

Abstract
Dualsteric ligands represent a novel mode of targeting G protein-coupled receptors (GPCRs). These compounds attach simultaneously to both, the orthosteric transmitter binding site and an additional allosteric binding area of a receptor protein. This approach allows the exploitation of favourable characteristics of the orthosteric and the allosteric site by a single ligand molecule. The orthosteric interaction provides high affinity binding and activation of receptors. The allosteric interaction yields receptor subtype-selectivity and, in addition, may modulate both, efficacy and intracellular signalling pathway activation. Insight into the spatial arrangement of the orthosteric and the allosteric site is far advanced in the muscarinic acetylcholine receptor, and the design of dualsteric muscarinic agonists has now been accomplished. Using the muscarinic receptor as a paradigm, this review summarizes the way from suggestive evidence for an orthosteric/allosteric overlap binding to the rational design and experimental validation of dualsteric ligands. As allosteric interactions are increasingly described for GPCRs and as insight into the spatial geometry of ligand/GPCR-complexes is growing impressively, the rational design of dualsteric drugs is a promising new approach to achieve fine-tuned GPCR-modulation.
This article is part of a themed section on Molecular Pharmacology of GPCR. To view the editorial for this themed section visit http://dx.doi.org/10.1111/j.1476-5381.2010.00695.x
Keywords: G protein-coupled receptors, allosteric, orthosteric, dualsteric, bitopic, multivalent, subtype selectivity, functional selectivity, muscarinic acetylcholine receptor
The promise
The superfamily of G protein-coupled receptors (GPCRs) has high relevance with regard to both, current drug treatment and new drug discovery (Chung et al., 2008; Heilker et al., 2009). A traditional approach in the design of new ligands is to target the receptor binding site of the endogenous messenger compound. This site is classified as ‘orthosteric’ site. Ligands that compete with the messenger compound for this site are named orthosteric ligands. It is increasingly recognized that GPCRs may contain additional, ‘allosteric’ binding sites that allow for simultaneous receptor binding of both, an orthosteric and an allosteric ligand (for recent reviews see Gao and Jacobson, 2006; May et al., 2007a; Conn et al., 2009; De Amici et al., 2009). Such ternary complex formation allows for novel qualities of drug action such as an enhancement of binding and action of the endogenous receptor activator. In addition, allosteric ligands typically display selectivity for receptor subtypes. Whereas the orthosteric site of the endogenous messenger molecule is usually conserved between the receptor subtypes, allosteric receptor areas may reveal considerable heterogeneity with respect to amino acid sequence and spatial conformation. Yet, allosteric ligands may not reach the level of affinity that is seen with ligands of the orthosteric site. Recently, a novel approach in drug design has been proposed that should allow the exploitation of the above-mentioned favourable consequences that arise from addressing both the orthosteric and the allosteric site of a receptor protein (Disingrini et al., 2006; Steinfeld et al., 2007). As a proof of concept, a new type of receptor activator has been introduced that is constructed to utilize simultaneously orthosteric and allosteric receptor areas to achieve both, receptor activation and selectivity with respect to receptor subtype binding and intracellular signalling pathway activation (Antony et al., 2009). This novel topography of drug/receptor-interaction has been referred to as dualsteric binding. Terms also used in this context are ‘bitopic orthosteric/allosteric’ (Tahtaoui et al., 2004; Valant et al., 2008) and ‘multivalent orthosteric/allosteric’ (Steinfeld et al., 2007). For a brief nomenclature we suggest ‘dualsteric’ to address concomitant orthosteric/allosteric site occupancy as well as the ligands that display this property. The rational design of dualsteric compounds requires detailed insight into the three-dimensional structure of the receptor protein of interest and into the topography of orthosteric and allosteric ligand binding. With the increasing number of crystallized drug/GPCR-complexes such insight will soon increase (Rosenbaum et al., 