Google Scholar]
- 5.Park K-H, Kurth MJ. Tetrahedron. 2002;58:8629–8659. [Google Scholar]
- 6.Gelmi ML, Pocar D. Org. Prep. Proced. Int. 2003;35:141–205. [Google Scholar]
- 7.Qiu X-L, Meng W-D, Qing F-L. Tetrahedron. 2004;60:6711–6745. [Google Scholar]
- 8.Perdih A, Dolenc MS. Curr. Org. Chem. 2007;11:801–832. [Google Scholar]
- 9.Galeazzi R, Mobbili G, Orena M. Curr. Org. Chem. 2004;8:1799–1829. [Google Scholar]
- 10.Lasa M, Cativiela C. Synlett. 2006:2517–2533. [Google Scholar]
- 11.Brackmann F, de Meijere A. Chem. Rev. 2007;107:4493–4537. doi: 10.1021/cr078376j. [DOI] [PubMed] [Google Scholar]
- 12.Brackmann F, de Meijere A. Chem. Rev. 2007;107:4538–4671. doi: 10.1021/cr0784083. [DOI] [PubMed] [Google Scholar]
- 13.Nájera C, Sansano JM. Chem. Rev. 2007;107:4584–4671. doi: 10.1021/cr050580o. [DOI] [PubMed] [Google Scholar]
- 14.(a) Tanaka M. Chem. Pharm. Bull. 2007;55:349–358. doi: 10.1248/cpb.55.349. [DOI] [PubMed] [Google Scholar]; (b) Maity P, König B. Biopolymers (Pept. Sci.) 2007;90:8–27. doi: 10.1002/bip.20902. [DOI] [PubMed] [Google Scholar]
- 15.(a) Vogt H, Bräse S. Org. Biomol. Chem. 2007;5:406–430. doi: 10.1039/b611091f. [DOI] [PubMed] [Google Scholar]; (b) Nájera C. Synlett. 2002:1388–1493. [Google Scholar]
- 16.Arduin M, Spagnolo B, Calò G, Guerrini R, Carrà G, Fischetti C, Trapell C, Marzola E, McDonald J, Lambert DG, Regoli D, Salvadori S. Bioorg. Med. Chem. 2007;15:4434–4443. doi: 10.1016/j.bmc.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 17.Prasad S, Mathur A, Jaggi M, Singh AT, Mukherjee R. J. Pept. Sci. 2007;13:544–548. doi: 10.1002/psc.886. [DOI] [PubMed] [Google Scholar]
- 18.Vijayalakshmi S, Rao RB, Karle IL, Balaram P. Biopolymers. 2000;53:84–98. doi: 10.1002/(SICI)1097-0282(200001)53:1<84::AID-BIP8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 19.Saviano M, Iacovino R, Menchise V, Benedetti E, Bonora GM, Gatos M, Graci L, Formaggio F, Crisma M, Toniolo C. Biopolymers. 2000;53:200–212. doi: 10.1002/(SICI)1097-0282(200002)53:2<200::AID-BIP10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Saviano M, Iacovino R, Benedetti E, Moretto V, Banzato A, Formaggio F, Crisma M, Toniolo C. J. Pept. Sci. 2000;6:571–583. doi: 10.1002/1099-1387(200011)6:11<571::AID-PSC290>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 21.Moretto A, Formaggio F, Crisma M, Toniolo C, Saviano M, Iacovino R, Vitale RM, Benedetti E. J. Pept. Res. 2001;57:307–315. doi: 10.1046/j.1397-002x.2000.00834.x. [DOI] [PubMed] [Google Scholar]
- 22.Romanelli A, Garella I, Menchise V, Iacovino R, Saviano M, Montesarchio D, Didierjean C, di Lello P, Rossi F, Benedetti E. J. Pept. Sci. 2001;7:15–26. doi: 10.1002/psc.278. [DOI] [PubMed] [Google Scholar]
- 23.Datta S, Rathore RNS, Vijayalakshmi S, Vasudev PG, Rao RB, Balaram P, Shamala DN. J. Pept. Sci. 2004;10:160–172. doi: 10.1002/psc.507. [DOI] [PubMed] [Google Scholar]
- 24.Ohwada T, Kojima D, Kiwada T, Futaki S, Sugiura Y, Yamaguchi K, Nishi Y, Kobayashi Y. Chem. Eur. J. 2004;10:617–626. doi: 10.1002/chem.200305492. [DOI] [PubMed] [Google Scholar]
- 25.Wolf WM, Stasiak M, Leplawy MT, Bianco A, Formaggio F, Crisma M, Toniolo C. J. Am. Chem. Soc. 1998;120:11558–11566. [Google Scholar]
- 26.Savrda J, Mazaleyrat J-P, Wakselman M, Formaggio F, Crisma M, Toniolo C. J. Pept. Sci. 1999;5:61–74. doi: 10.1002/(SICI)1099-1387(199902)5:2<61::AID-PSC173>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Crisma M, Formaggio F, Mezzato S, Toniolo C, Savrda J, Mazaleyrat J-P, Wakselman M. Lett. Pept. Sci. 2000;7:123–131. [Google Scholar]
- 28.Mazaleyrat J-P, Wakselman M, Formaggio F, Crisma M, Toniolo C. Tetrahedron Lett. 1999;40:6245–6248. [Google Scholar]
- 29.Peggion C, Crisma M, Formaggio F, Toniolo C, Wright K, Wakselman M, Mazaleyrat J-P. Biopolymers. 2002;63:314–324. doi: 10.1002/bip.10071. [DOI] [PubMed] [Google Scholar]
- 30.Aschi M, Lucente G, Mazza F, Mollica A, Morera E, Nalli M, Paradisi MP. Org. Biomol. Chem. 2003;1:1980–1988. doi: 10.1039/b212247b. [DOI] [PubMed] [Google Scholar]
- 31.Formaggio F, Crisma M, Toniolo C, Tchertanov L, Guilhem J, Mazaleyrat J-P, Gaucher A, Wakselman M. Tetrahedron. 2000;56:8721–8734. [Google Scholar]
- 32.Formaggio F, Peggion C, Crisma M, Toniolo C, Tchertanov L, Guilhem J, Mazaleyrat J-P, Goubard Y, Gaucher A, Wakselman M. Helv. Chim. Acta. 2001;84:481–501. [Google Scholar]
- 33.Mazaleyrat J-P, Wright K, Gaucher A, Wakselman M, Oancea S, Formaggio F, Toniolo C, Setnička V, Kapitán J, Keiderling TA. Tetrahedron: Asymmetry. 2003;14:1879–1893. [Google Scholar]
- 34.Mazaleyrat J-P, Wright K, Gaucher A, Toulemonde N, Wakselman M, Oancea S, Peggion C, Formaggio F, Setnička V, Keiderling TA, Toniolo C. J. Am. Chem. Soc. 2004;126:12874–12879. doi: 10.1021/ja040100v. [DOI] [PubMed] [Google Scholar]
- 35.Mazaleyrat J-P, Wright K, Gaucher A, Toulemonde N, Dutot L, Wakselman M, Broxterman QB, Kaptein B, Oancea S, Peggion C, Crisma M, Formaggio F, Toniolo C. Chem. Eur. J. 2005;11:6921–6929. doi: 10.1002/chem.200500187. [DOI] [PubMed] [Google Scholar]
- 36.Dutot L, Gaucher A, Wright K, Wakselman M, Mazaleyrat J-P, Oancea S, Peggion C, Formaggio F, Toniolo C. Tetrahedron: Asymmetry. 2006;17:363–371. [Google Scholar]
- 37.Gaucher A, Dutot L, Barbeau O, Wakselman M, Mazaleyrat J-P, Peggion C, Oancea S, Formaggio F, Crisma M, Toniolo C. Tetrahedron: Asymmetry. 2006;17:30–39. [Google Scholar]
- 38.Mazaleyrat J-P, Goubard Y, Azzini M-V, Wakselman M, Peggion C, Formaggio F, Toniolo C. Eur. J. Org. Chem. 2002:1232–1247. [Google Scholar]
- 39.Lohier J-F, Wright K, Peggion C, Formaggio F, Toniolo C, Wakselman M, Mazaleyrat J-P. Tetrahedron. 2006;62:6203–6213. [Google Scholar]
- 40.Wright K, Lohier J-F, Wakselman M, Mazaleyrat J-P, Peggion C, Formaggio F, Toniolo C. Biopolymers (Pept. Sci.) 2007;88:797–806. doi: 10.1002/bip.20841. [DOI] [PubMed] [Google Scholar]
- 41.Gómez-Catalán J, Alemán C, Pérez JJ. Theor. Chem. Acc. 2000;103:380–389. [Google Scholar]
- 42.Casanovas J, Zanuy D, Nussinov R, Alemán C. Chem. Phys. Lett. 2006;429:558–562. [Google Scholar]
- 43.Alemán C, Zanuy D, Casanovas J, Cativiela C, Nussinov R. J. Phys. Chem. B. 2006;110:21264–21271. doi: 10.1021/jp062804s. [DOI] [PubMed] [Google Scholar]
- 44.Alemán C, Jiménez AI, Cativiela C, Pérez. JJ, Casanovas J. J. Phys. Chem. B. 2002;106:11849–11858. [Google Scholar]
- 45.Jiménez AI, Vanderesse R, Marraud M, Aubry A, Cativiela C. Tetrahedron Lett. 1997;38:7559–7562. [Google Scholar]
- 46.Jiménez AI, Cativiela C, Aubry A, Marraud M. J. Am. Chem. Soc. 1998;120:9452–9459. [Google Scholar]
- 47.Jiménez AI, Cativiela C, Marraud M. Tetrahedron Lett. 2000;41:5353–5356. [Google Scholar]
- 48.Casanovas J, Jiménez AI, Cativiela C, Pérez JJ, Alemán C. J. Org. Chem. 2003;68:7088–7091. doi: 10.1021/jo034720a. [DOI] [PubMed] [Google Scholar]
- 49.Jiménez AI, Ballano G, Cativiela C. Angew. Chem. Int. Ed. 2005;44:396–399. doi: 10.1002/anie.200461230. [DOI] [PubMed] [Google Scholar]
- 50.Casanovas J, Jiménez AI, Cativiela C, Pérez JJ, Alemán C. J. Phys. Chem. B. 2006;110:5762–5766. doi: 10.1021/jp0542569. [DOI] [PubMed] [Google Scholar]
- 51.Jiménez AI, Marraud M, Cativiela C. Tetrahedron Lett. 2003;44:3147–3150. [Google Scholar]
- 52.Peggion C, Formaggio F, Crisma M, Toniolo C, Jiménez AI, Cativiela C, Kaptein B, Broxterman QB, Saviano M, Benedetti E. Biopolymers. 2003;68:178–191. doi: 10.1002/bip.10295. [DOI] [PubMed] [Google Scholar]
- 53.Casanovas J, Jiménez AI, Cativiela C, Nussinov R, Alemán C. J. Org. Chem. 2008;73:644–651. doi: 10.1021/jo702107s. [DOI] [PubMed] [Google Scholar]
- 54.Jiménez. AI, Cativiela C, Gómez-Catalán J, Pérez JJ, Aubry A, París M, Marraud M. J. Am. Chem. Soc. 2000;122:5811–5821. [Google Scholar]
- 55.Gómez-Catalán J, Jiménez AI, Cativiela C, Pérez JJ. J. Pept. Res. 2001;57:435–446. doi: 10.1034/j.1399-3011.2001.00840.x. [DOI] [PubMed] [Google Scholar]
- 56.Lasa M, Jiménez AI, Zurbano MM, Cativiela C. Tetrahedron Lett. 2005;46:8377–8380. [Google Scholar]
- 57.Bisetty K, Corcho FJ, Canto J, Kruger HG, Pérez JJ. J. Pept. Sci. 2006;12:92–105. doi: 10.1002/psc.692. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka M, Demizu Y, Doi M, Kurihara M, Suemune H. Angew. Chem. Int. Ed. 2004;43:5360–5363. doi: 10.1002/anie.200460420. [DOI] [PubMed] [Google Scholar]
- 59.Royo S, De Borggraeve WM, Peggion C, Formaggio F, Crisma M, Jiménez AI, Cativiela C, Toniolo C. J. Am. Chem. Soc. 2005;127:2036–2037. doi: 10.1021/ja043116u. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka M, Anan K, Demizu Y, Kurihara M, Doi M, Suemune H. J. Am. Chem. Soc. 2005;127:11570–11571. doi: 10.1021/ja053842c. [DOI] [PubMed] [Google Scholar]
- 61.Grieco P, Lavecchia A, Cai M, Trivedi D, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. J. Med. Chem. 2002;45:5287–5294. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 62.Horvat Š, Mlinarić-Majerski K, Glavaš-Obrovac L, Jakas A, Veljković J, Marczi S, Kragol G, Roščić M, Matković M, Milostić-Srb A. J. Med. Chem. 2006;49:3136–3142. doi: 10.1021/jm051026+. [DOI] [PubMed] [Google Scholar]
- 63.Ruzza P, Cesaro L, Tourwé D, Calderan A, Biondi B, Maes V, Menegazzo I, Osler A, Rubini C, Guiotto A, Pinna LA, Borin G, Donella-Deana A. J. Med. Chem. 2006;49:1916–1924. doi: 10.1021/jm051080q. [DOI] [PubMed] [Google Scholar]
- 64.Strecker D. Ann. Chem. Pharm. 1850;75:27–45. [Google Scholar]
- 65.For specific reviews of the Strecker reaction and analogous synthesis, see: Vachal P, Jacobsen EN. In: In Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Berlin: Springer; 2004. p. 117. Ohfune Y, Shinada T. Bull. Chem. Soc. Jpn. 2003;76:1115–1129. Gröger H. Chem. Rev. 2003;103:2795–2828. doi: 10.1021/cr020038p. Enders D, Shilvock JP. Chem. Soc. Rev. 2000;29:359–373. Mori A, Inoue S. In: In Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. vol. II. Berlin: Springer; 1999. p. 983.
- 66.(a) Fadel A, Khesrani A. Tetrahedron: Asymmetry. 1998;9:305–320. [Google Scholar]; (b) Fadel A. Synlett. 1993:503–505. [Google Scholar]; (c) Subramanian PK, Woodard RW. Synth. Commun. 1986;16:337–342. [Google Scholar]; (d) Weinges K, Gries K, Stemmle B, Schrank W. Chem. Ber. 1977;110:2098–2105. [Google Scholar]
- 67.(a) Tang G, Tian H, Ma D. Tetrahedron. 2004;60:10547–10552. [Google Scholar]; (b) Ma D, Tian H, Zou G. J. Org. Chem. 1999;64:120–125. doi: 10.1021/jo981297a. [DOI] [PubMed] [Google Scholar]; (c) Ma D, Tang G, Tian H, Zou G. Tetrahedron Lett. 1999;40:5753–5756. [Google Scholar]; (d) Chakraborty TK, Hussain KA, Reddy GV. Tetrahedron. 1995;51:9179–9190. [Google Scholar]
- 68.Boesten WHJ, Seerden J-PG, Lange B, Dielemans HJA, Elsenberg HLM, Kaptein B, Moody HM, Kellogg RM, Broxterman QB. Org. Lett. 2001;3:1121–1124. doi: 10.1021/ol007042c. [DOI] [PubMed] [Google Scholar]
- 69.(a) Avenoza A, Busto JH, Corzana F, Peregrina JM, Sucunza D, Zurbano MM. Synthesis. 2005:575–578. [Google Scholar]; (b) Davis FA, Lee S, Zhang H, Fanelli DL. J. Org. Chem. 2000;65:8704–8708. doi: 10.1021/jo001179z. [DOI] [PubMed] [Google Scholar]; (c) Davis FA, Portonovo PS, Reddy RE, Chiu Y. J. Org. Chem. 1996;61:440–441. doi: 10.1021/jo9519928. [DOI] [PubMed] [Google Scholar]; (d) Bravo P, Capelli S, Meille SV, Seresini P, Volonterio A, Zanda M. Tetrahedron: Asymmetry. 1996;7:2321–2332. [Google Scholar]; (e) Davis FA, Reddy RE, Portonovo PS. Tetrahedron Lett. 1994;35:9351–9354. [Google Scholar]
- 70.(a) Ros A, Díez E, Marqués-López E, Martín-Zamora E, Vázquez J, Iglesias-Sigüenza J, Pappalardo RR, Alvarez E, Lassaletta JM, Fernández R. Tetrahedron: Asymmetry. 2008;19:998–1004. [Google Scholar]; (b) Enders D, Moser M. Tetrahedron Lett. 2003;44:8479–8481. [Google Scholar]
- 71.Choi JY, Kim YH. Tetrahedron Lett. 1996;37:7795–7796. [Google Scholar]
- 72.(a) Kunz H, Sager W, Pfrengle W, Schanzenbach D. Tetrahedron Lett. 1988;29:4397–4400. [Google Scholar]; (b) Kunz H, Sager W. Angew. Chem. Int. Ed. 1987;26:557–559. [Google Scholar]
- 73.For the synthesis of chiral 2-alkylcyclobutanones of type 6, see: Hazelard D, Fadel A. Tetrahedron: Asymmetry. 2005;16:2067–2070.
- 74.Truong M, Lecornué F, Fadel A. Tetrahedron: Asymmetry. 2003;14:1063–1072. [Google Scholar]
- 75.Volk F-J, Wagner M, Frahm AW. Tetrahedron: Asymmetry. 2003;14:497–502. [Google Scholar]
- 76.(a) Hazelard D, Fadel A, Girard C. Tetrahedron: Asymmetry. 2006;17:1457–1464. [Google Scholar]; (b) Hazelard D, Fadel A, Guillot R. Tetrahedron: Asymmetry. 2008;19:2063–2067. [Google Scholar]
- 77.For the synthesis of ketones of type 16, see: Cornubert R, Borrel C. Bull. Soc. Chim. Fr. 1930;47:301–322. Wiehl W, Frahm AW. Chem. Ber. 1986;119:2668–2677. Reetz MT, Maier WF. Angew. Chem. 1978;90:50.
- 78.(a) Meyer U, Breitling E, Bisel P, Frahm AW. Tetrahedron: Asymmetry. 2004;15:2029–2037. [Google Scholar]; (b) Wede J, Volk F-J, Frahm AW. Tetrahedron: Asymmetry. 2000;11:3231–3252. [Google Scholar]
- 79.Ma D, Tian H, Zou G. J. Org. Chem. 1999;64:120–125. doi: 10.1021/jo981297a. [DOI] [PubMed] [Google Scholar]
- 80.Schann S, Menet C, Arvault P, Mercier G, Frauli M, Mayer S, Hubert N, Triballeau N, Bertrand H-O, Acher F, Neuville P. Bioorg. Med. Chem. Lett. 2006;16:4856–4860. doi: 10.1016/j.bmcl.2006.06.062. [DOI] [PubMed] [Google Scholar]
- 81.For the preparation of peptides from 41 and 42, see: Levins CG, Schafmeister CE. J. Org. Chem. 2005;70:9002–9008. doi: 10.1021/jo051639u.