2009). Although not having at hand crystallized complexes until now, knowledge about the topography of ligand binding is well advanced in the field of muscarinic acetylcholine receptors (M-receptors). There are five M-receptor subtypes that differ in tissue distribution, G protein-coupling preference and physiological function (Wess et al., 2007; Alexander et al., 2008). The M2 subtype was the first GPCR to be recognized as being sensitive to allosteric modulation (Lüllmann et al., 1969; Clark and Mitchelson, 1976). Since then, intensive efforts were spent by various groups to gain insight into the molecular determinants governing allosteric/orthosteric interactions at muscarinic receptors. Meanwhile muscarinic receptors have evolved as prototypes for the investigation of allosteric GPCR modulation (for review see Birdsall and Lazareno, 2005; De Amici et al., 2009). In the following we will review how the growing insight into the molecular events underlying muscarinic allosteric/orthosteric receptor interactions paved the way for the rational design of dualsteric ligands that activate muscarinic receptors in a subtype- and signalling pathway-selective fashion. We will discuss experimental approaches to validate that the intended dualsteric receptor targeting is in fact attained. In this context, aspects of GPCR structure and function will be mentioned that await clarification before a fully advanced rational design of dualsteric ligands can be achieved that promises high affinity and subtype-selectivity in conjunction with well adjusted efficacy and signalling pathway selectivity.
A settled quest: spatial vicinity between the allosteric and orthosteric site
The best-studied allosteric site of M-receptors is located in the entrance of the ligand binding pocket that harbours the orthosteric receptor binding site further down in the region of the transmembrane helices (Figure 1A). Worth mentioning, there is evidence for more than one allosteric site on muscarinic receptors (for an overview about nomenclature and ligands see Alexander et al., 2008). First evidence for an allosteric binding topography arose from kinetic binding studies using radiolabelled orthosteric antagonists (Stockton et al., 1983; Nedoma et al., 1986; Jepsen et al., 1988; Potter et al., 1989). Muscarinic allosteric ligands typically retard the dissociation and the association of orthosteric radioligands. It was suggested that the allosteric site is located extracellular to the orthosteric site (Jakubík and Tuček, 1994). Allosteric inhibition of orthosteric ligand association results from allosteric ligand binding to the orthosterically free receptor. Inhibition of orthosteric ligand dissociation is a consequence of ternary complex formation, i.e. allosteric agent binding to the orthosterically liganded receptor. Noteworthy, in other GPCRs such as the α2-adrenergic receptor (Leppik et al., 1998) orthosteric ligand dissociation is accelerated by allosteric modulators. The allosteric inhibitive actions on muscarinic orthosteric ligand association and dissociation often differ, as the binding affinity of the allosteric ligands is sensitive to the differences in conformation between the free and the orthosterically liganded receptor. Depending on the affinity ratio or, in other words, the relative extent by which orthosteric ligand association and dissociation are inhibited, the allosteric ligand may change equilibrium binding of the orthosteric ligand:
Figure 1.
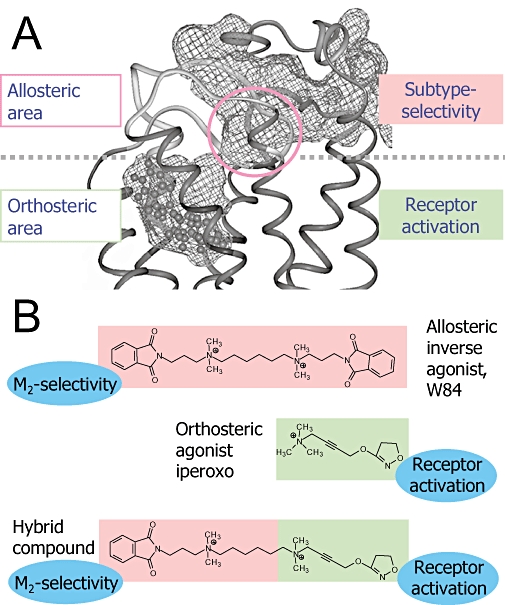
‘Blue-print’ for the design of dualsteric receptor activators with subtype-selectivity. (A) Side view on the upper part of the muscarinic M2 acetylcholine receptor. The free volume of the ligand binding pocket is lined by the grid structure. The model is based on the crystal structure of bovine rhodopsin in the inactive state. An inverse agonist (N-methylscopolamine) is docked into the orthosteric site to stabilize an inactive receptor conformation in the molecular dynamics simulation (modified from Voigtländer et al., 2003). The circle marks the allosteric core region which harbours non-conserved subtype-selectivity providing amino acids. (B) Design concept for subtype-selective allosteric/orthosteric hybrid agonists. The allosteric building block is derived from the alkane-bis-ammonio (ABA)-type compound W84, iperoxo (agonist d) is the orthosteric building block, the resulting hybrid compound is 1d.