- 82. Bucherer HT, Brandt W. J. Prakt. Chem. 1934;140:129–150. Bucherer HT, Steiner W. J. Prakt. Chem. 1934;140:291–316. Ware E. Chem. Rev. 1950;46:403–470. doi: 10.1021/cr60145a001. Rousset A, Lasperas M, Taillades J, Commeyras A. Tetrahedron. 1980;36:2649–2661. Sarges R, Goldstein SW, Welch WM, Swindell AC, Siegel TW, Beyerpp TA. J. Med. Chem. 1990;33:1859–1865. doi: 10.1021/jm00169a005. For recent applications, see: (f) Pesquet A, Daïch A, Van Hijfte L. J. Org. Chem. 2006;71:5303–5311. doi: 10.1021/jo060616s. Averina EB, Yashin NV, Grishin YK, Kuznetsova TS, Zefirov NS. Synthesis. 2006:880–884.
- 83.(a) Nguyen Van Nhien A, Ducatel H, Len C, Postel D. Tetrahedron Lett. 2002;43:3805–3808. [Google Scholar]; (b) Postel D, Nguyen Van Nhien A, Villa P, Ronco G. Tetrahedron Lett. 2001;42:1499–1502. [Google Scholar]
- 84.Conti P, Pinto A, Roda G, Tamborini L, Arosio D, De Micheli C. Synthesis. 2007:2145–2148. [Google Scholar]
- 85.For the synthesis of LY354740, see: Ohfune Y, Demura T, Iwama S, Matsuda H, Namba K, Shimamoto K, Shinada T. Tetrahedron Lett. 2003;44:5431–5434. ; (b) Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP, Harkness AR, Grutsch JL, Writht RA, Johnson BG, Andis SL, Kingston A, Tomlinson R, Lewis R, Griffey KR, Tizzano JP, Schoepp DD. J. Med. Chem. 1999;42:1027–1040. doi: 10.1021/jm980616n. [DOI] [PubMed] [Google Scholar]; (c) Monn JA, Valli MJ, Massey SM, Writht RA, Salhoff CR, Johnson BG, Howe T, Alt CA, Rhodes GA, Robey RL, Griffey KR, Tizzano JP, Kallman MJ, Helton DR, Schoepp DD. J. Med. Chem. 1997;40:528–537. doi: 10.1021/jm9606756. [DOI] [PubMed] [Google Scholar]; (d) Ohfune Y, Demura T, Iwama S, Matsuda H, Namba K, Shimamoto K, Shinada T. Tetrahedron Lett. 2003;44:5431–5434. [Google Scholar]
- 86.Gereau RW, Swanson GT, editors. The Glutamate Receptors. Portland, OR: Humana Press Inc.; 2008. [Google Scholar]
- 87.(a) Fennell JW, Semo MJ, Wirth DD, Vaid RK. Synthesis. 2006:2659–2664. [Google Scholar]; (b) Bueno AB, Collado I, de Dios A, Domínguez C, Martín JA, Martín LM, Martínez-Grau MA, Montero C, Pedregal C, Catlow J, Coffey DS, Clay MP, Dantzig AH, Lindstrom T, Monn JA, Jiang H, Shoepp DD, Stratford RE, Tabas LB, Tizzano JP, Wright RA, Herin MF. J. Med. Chem. 2005;48:5305–5320. doi: 10.1021/jm050235r. [DOI] [PubMed] [Google Scholar]; (c) Coffey DS, Hawk MK, Pedersen SW, Vaid RK. Tetrahedron Lett. 2005;46:7299–7302. [Google Scholar]
- 88.Dominguez C, Prieto L, Valli MJ, Massey SM, Bures M, Wright RA, Johnson BG, Andis SL, Kingston A, Schoepp DD, Monn JA. J. Med. Chem. 2005;48:3605–3612. doi: 10.1021/jm040222y. [DOI] [PubMed] [Google Scholar]
- 89.For the synthesis of 52, see: Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Johnson BG, Howe T, Alt CA, Rhodes GA, Robey RL, Griffey KR, Tizzano JP, Kallan MJ, Helton DR, Schoepp DD. J. Med. Chem. 1997;40:528–537. doi: 10.1021/jm9606756.
- 90.Lee W, Miller MJ. J. Org. Chem. 2004;69:4516–4519. doi: 10.1021/jo0495034. [DOI] [PubMed] [Google Scholar]
- 91.For the cyclopropanation using sulfonium ylides, see: Zhang F, Moher ED, Zhang TY. Tetrahedron Lett. 2007;48:3277–3279. Aggarwal VK, Grange E. Chem. Eur. J. 2006;12:568–575. doi: 10.1002/chem.200500693. Domínguez C, Ezquerra J, Prieto L, Espada M, Pedregal C. Tetrahedron: Asymmetry. 1997;8:511–514.
- 92.Bessières B, Schoenfelder A, Verrat C, Mann A, Ornstein P, Pedregal C. Tetrahedron Lett. 2002;43:7659–7662. [Google Scholar]
- 93. Sibille P, López S, Brabet I, Valenti O, Oueslati N, Gaven F, Goudet C, Bertrand H-O, Neyton J, Marino MJ, Amalric M, Pin J-P, Acher FC. J. Med. Chem. 2007;50:3585–3595. doi: 10.1021/jm070262c. Reyes-Rangel G, Marañon V, Avila-Ortiz CG, Anaya de Parrodi C, Quintero L, Juaristi E. Tetrahedron. 2006;62:8404–8409. Fernández MC, Díaz A, Guillín JJ, Blanco O, Ruiz M, Ojea V. J. Org. Chem. 2006;71:6958–6974. doi: 10.1021/jo061072x. and references therein.; (d) Fernández MC, Quintela JM, Ruiz M, Ojea V. Tetrahedron: Asymmetry. 2002;13:233–237. [Google Scholar]; (e) Jiao X, Chen W, Hu B. Synth. Commun. 1992;22:1179–1186. [Google Scholar]
- 94.Tan L, Yasuda N, Yoshikawa N, Hartner FW, Eng KK, Leonard WR, Tsay F-R, Volante RP, Tillyer RD. J. Org. Chem. 2005;70:8027–8034. doi: 10.1021/jo0511187. [DOI] [PubMed] [Google Scholar]
- 95.Nakazato A, Sakagami K, Yasuhara A, Ohta H, Yoshikawa R, Itoh M, Nakamura M, Chaki S. J. Med. Chem. 2004;47:4570–4587. doi: 10.1021/jm0400294. [DOI] [PubMed] [Google Scholar]
- 96.(a) Yasuhara A, Sakagami K, Yoshikawa R, Chaki S, Nakamura M, Nakazato A. Bioorg. Med. Chem. 2006;14:3405–3420. doi: 10.1016/j.bmc.2005.12.061. [DOI] [PubMed] [Google Scholar]; (b) Yasuhara A, Nakamura M, Sakagami K, Shimazaki T, Yoshikawa R, Chaki S, Ohta H, Nakazato A. Bioorg. Med. Chem. 2006;14:4193–4207. doi: 10.1016/j.bmc.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 97. Gololobov YG, Kasukhin LF. Tetrahedron. 1992;48:1353–1406. Tian WQ, Wang YA. J. Org. Chem. 2004;69:4299–4308. doi: 10.1021/jo049702n. and references therein.
- 98.Woltering TJ, Adam G, Huguenin P, Wichmann J, Kolczewski S, Gatti S, Bourson A, Kew JNC, Richards G, Kemp JA, Mutel V, Knoflach F. ChemMedChem. 2008;3:323–335. doi: 10.1002/cmdc.200700226. [DOI] [PubMed] [Google Scholar]
- 99.Martins FJC, Viljoen AM, Kruger HG, Fourie L, Roscher J, Joubert AJ, Wessels PL. Tetrahedron. 2001;57:1601–1607. [Google Scholar]
- 100.Fondekar KP, Volk F-J, Frahm AW. Tetrahedron: Asymmetry. 1999;10:727–735. [Google Scholar]
- 101.Fondekar KP, Volk F-J, Khaliq-uz-Zaman SM, Bisel P, Frahm AW. Tetrahedron: Asymmetry. 2002;13:2241–2249. [Google Scholar]
- 102.Bisel P, Fondekar KP, Volk F-J, Frahm AW. Tetrahedron. 2004;60:10541–10545. [Google Scholar]
- 103.For the synthesis of 103, see: Pellicciari R, Natalini B, Luneia R, Marinozzi M, Roberti M, Rosato GC, Sadeghpour BM, Synyder JP, Monahan JB, Moroni F. Med. Chem. Res. 1992;2:491–496.
- 104.Warmuth R, Munsch TE, Stalker RA, Li B, Beatty A. Tetrahedron. 2001;57:6383–6397. [Google Scholar]
- 105.Gupta S, Das BC, Schafmeister CE. Org. Lett. 2005;7:2861–2864. doi: 10.1021/ol0507672. [DOI] [PubMed] [Google Scholar]
- 106.Stalker RA, Munsch TE, Tran JD, Nie X, Warmuth R, Beatty A, Aakeröy CB. Tetrahedron. 2002;58:4837–4849. [Google Scholar]
- 107.Ito H, Saito A, Kakuuchi A, Taguchi T. Tetrahedron. 1999;55:12741–12750. [Google Scholar]
- 108.(a) Shinada T, Kawakami T, Sakai H, Matsuda H, Omezawa T, Kawasaki M, Namba K, Ohfune Y. Bull. Chem. Soc. Jpn. 2006;79:768–774. [Google Scholar]; (b) Namba K, Kawasaki M, Takada I, Iwama S, Izumida M, Shinada T, Ohfune Y. Tetrahedron Lett. 2001;42:3733–3736. [Google Scholar]
- 109.For reviews, see: Ohfune Y, Shinada T. Eur. J. Org. Chem. 2005:5127–5143. doi: 10.1021/jo0477244. and references therein. (b) ref. 65b.
- 110.Reviews: Janey JM. Angew. Chem., Int. Ed. 2005;44:4292–4300. doi: 10.1002/anie.200462314. Erdik E. Tetrahedron. 2004;60:8747–8782. Greck C, Drouillat B, Thomassiang C. Eur. J. Org. Chem. 2004:1377–1385. Genet J-P, Greck C, Lavergne CD. In: Modern Amination Methods. Ricci A, editor. Weinheim: Wiley-VCH; 2000. ch. 3. Greck C, Genet J-P. Synlett. 1997;7:741–748. Krohn K. Organic Synthesis Highlights. Weinheim: VCH; 1991. pp. 45–53.
- 111.Felice E, Fioravanti S, Pellacani L, Tardella PA. Tetrahedron Lett. 1999;40:4413–4416. [Google Scholar]
- 112.Saaby S, Bella M, Jørgensen KA. J. Am. Chem. Soc. 2004;126:8120–8121. doi: 10.1021/ja047704j. [DOI] [PubMed] [Google Scholar]
- 113.Xu X, Yabuta T, Yuan P, Takemoto Y. Synlett. 2006:137–140. [Google Scholar]
- 114.Pihko PM, Pohjakallio A. Synlett. 2004:2115–2118. [Google Scholar]
- 115.Terada M, Nakano M, Ube H. J. Am. Chem. Soc. 2006;128:16044–16045. doi: 10.1021/ja066808m. [DOI] [PubMed] [Google Scholar]
- 116.Kang YK, Kim DY. Tetrahedron Lett. 2006;47:4565–4568. [Google Scholar]
- 117.Comelles J, Pericás A, Moreno-Mañas M, Vallribera A, Drudis-Solé G, Lledós A, Parella T, Roglans A, Santiago García-Granda S, Roces-Fernández L. J. Org. Chem. 2007;72:2077–2087. doi: 10.1021/jo0622678. [DOI] [PubMed] [Google Scholar]
- 118.For recent reviews, see: Chem. Rev. 2007;107 special issue on organocatalysis. Enders D, Grondal C, Hüttl MRM. Angew. Chem., Int. Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. Gaunt MJ, Johansson CCC, McNally A, Vo NT. Drug Discovery Today. 2007;12:8–27. doi: 10.1016/j.drudis.2006.11.004. Acc. Chem. Res. 2004;37 special issue on organocatalysis. Dalko PI, Moisan L. Angew. Chem., Int. Ed. 2004;43:5138–5175. doi: 10.1002/anie.200400650. Dalko PI, editor. Enantioselective Organocatalysis: Reactions and Experimental Procedures. New York: Wiley-VCH; 2007. Berkessel A, Gröger H, editors. Asymmetric Organocatalysis – From Biomimetic Concepts to Applications in Asymmetric Synthesis. New York: Wiley-VCH; 2005.
- 119.Suri JT, Steiner DD, Barbas CF., III Org. Lett. 2005;7:3885–3888. doi: 10.1021/ol0512942. [DOI] [PubMed] [Google Scholar]
- 120.Mashiko T, Hara K, Tanaka D, Fujiwara Y, Kumagai N, Shibasaki M. J. Am. Chem. Soc. 2007;129:11342–11343. doi: 10.1021/ja0752585. [DOI] [PubMed] [Google Scholar]
- 121.Mashiko T, Kumagai N, Shibasaki M. Org. Lett. 2008;10:2725–2728. doi: 10.1021/ol8008446. [DOI] [PubMed] [Google Scholar]
- 122.(a) Giannoukakis N. Expert Opin. Invest. Drugs. 2008;12:575–581. doi: 10.1517/13543784.17.4.575. [DOI] [PubMed] [Google Scholar]; (b) Kurono M, Fujii A, Murata M, Fujitani B, Negoro T. Biochem. Pharmacol. 2006;71:338–353. doi: 10.1016/j.bcp.2005.10.036. [DOI] [PubMed] [Google Scholar]; (c) Giannoukakis N. Curr. Opin. Invest. Drugs. 2006;7:916–923. [PubMed] [Google Scholar]; (d) Negoro T, Murata M, Ueda S, Fujitani B, Ono Y, Kuromiya A, Suzuki K, Matsumoto J-I. J. Med. Chem. 1998;41:4118–4129. doi: 10.1021/jm9802968. [DOI] [PubMed] [Google Scholar]; (e) Ishii A, Kotani T, Nagaki Y, Shibayama Y, Toyomaki Y, Okukada N, Ienaga K, Okamoto K. J. Med. Chem. 1996;39:1924–1927. doi: 10.1021/jm9508393. [DOI] [PubMed] [Google Scholar]; (f) Lee YS, Pearlstein R, Kador PF. J. Med. Chem. 1994;37:787–792. doi: 10.1021/jm00032a011. [DOI] [PubMed] [Google Scholar]; (g) Kador PF, Kinoshita JH, Sharpless NE. J. Med. Chem. 1985;28:841–849. doi: 10.1021/jm00145a001. [DOI] [PubMed] [Google Scholar]
- 123.(a) Satoh T, Hirano M, Kuroiwa A, Kaneko Y. Tetrahedron. 2006;62:9268–9279. [Google Scholar]; (b) Satoh T, Hirano M, Kuroiwa A. Tetrahedron Lett. 2005;46:2659–2662. [Google Scholar]
- 124.Katoh M, Hisa C, Honda T. Tetrahedron Lett. 2007;48:4691–4694. [Google Scholar]
- 125.(a) Domínguez C, Ezquerra J, Baker SR, Borrelly S, Prieto L, Espada M, Pedregal C. Tetrahedron Lett. 1998;39:9305–9308. [Google Scholar]; (b) Corey EJ, Link JO. J. Am. Chem. Soc. 1992;114:1906–1908. [Google Scholar]
- 126.For the transformation of proline derivatives into δ-lactams, see: Katoh M, Inoue H, Suzuki A, Honda T. Synlett. 2005:2820–2822. Katoh M, Mizutani H, Honda T. Tetrahedron Lett. 2005;46:5161–5163. Honda T, Takahashi R, Namiki H. J. Org. Chem. 2005;70:499–504. doi: 10.1021/jo048365f. Katoh M, Matsune R, Nagase H, Honda T. Tetrahedron Lett. 2004;45:6221–6223.
- 127.Achatz O, Grandl A, Wanner KT. Eur. J. Org. Chem. 1999:1967–1978. [Google Scholar]
- 128.Koch C-J, Šimonyiová S, Pabel J, Kärtner A, Polborn K, Wanner KT. Eur. J. Org. Chem. 2003:1244–1263. [Google Scholar]
- 129.(a) Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]; (b) Castro BR. Org. React. 1983;29:1–162. [Google Scholar]; (c) Hughess DL. Org. React. 1992;42:335–356. [Google Scholar]; (d) Hughess DL. Org. Prep. Proced. Int. 1996;28:127–164. [Google Scholar]; (e) Dodge JA, Jones SA. Rec. Res. Dev. Org. Chem. 1997;1:273–283. [Google Scholar]; (f) Dembinski R. Eur. J. Org. Chem. 2004:2763–2772. [Google Scholar]; (g) Dandapani S, Curran DP. Tetrahedron. 2002;58:3855–3864. [Google Scholar]; (h) Lipshutz BH, Chung DW, Rich B, Corral R. Org. Lett. 2006;8:5069–5072. doi: 10.1021/ol0618757. [DOI] [PubMed] [Google Scholar]
- 130.Sibille P, López S, Brabet I, Valenti O, Oueslati N, Gaven F, Goudet C, Bertrand H-O, Neyton J, Marino MJ, Almalric M, Pin J-P, Acher FC. J. Med. Chem. 2007;50:3585–3595. doi: 10.1021/jm070262c. [DOI] [PubMed] [Google Scholar]
- 131.For the synthesis of 180 and 185, see: Burgess K, Ho K-K. Tetrahedron Lett. 1992;33:5677–5680. Burgess K, Ho K-K. J. Org. Chem. 1992;57:5931–5936.
- 132.For the synthesis of racemic 184 and 186, see: Johnson RL, Rao KSSP. Bioorg. Med. Chem. Lett. 2005;15:57–60. doi: 10.1016/j.bmcl.2004.10.040.
- 133.Bhattacharya AK, Thyagarajan G. Chem. Rev. 1981;81:415–430. [Google Scholar]
- 134.Debache A, Collet S, Bauchat P, Danion D, Euzenat L, Hercouet A, Carboni B. Tetrahedron: Asymmetry. 2001;12:761–764. [Google Scholar]
- 135.Fox ME, Lennon IC, Farina V. Tetrahedron Lett. 2007;48:945–948. [Google Scholar]
- 136.For the epimerization of (1R,2S)-N-Boc-dehydrocoronamic acid methyl ester catalyzed by ruthenium carbenes, see: Zeng X, Wei X, Farina V, Napolitano E, Xu Y, Zhang L, Haddad N, Yee NK, Grinberg N, Shen S, Senanayake CH. J. Org. Chem. 2006;71:8864–8875. doi: 10.1021/jo061587o.
- 137.(a) Katagiri T, Irie M, Uneyama K. Org. Lett. 2000;2:2423–2425. doi: 10.1021/ol0059955. [DOI] [PubMed] [Google Scholar]; (b) Katagiri T, Uneyama K. Chirality. 2003;15:4–9. doi: 10.1002/chir.10160. [DOI] [PubMed] [Google Scholar]
- 138. Wallis ES, Lane JF. Org. React. 1946:267–306. Gogoi P, Konwar D. Tetrahedron Lett. 2007;48:531–533. and references therein.
- 139.Donkor IO, Zheng X, Han J, Miller DD. Chirality. 2000;12:551–557. [PubMed] [Google Scholar]
- 140.(a) Lebel H, Leogane O. Org. Lett. 2006;8:5717–5720. doi: 10.1021/ol0622920. [DOI] [PubMed] [Google Scholar]; (b) Lebel H, Leogane O. Org. Lett. 2005;7:4107–4110. doi: 10.1021/ol051428b. [DOI] [PubMed] [Google Scholar]; (c) Marinescu L, Thinggaard J, Thomsen IB, Bols M. J. Org. Chem. 2003;68:9453–9455. doi: 10.1021/jo035163v. [DOI] [PubMed] [Google Scholar]; (d) Scriven EF, Turnbull K. Chem. Rev. 1988;88:297–368. [Google Scholar]; (e) Smith PAS. Org. React. 1946;3:337–449. [Google Scholar]
- 141.Frick JA, Klassen JB, Rapoport H. Synthesis. 2005:1751–1756. [Google Scholar]
- 142.Chang HS, Bergmeier SC, Frick JA, Bathe A, Rapoport H. J. Org. Chem. 1994;59:5336–5342. [Google Scholar]
- 143.(a) Chinchilla R, Falvello LR, Galindo N, Nájera C. J. Org. Chem. 2000;65:3034–3041. doi: 10.1021/jo991736l. [DOI] [PubMed] [Google Scholar]; (b) Abellán T, Chinchilla R, Galindo N, Guillena G, Nájera C, Sansano JM. Eur. J. Org. Chem. 2000:2689–2697. [Google Scholar]
- 144.(a) Abellán T, Balbino M, Nájera C, Sansano JM. Tetrahedron. 2001;57:6627–6640. [Google Scholar]; (b) Abellán T, Nájera C, Sansano JM. Tetrahedron: Asymmetry. 2000;11:1051–1055. [Google Scholar]; (c) Abellán T, Chinchilla R, Galindo N, Nájera C, Sansano JM. J. Heterocyclic Chem. 2000;37:467–479. [Google Scholar]
- 145.Buñuel E, Bull SD, Davies SG, Garner AC, Savory ED, Smith AD, Vickers RJ, Watkin DJ. Org. Biomol. Chem. 2003;1:2531–2542. doi: 10.1039/b303348a. [DOI] [PubMed] [Google Scholar]
- 146.Papageorgiou CD, Cubillo de Dios MA, Ley SV, Gaunt MJ. Angew. Chem. Int. Ed. 2004;43:4641–4644. doi: 10.1002/anie.200460234. [DOI] [PubMed] [Google Scholar]
- 147.Badorrey R, Cativiela C, Díaz-de-Villegas MD, Gálvez JA. Tetrahedron: Asymmetry. 2000;11:1015–1025. [Google Scholar]
- 148.Koch C-J, Höfner G, Polborn K, Wanner KT. Eur. J. Org. Chem. 2003:2233–2242. [Google Scholar]
- 149.Howarth NM, Wakelin LPG, Walker DM. Tetrahedron Lett. 2003;44:695–698. [Google Scholar]
- 150.Tanaka M, Anan K, Demizu Y, Kurihara M, Doi M, Suemune H. J. Am. Chem. Soc. 2005;127:11570–11571. doi: 10.1021/ja053842c. [DOI] [PubMed] [Google Scholar]
- 151.Ma D, Ding K, Tian H, Wang B, Cheng D. Tetrahedron: Asymmetry. 2002;13:961–969. [Google Scholar]
- 152.Gabaitsekgosi R, Hayes CJ. Tetrahedron Lett. 1999;40:7713–7716. [Google Scholar]
- 153.Bradley DM, Mapitse R, Thompson NM, Hayes CJ. J. Org. Chem. 2002;67:7613–7617. doi: 10.1021/jo025892v. [DOI] [PubMed] [Google Scholar]
- 154.Soengas RG, Estévez JC, Estévez RJ. Tetrahedron: Asymmetry. 2003;14:3955–3963. [Google Scholar]
- 155.For the synthesis of racemic quaternary tetrahydrofuran α-amino acids derivatives, see: Maity P, Zabel M, König B. J. Org. Chem. 2007;72:8046–8053. doi: 10.1021/jo701423w.
- 156.Solladié-Cavallo A, Martín-Cabrejas LM, Caravatti G, Lang M. Tetrahedron: Asymmetry. 2001;12:967–969. [Google Scholar]
- 157.Saaby S, Nakama K, Lie MA, Hazell RG, Jørgensen KA. Chem. Eur. J. 2003;9:6145–6154. doi: 10.1002/chem.200305302. [DOI] [PubMed] [Google Scholar]
- 158.Zhuang W, Saaby S, Jørgensen KA. Angew. Chem. Int. Ed. 2004;43:4476–4478. doi: 10.1002/anie.200460158. [DOI] [PubMed] [Google Scholar]
- 159.For recent reviews, see: Schrodi Y, Pederson RL. Aldrichimca Acta. 2007;40:45–52. ; (b) Adv. Synth. Catal. 2007;349:1–268. doi: 10.1002/adsc.200600264. Olefin Metathesis Special Issue. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chauvin Y. Angew. Chem., Int. Ed. 2006;45:3740–3747. doi: 10.1002/anie.200601234. [DOI] [PubMed] [Google Scholar]; (d) Schrock RR. Angew. Chem. Int. Ed. 2006;45:3748–3759. doi: 10.1002/anie.200600085. [DOI] [PubMed] [Google Scholar]; (e) Grubbs RH. Angew. Chem. Int. Ed. 2006;45:3760–3765. doi: 10.1002/anie.200600680. [DOI] [PubMed] [Google Scholar]; (f) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; (g) Grubbs RH. Tetrahedron. 2004;60:7117–7140. [Google Scholar]; (h) Grubbs RH, editor. Handbook of Metathesis. Vols 1–3. Weinheim: Wiley-VCH; 2003. [Google Scholar]; (i) Connon SJ, Bhechert S. Angew. Chem. Int. Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]; (j) Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (k) Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; (l) Buchmeiser MR. Chem. Rev. 2000;100:1565–1604. doi: 10.1021/cr990248a. [DOI] [PubMed] [Google Scholar]; (m) Fürstner A. Angew. Chem. Int. Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]
- 160.(a) Krikstolaitytè S, Hammer K, Undheim K. Tetrahedron Lett. 1998;39:7595–7598. [Google Scholar]; (b) Hammer K, Rømming C, Undheim K. Tetrahedron. 1998;54:10837–10850. [Google Scholar]; (c) Hammer K, Undheim K. Tetrahedron: Asymmetry. 1998;9:2359–2368. [Google Scholar]
- 161.Hammer K, Wang J, Falck-Pedersen ML, Rømming C, Undheim K. J. Chem. Soc., Perkin Trans 1. 2000:1691–1695. [Google Scholar]
- 162.Jam F, Tullberg M, Luthman K, Grøtli M. Tetrahedron. 2007;63:9881–9889. [Google Scholar]
- 163.Undheim K, Efskind J. Tetrahedron. 2000;56:4847–4857. [Google Scholar]
- 164.Efskind J, Römming C, Undheim K. J. Chem. Soc., Perkin Trans 1. 2001:2697–2703. [Google Scholar]
- 165.Hoven GB, Efskind J, Rømming C, Undheim K. J. Org. Chem. 2002;67:2459–2463. doi: 10.1021/jo010888p. [DOI] [PubMed] [Google Scholar]
- 166.Efskind J, Undheim K. Tetrahedron Lett. 2003;44:2837–2839. [Google Scholar]
- 167.Undheim K, Efskind J, Hoven GB. Pure Appl. Chem. 2003;75:279–292. [Google Scholar]
- 168.Andrei M, Efskind J, Undheim K. Tetrahedron. 2007;63:4347–4355. [Google Scholar]
- 169.Trost BM, Dogra K. J. Am. Chem. Soc. 2002;124:7256–7257. doi: 10.1021/ja020290e. [DOI] [PubMed] [Google Scholar]
- 170.(a) Fustero S, Sánchez-Roselló M, Rodrigo V, del Pozo C, Sanz-Cervera JF, Simón-Fuentes A. Org. Lett. 2006;8:4129–4132. doi: 10.1021/ol061733c. [DOI] [PubMed] [Google Scholar]; (b) Fustero S, Sánchez-Roselló M, Rodrigo V, Sanz-Cervera JF, Piera J, Simón-Fuentes A, del Pozo C. Chem. Eur. J. 2008;14:7019–7029. doi: 10.1002/chem.200702009. [DOI] [PubMed] [Google Scholar]
- 171.Krikstolaityté S, Sackus A, Rømming C, Undheim K. Tetrahedron: Asymmetry. 2001;12:393–398. [Google Scholar]
- 172.(a) Møller B, Undheim K. Eur. J. Org. Chem. 2003:332–336. [Google Scholar]; (b) Efskind J, Rømming C, Undheim K. J. Chem. Soc., Perkin Trans. 1. 1999:1677–1684. [Google Scholar]
- 173.Andrei M, Undheim K. Tetrahedron: Asymmetry. 2004;15:53–63. [Google Scholar]
- 174.For the non-asymmetric cyclopropanation of α-nitro-α-diazocarbonyl derivatives, see: Wurz RP, Charette AB. J. Org. Chem. 2004;69:1262–1269. doi: 10.1021/jo035596y. ; Charette AB, Wurz RP, Ollivier T. Helv. Chim. Acta. 2002;85:4468–4484. [Google Scholar]
- 175.For review on the chiral catalyst for asymmetric transformations of vinyl- and aryldiazoacetates, see: Davies HML. Eur. J. Org. Chem. 1999:2459–2469.
- 176. Charette AB, Wurz RP. J. Mol. Catal. A: Chem. 2003;196:83–91. For cobalt-catalyzed asymmetric cyclopropanation of alkenes with α-nitrodiazoacetates, see: Zhu S, Perman JA, Zhang XP. Angew. Chem. Int. Ed. 2008;47:1–5. doi: 10.1002/anie.200803857.
- 177.Moreau B, Charette AB. J. Am. Chem. Soc. 2005;127:18014–18015. doi: 10.1021/ja056192l. [DOI] [PubMed] [Google Scholar]
- 178.Barluenga J, Aznar F, Gutiérrez I, García-Granda S, Llorca-Baragaño MA. Org. Lett. 2002;4:4273–4276. doi: 10.1021/ol026896p. [DOI] [PubMed] [Google Scholar]
- 179.Clerici F, Gelmi ML, Pocar D, Pilati T. Tetrahedron: Asymmetry. 2001;12:2663–2669. [Google Scholar]
- 180.For the origin of the π-facial diastereoselection of this reaction, see: Muray E, Alvarez-Larena A, Piniella JF, Branchadell V, Ortuño RM. J. Org. Chem. 2000;65:388–396. doi: 10.1021/jo991227j.
- 181.Rifé J, Ortuño RM. Tetrahedron: Asymmetry. 1999;10:4245–4260. [Google Scholar]
- 182.Illescas B, Rifé J, Ortuño RM, Martín N. J. Org. Chem. 2000;65:6246–6248. doi: 10.1021/jo0003955. [DOI] [PubMed] [Google Scholar]
- 183.Moglioni AG, García-Expósito E, Alvarez-Larena A, Branchadell V, Moltrasio GY, Ortuño RM. Tetrahedron: Asymmetry. 2000;11:4903–4914. [Google Scholar]
- 184.Avenoza A, Busto JH, Canal N, Peregrina JM, Pérez-Fernández M. Org. Lett. 2005;7:3597–3600. doi: 10.1021/ol0514707. [DOI] [PubMed] [Google Scholar]
- 185.For the synthesis of non-chiral 1-aminocyclobutanecarboxylic acid 2-substituted via [2+2] cycloaddition of 2-aminoacrylates, see: Avenoza A, Busto JH, Mata L, Peregrina JM, Pérez-Fernández M. Synthesis. 2008:743–746. ; (b) Jiménez-Osés G, Corzana F, Busto JH, Pérez-Fernández M, Peregrina JM, Avenoza A. J. Org. Chem. 2006;71:1869–1878. doi: 10.1021/jo0521955. [DOI] [PubMed] [Google Scholar]; (c) Avenoza A, Busto JH, Canal N, Peregrina JM. J. Org. Chem. 2005;70:330–333. doi: 10.1021/jo048943s. [DOI] [PubMed] [Google Scholar]; (d) Avenoza A, Busto JH, Peregrina JM, Pérez-Fernández M. Tetrahedron. 2005;61:4165–4172. [Google Scholar]; (e) Avenoza A, Busto JH, Canal N, Peregrina JM. Chem. Commun. 2003:1376–1377. [PubMed] [Google Scholar]
- 186.Tanaka K, Takahashi M, Imase H, Osaka T, Noguchi K, Hirano M. Tetrahedron. 2008;64:6289–6293. [Google Scholar]
- 187.Ung AT, Schafer K, Lindsay KB, Pyne SG, Amornraksa K, Wouters R, Van der Linden I, Biesmans I, Lesage ASJ, Skelton BW, White AH. J. Org. Chem. 2002;67:227–233. doi: 10.1021/jo010864i. [DOI] [PubMed] [Google Scholar]
- 188.Chinchilla R, Falvello LR, Galindo N, Nájera C. Tetrahedron: Asymmetry. 1999;10:821–825. [Google Scholar]
- 189.Burkett BA, Chai CLL. Tetrahedron Lett. 2001;42:2239–2242. [Google Scholar]
- 190.Abbiati G, Clerici F, Gelmi ML, Gambini A, Pilati T. J. Org. Chem. 2001;66:6299–6304. doi: 10.1021/jo010374q. [DOI] [PubMed] [Google Scholar]
- 191.For the Diels-Alder cycloaddition of 424 with cyclopentadiene, see: Clerici F, Gelmi ML, Pellegrino S, Pilati T. J. Org. Chem. 2003;68:5286–5291. doi: 10.1021/jo030085x. ; (b) Clerici F, Gelmi ML, Gambini A. J. Org. Chem. 2001;66:4941–4944. doi: 10.1021/jo010257v. [DOI] [PubMed] [Google Scholar]; (c) Clerici F, Gelmi ML, Gambini A. J. Org. Chem. 2000;65:6138–6141. doi: 10.1021/jo000595g. [DOI] [PubMed] [Google Scholar]
- 192.Caputo F, Clerici F, Gelmi ML, Pellegrino S, Pilati T. Tetrahedron: Asymmetry. 2006;17:61–67. [Google Scholar]
- 193.Caputo F, Clerici F, Gelmi ML, Pellegrino S, Pocar D. Tetrahedron: Asymmetry. 2006;17:1430–1436. [Google Scholar]
- 194.Gelmi ML, Caputo F, Clerici F, Pellegrino S, Gianaccini G, Betti L, Fabbrini L, Schmid L, Palego L, Lucacchini A. Bioorg. Med. Chem. 2007;15:7581–7589. doi: 10.1016/j.bmc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 195.For the application of diol exo-442 in the synthesis of cyclopentylglycine derivatives, see: Pellegrino S, Clerici F, Gelmi ML. Tetrahedron. 2008;64:5657–5665.
- 196.Caputo F, Cattaneo C, Clerici F, Gelmi ML, Pellegrino S. J. Org. Chem. 2006;71:8467–8472. doi: 10.1021/jo061391o. [DOI] [PubMed] [Google Scholar]
- 197.Gelmi ML, Cattaneo C, Pellegrino S, Clerici F, Montali M, Martini C. J. Org. Chem. 2007;72:9811–9814. doi: 10.1021/jo7019702. [DOI] [PubMed] [Google Scholar]
- 198.Williams LJ, Jagadish B, Lansdown MG, Carducci MD, Mash EA. Tetrahedron. 1999;55:14301–14322. [Google Scholar]
- 199.Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP, Harkness AR, Grutsch JL, Jr, Wright RA, Johnson BG, Andis SL, Kingston A, Tomlinson R, Lewis R, Griffey KR, Tizzano JP, Schoepp DD. J. Med. Chem. 1999;42:1027–1040. doi: 10.1021/jm980616n. [DOI] [PubMed] [Google Scholar]
- 200.Monn JA, Massey SM, Valli MJ, Henry SS, Stephenson GA, Bures M, Hérin M, Catlow J, Giera D, Wright RA, Johnson BG, Andis SL, Kingston A, Schoepp DD. J. Med. Chem. 2007;50:233–240. doi: 10.1021/jm060917u. [DOI] [PubMed] [Google Scholar]
- 201.Oishi S, Kang S-U, Liu H, Zhang M, Yang D, Deschamps JR, Burke TR., Jr Tetrahedron. 2004;60:2971–2977. [Google Scholar]
- 202.Avenoza A, Barriobero JI, Cativiela C, Fernández-Recio MA, Peregrina JM, Rodríguez F. Tetrahedron. 2001;57:2745–2755. [Google Scholar]
- 203.Mazaleyrat J-P, Boutboul A, Lebars Y, Gaucher A, Wakselman M. Tetrahedron: Asymmetry. 1998;9:2701–2713. [Google Scholar]
- 204.Ridvan L, Buděšínsky M, Tichý M, Maloň P, Závada J, Podlaha J, Císařová I. Tetrahedron. 1999;55:12331–12348. [Google Scholar]
- 205.(a) Mazaleyrat J-P, Wright K, Azzini M-V, Gaucher A, Wakselman M. Tetrahedron Lett. 2003;44:1741–1745. [Google Scholar]; (b) Wright K, Lohier J-F, Wakselman M, Mazaleyrat J-P, Formaggio F, Peggion C, De Zotti M, Toniolo C. Tetrahedron. 2008;64:2307–2320. [Google Scholar]
- 206.For the application of derivatives of (R)-502 in the assignment of the absolute configuration of β-amino acids, see: Dutot L, Wright K, Gaucher A, Wakselman M, Mazaleyrat J-P, De Zotti M, Peggion C, Formaggio F, Toniolo C. J. Am. Chem. Soc. 2008;130:5986–5992. doi: 10.1021/ja800059d.
- 207.Hodgson DM, Thompson AJ, Wadman S, Keats CJ. Tetrahedron. 1999;55:10815–10834. [Google Scholar]
- 208.Varie DL, Beck C, Borders SK, Brady MD, Cronin JS, Ditsworth TK, Hay DA, Hoard DW, Hoying RC, Linder RJ, Miller RD, Moher ED, Remacle JR, Rieck JA., III Org. Proc. Res. Develop. 2007;11:546–559. [Google Scholar]
- 209.Salgado A, Huybrechts T, Eeckhaut A, Van der Eycken J, Szakonyi Z, Fülöp F, Tkachev A, De Kimpe N. Tetrahedron. 2001;57:2781–2786. [Google Scholar]
- 210.Beaulieu PL, Gillard J, Bailey MD, Boucher C, Duceppe J-S, Simoneau B, Wang X-J, Zhang L, Grozinger K, Houpis I, Farina V, Heimroth H, Krueger T, Schnaubelt J. J. Org. Chem. 2005;70:5869–5879. doi: 10.1021/jo050468q. [DOI] [PubMed] [Google Scholar]
- 211.For the epimerization of 515, see: Zeng X, Wei X, Farina V, Napolitano E, Xu Y, Zhang L, Haddad N, Yee NK, Grinberg N, Shen S, Senanayake CH. J. Org. Chem. 2006;71:8864–8875. doi: 10.1021/jo061587o.
- 212.For selected examples, see: Shu C, Zeng X, Hao M-H, Wei X, Yee NK, Busacca CA, Han Z, Farina V, Senanayake CH. Org. Lett. 2008;10:1303–1306. doi: 10.1021/ol800183x. ; (b) Liverton NJ, Holloway MK, McCauley JA, Rudd MT, Butcher JW, Carroll SS, DiMuzio J, Fandozzi C, Gilbert KF, Mao S-S, McIntyre CJ, Nguyen KT, Romano JJ, Sthlhut M, Wan B-L, Olsen DB, Vacca JP. J. Am. Chem. Soc. 2008;130:4607–4609. doi: 10.1021/ja711120r. [DOI] [PubMed] [Google Scholar]; (c) Randolph JT, Zhang X, Huang PP, Klein LL, Kurtz KA, Konstantinidis AK, He W, Kati WM, Kempf DJ. Bioorg. Med. Chem. Lett. 2008;18:2745–2750. doi: 10.1016/j.bmcl.2008.02.053. [DOI] [PubMed] [Google Scholar]; (d) Naud J, Lemke C, Goudreau N, Beualieu E, White PD, Llinàs-Brunet M, Forgione P. Bioorg. Med. Chem. Lett. 2008;18:3400–3404. doi: 10.1016/j.bmcl.2008.04.012. [DOI] [PubMed] [Google Scholar]; (e) Poirier M, Aubry N, Boucher C, Ferland J-M, LaPlante S, Tsantrizos YS. J. Org. Chem. 2005;70:10765–10773. doi: 10.1021/jo051706k. [DOI] [PubMed] [Google Scholar]
- 213.Kirihara M, Kawasaki M, Takuwa T, Kakuda H, Wakikawa T, Takeuchi Y, Kirk KL. Tetrahedron: Asymmetry. 2003;14:1753–1761. [Google Scholar]
- 214.Roda G, Conti P, De Amici M, He J, Polavarapu PL, De Micheli C. Tetrahedron: Asymmetry. 2004;15:3079–3090. [Google Scholar]
- 215.For the aplication of quaternary α-amino acids 525 and 528 as ionotropic and metabotropic glutamate receptor subtypes, see: Conti P, De Amici M, Grazioso G, Roda G, Pinto A, Hansen KB, Nielsen B, Madsen U, Bräuner-Osborne H, Egebjerg J, Vestri V, Pellegrini-Giampietro DE, Sibille P, Acher FC, De Micheli C. J. Med. Chem. 2005;48:6315–6325. doi: 10.1021/jm0504499.
- 216.(a) Lane JW, Halcomb RL. J. Org. Chem. 2003;68:1348–1357. doi: 10.1021/jo020532t. [DOI] [PubMed] [Google Scholar]; (b) Lane JW, Halcomb RL. Tetrahedron. 2001;57:6531–6538. [Google Scholar]
- 217.Tanaka M, Demizu Y, Nagano M, Hama M, Yoshida Y, Kurihara M, Suemune H. J. Org. Chem. 2007;72:7750–7756. doi: 10.1021/jo701317d. [DOI] [PubMed] [Google Scholar]
- 218.Jiménez AI, López P, Oliveros L, Cativiela C. Tetrahedron. 2001;57:6019–6026. [Google Scholar]
- 219.Jiménez AI, López P, Cativiela C. Chirality. 2005;17:22–29. doi: 10.1002/chir.20091. [DOI] [PubMed] [Google Scholar]
- 220.Lasa M, López P, Cativiela C. Tetrahedron: Asymmetry. 2005;16:4022–4033. [Google Scholar]
- 221.Cativiela C, Lasa M, López P. Tetrahedron: Asymmetry. 2005;16:2613–2623. [Google Scholar]
- 222.Natalini B, Marinozzi M, Bade K, Sardella R, Thomsen C, Pellicciari R. Chirality. 2004;16:314–317. doi: 10.1002/chir.20027. [DOI] [PubMed] [Google Scholar]
- 223.Alías M, Cativiela C, Jiménez AI, López P, Oliveros L, Marraud M. Chirality. 2001;13:48–55. doi: 10.1002/1520-636X(2001)13:1<48::AID-CHIR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 224.Avenoza A, París M, Peregrina JM, Alías M, López MP, García JI, Cativiela C. Tetrahedron. 2002;58:4899–4905. [Google Scholar]
- 225.Cativiela C, López P, Lasa M. Eur. J. Org. Chem. 2004:3898–3908. [Google Scholar]
- 226.Alías M, Lasa M, López MP, García JI, Cativiela C. Tetrahedron. 2005;61:2913–2919. [Google Scholar]
- 227.Patwardhan AP, Pulgam VR, Zhang Y, Wulff WD. Angew. Chem. Int. Ed. 2005;44:6169–6172. doi: 10.1002/anie.200500923. [DOI] [PubMed] [Google Scholar]
- 228.For the synthesis of aziridine-2-carboxylic acids alkyl esters, see: Hansen KB, Finney NS, Jacobsen EN. Angew. Chem., Int. Ed. Engl. 1995;34:676–678. Osborn H, Sweeney MIJ. Tetrahedron: Asymmetry. 1997;8:1693–1715. Muller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. Bew SP, Hughes DL, Savic V, Soapi KM, Wilson MA. Chem. Commun. 2006:3513–3515. doi: 10.1039/b606033a.
- 229.Lu Z, Zhang Y, Wulff WD. J. Am. Chem. Soc. 2007;129:7185–7194. doi: 10.1021/ja069371r. [DOI] [PubMed] [Google Scholar]
- 230.Calaza MI, Cativiela C. Eur. J. Org. Chem. 2008:3427–3448. doi: 10.1002/ejoc.200800225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sommer K, Williams RM. Tetrahedron. 2008;64:7106–7111. doi: 10.1016/j.tet.2008.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chandan N, Moloney MG. Org. Biomol. Chem. 2008;6:3664–3666. doi: 10.1039/b811642c. [DOI] [PubMed] [Google Scholar]
- 233.Kazmaier U, Mues H, Krebs A. Chem. Eur. J. 2002;8:1850–1855. doi: 10.1002/1521-3765(20020415)8:8<1850::AID-CHEM1850>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 234.Elliott MC, Wood JL, Wordinghan SV. Trends Heterocycl. Chem. 2005;10:7395. [Google Scholar]
- 235.(a) Seebach D, Naef R. Helv. Chim. Acta. 1981;64:2704–2708. [Google Scholar]; (b) Seebach D, Boes M, Naef R, Schweizer WJ. J. Am. Chem. Soc. 1983;105:5390–5398. [Google Scholar]; (c) Beck AK, Blank S, Job K, Seebach D. Sommerfeld. Org. Synth. 1995;72:62–73. [Google Scholar]
- 236.Seebach D, Sting AR, Hoffmann M. Angew. Chem., Int. Ed. Engl. 1996;35:2708–2748. [Google Scholar]
- 237.For the preparation of α-alkyl prolines from alkylation of (3S,7aR)-586, see: Bittermann H, Gmeiner P. J. Org. Chem. 2006;71:97–102. doi: 10.1021/jo0517287. Bittermann H, Böckler F, Einsiedel J, Gmeiner P. Chem. Eur. J. 2006;12:6315–6322. doi: 10.1002/chem.200600432. (c) See also ref. 230
- 238.Pisaneschi F, Cordero FM, Lumini M, Brandi A. Synlett. 2007:2882–2884. [Google Scholar]
- 239.(a) Garner P, Park J-M. J. Org. Chem. 1987;52:2361–2364. [Google Scholar]; (b) Garner P, Park J-M. Org. Synth. 1992;70:18–28. [Google Scholar]; (c) Marshall JA, Beaudoin S. J. Org. Chem. 1996;61:581–586. doi: 10.1021/jo9517914. [DOI] [PubMed] [Google Scholar]
- 240.Okue M, Kobayashi H, Shin-ya K, Furihata K, Hayakawa Y, Seto H, Watanabe H, Kitahara T. Tetrahedron Lett. 2002;43:857–860. [Google Scholar]
- 241.Watanabe H, Okue M, Kobayashi H, Kitahara T. Tetrahedron Lett. 2002;43:861–864. [Google Scholar]
- 242. Dondoni A, Perrone D. Org. Synth. 2000;77:64–77. (b) Ref. 216
- 243.Vaswani RG, Chamberlin AR. J. Org. Chem. 2008;73:1661–1681. doi: 10.1021/jo702329z. [DOI] [PubMed] [Google Scholar]
- 244.Sayago FJ, Calaza MI, Jiménez AI, Cativiela C. Tetrahedron. 2008;64:84–91. doi: 10.1016/j.tet.2007.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Alonso DA, Nordin SJM, Andersson PG. Org. Lett. 1999;1:1595–1597. [Google Scholar]
- 246.Goldspink NJ, Simpkins NS, Beckmann M. Synlett. 1999:1292–1294. [Google Scholar]
- 247.Hou D-R, Hung S-Y, Hu C-C. Tetrahedron: Asymmetry. 2005;16:3858–3864. [Google Scholar]
- 248.Balducci D, Grandi A, Porzi G, Sandri S. Tetrahedron: Asymmetry. 2005;16:1453–1462. [Google Scholar]
- 249.Palomo C, Aizpurua JM, Balentová E, Jiménez A, Oyarbide J, Fratila RM, Miranda JI. Org. Lett. 2007;9:101–104. doi: 10.1021/ol0626241. [DOI] [PubMed] [Google Scholar]
- 250.Balducci D, Porzi G, Sandri S. Tetrahedron: Asymmetry. 2004;15:1085–1093. [Google Scholar]
- 251.Ooi T, Takeuchi M, Maruoka K. Synthesis. 2001:1716–1718. [Google Scholar]
- 252.Andrei M, Efskind J, Viljugrein T, Römming C, Undheim K. Tetrahedron: Asymmetry. 2004;15:1301–1313. [Google Scholar]
- 253.Andrei M, Römming C, Undheim K. Tetrahedron: Asymmetry. 2004;15:1359–1370. [Google Scholar]
- 254.Buñuel E, Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 1996;7:1431–1436. [Google Scholar]
- 255.Buñuel E, Gil AM, Díaz-de-Villegas MD, Cativiela C. Tetrahedron. 2001;57:6417–6427. [Google Scholar]
- 256.(a) Corey EJ, Winter RAE. J. Am. Chem. Soc. 1963;85:2677–2678. [Google Scholar]; (b) Corey EJ, Carey FA, Winter RAE. J. Am. Chem. Soc. 1965;87:934–935. [Google Scholar]; (c) Corey EJ, Hopkins PB. Tetrahedron Lett. 1982;23:1979–1982. [Google Scholar]
- 257.Gil AM, Buñuel E, Díaz-de-Villegas MD, Cativiela C. Tetrahedron: Asymmetry. 2003;14:1479–1488. [Google Scholar]
- 258.(a) Gil AM, Orús E, López-Carrillo V, Buñuel E, Cativiela C. Tetrahedron: Asymmetry. 2005;16:3115–3123. [Google Scholar]; (b) Gil AM, Buñuel E, Cativiela C, Attanasi OA, Spinelli D. Targets in Heterocyclic Systems. Chemistry and Properties (Volume 8) Cambridge: Royal Society of Chemistry; 2005. pp. 56–86. [Google Scholar]
- 259.(a) Zhao H, Hsu DC, Carlier PR. Synthesis. 2005:1–16. [Google Scholar]; (b) Kawabata T, Fuji K. Top. Stereochem. 2003;23:175–205. [Google Scholar]; (c) Kawabata T, Chen J, Suzuki H, Nagae Y, Kinoshita T, Chancharunee S, Fuji K. Org. Lett. 2000;2:3883–3885. doi: 10.1021/ol0066274. [DOI] [PubMed] [Google Scholar]; (d) Fuji K, Kawabata T. Chem. Eur. J. 1998;4:373–376. [Google Scholar]; (e) Kawabata T, Wirth T, Yahiro K, Suzuki H, Fuji K. J. Am. Chem. Soc. 1994;116:10809–10810. [Google Scholar]
- 260.Kawabata T, Kawakami S, Majumdar S. J. Am. Chem. Soc. 2003;125:13012–13013. doi: 10.1021/ja0378299. [DOI] [PubMed] [Google Scholar]
- 261.Kawabata T, Matsuda S, Kawakami S, Monguchi D, Moriyama K. J. Am. Chem. Soc. 2006;128:15394–15395. doi: 10.1021/ja0670761. [DOI] [PubMed] [Google Scholar]
- 262.Kawabata T, Moriyama K, Kawakami S, Tsubaki K. J. Am. Chem. Soc. 2008;130:4153–4157. doi: 10.1021/ja077684w. [DOI] [PubMed] [Google Scholar]
- 263.Moriyama K, Sakai H, Kawabata T. Org. Lett. 2008;10:3883–3886. doi: 10.1021/ol801213c. [DOI] [PubMed] [Google Scholar]
- 264.Kawabata T, Majundar S, Tsubaki K, Monguchi D. Org. Biomol. Chem. 2005;3:1609–1611. doi: 10.1039/b416535g. [DOI] [PubMed] [Google Scholar]
- 265.Gardiner J, Abell AD. Tetrahedron Lett. 2003;44:4227–4230. [Google Scholar]
- 266.Gardiner J, Abell AD. Org. Biomol. Chem. 2004;2:2365–2370. doi: 10.1039/B406450J. [DOI] [PubMed] [Google Scholar]
- 267.For the one-pot aza-Darzens reaction of sulfinimines with lithium α-bromoenolates, see: Davis FA, Liang C-H, Liu H. J. Org. Chem. 1997;62:3796–3797. Davis FA, Liu H, Zhou P, Fang T, Reddy GV, Zhang Y. J. Org. Chem. 1999;64:7559–7567. doi: 10.1021/jo991389f.
- 268.Davis FA, Deng J, Zhang Y, Haltiwanger RC. Tetrahedron. 2002;58:7135–7143. [Google Scholar]
- 269.Anelli PL, Beltrami A, Lolli M, Uggeri F. Synth. Commun. 1993;23:2639–2645. [Google Scholar]
- 270.Davis FA, Zhang Y, Rao A, Zhang Z. Tetrahedron. 2001;58:6345–6352. [Google Scholar]
- 271.Agbodjan AA, Cooley BE, Copley RCB, Corfield JA, Flanagan RC, Glover BN, Guidetti R, Haigh D, Howes PD, Jackson MM. Matsuoka RT, Medhurst KJ, Millar A, Sharp MJ, Slater MJ, Toczko JF, Xie S. J. Org. Chem. 2008;73:3094–3102. doi: 10.1021/jo800062c. [DOI] [PubMed] [Google Scholar]
- 272.Tsubogo T, Saito S, Seki K, Yamashita Y, Kobayashi S. J. Am. Chem. Soc. 2008;130:13321–13332. doi: 10.1021/ja8032058. [DOI] [PubMed] [Google Scholar]
- 273.Nájera C, Retamosa MG, Sansano JM, Cozar A, Cossío FP. Eur. J. Org. Chem. 2007:5038–5049. [Google Scholar]
- 274.Nájera C, Retamosa MG, Sansano JM. Angew. Chem. Int. Ed. 2008;47:6055–6058. doi: 10.1002/anie.200801690. [DOI] [PubMed] [Google Scholar]
- 275.For the synthesis of highly substituted conformationally constrained and spiro nitroprolines via Ag(I) catalyzed 1,3-cycloadditions, see: Grigg R, Kilner C, Sarker MAB, Orgaz de la Cierva C, Dondas HA. Tetrahedron. 2008;64:8974–8991.
- 276.Fukuzawa S-I, Oki H. Org. Lett. 2008;10:1747–1750. doi: 10.1021/ol8003996. [DOI] [PubMed] [Google Scholar]
- 277.López-Pérez A, Adrio J, Carretero JC. J. Am. Chem. Soc. 2008;130:10084–10085. doi: 10.1021/ja804021m. [DOI] [PubMed] [Google Scholar]
- 278.Matsumura Y, Inoue M, Nakamura Y, Talib IL, Maki T, Onomura O. Tetrahedron Lett. 2000;41:4619–4622. [Google Scholar]
- 279.Avenoza A, Cativiela C, Busto JH, Fernández-Recio MA, Peregrina JM, Rodríguez F. Tetrahedron. 2001;57:545–548. [Google Scholar]
- 280.Avenoza A, Barriobero JI, Busto JH, Cativiela C, Peregrina JM. Tetrahedron: Asymmetry. 2002;13:625–632. [Google Scholar]
- 281.Gerona-Navarro G, Bonache MA, Alías M, Pérez-de-Vega MJ, García-López MT, López P, Cativiela C, Gonzalez-Muñiz R. Tetrahedron Lett. 2004;45:2193–2196. [Google Scholar]
- 282.(a) Gil AM, Buñuel E, López P, Cativiela C. Tetrahedron: Asymmetry. 2004;15:811–819. [Google Scholar]; (b) (a) Gil AM, Buñuel E, Cativiela C. Arkivoc. 2007;iv:157–159. [Google Scholar]
- 283.Silveira CC, Bernardi CR, Braga AL, Kaufman TS. Tetrahedron Lett. 1999;40:4969–4972. [Google Scholar]
- 284.Alezra V, Bonin M, Micouin L, Husson H-P. Tetrahedron Lett. 2001;42:2111–2113. [Google Scholar]
- 285.Kawabata T, Oztürk O, Suzuki H, Fuji K. Synthesis. 2003:505–508. [Google Scholar]
- 286.Rutjes FPJT, Veerman JJN, Meester WJN, Hiemstra H, Schoemaker HE. Eur. J. Org. Chem. 1999:1127–1135. [Google Scholar]
- 287.Clayden J, Knowles FE, Menet CJ. Tetrahedron Lett. 2003;44:3397–3400. [Google Scholar]
- 288.Makosza M, Sulikowski D, Maltsev O. Synlett. 2008:1711–1713. [Google Scholar]