Association inhibition > dissociation inhibition → decrease of orthoster binding
Association inhibition < dissociation inhibition → increase of orthoster binding
Association inhibition = dissociation inhibition → unchanged orthoster binding
This is the kinetic basis of negative, positive and neutral cooperativity which means a reciprocal inhibiting, enhancing or neutral action on the equilibrium binding between allosteric and orthosteric ligand at muscarinic receptors (Mohr et al., 2003).
The location of the allosteric site on the receptor protein was narrowed down by making use of the subtype selective receptor interaction that is a characteristic of muscarinic allosteric ligands (e.g. Ellis et al., 1991; Lee and El-Fakahany, 1991). This results from a less well conserved amino acid sequence between the muscarinic receptor subtypes in their extracellular regions. Chimeric receptor constructs helped to identify relevant receptor areas (Ellis et al., 1993), point mutated receptors allowed to identify key amino acids for allosteric subtype selectivity. These are contained in the second outer loop (EDGE sequence, 172Glu-173Asp-174Gly-175Glu and 177Tyr in M2; Leppik et al., 1994; Voigtländer et al., 2003; Huang et al., 2005) and in the region of the end of the third outer loop/beginning of transmembrane helix 7 [TM7 (Gnagey et al., 1999; Krejcí and Tuček, 2001; Jakubík et al., 2005), in particular M2423Thr (Buller et al., 2002), position 7.36 according to Ballesteros and Weinstein, 1995]. Subtype-independent allosteric base-line affinity is provided by Trp 7.35 that is conserved among the five muscarinic subtypes and located in close vicinity to M2177Tyr (Prilla et al., 2006). Furthermore, the conserved disulfide bridge between transmembrane helix TM3 and M2176Cys of the second outer loop is relevant for allosteric and orthosteric ligand binding (Avlani et al., 2007; Huang and Ellis, 2007). Modelling of the [3H]NMS-bound receptor and allosteric docking simulations revealed that the above-mentioned receptor epitopes are clustered in the so-called allosteric core region that is located at the bottom of the allosteric binding area next to the orthosteric site (Figure 1A; Jöhren and Höltje, 2002; Voigtländer et al., 2003; Prilla et al., 2006). This insight into the allosteric/orthosteric topography provides the rational fundament for the design of dualsteric ligands. The concept is to fuse an orthosteric ligand intended to activate the receptor with an appropriate allosteric anchor to achieve muscarinic subtype-selectivity (Figure 1B).
A closer look at binding modes
When the binding topography of a ligand is to be classified, some additional aspects are worth to be considered with respect to orthosteric, allosteric and dualsteric binding.
Orthosteric binding in muscarinic receptors requires that the ligand has to pass the allosteric region which may modulate both, access to and egress from the orthosteric site. Worth mentioning, the rather fast binding kinetics of [3H]N-methylscopolamine observed in the M2 subtype are slowed down without a relevant change of the binding affinity of [3H]NMS, when amino acids of the M2 allosteric core region are replaced by their corresponding counterparts from the M5-subtype. The M5 receptor subtype is characterized by very slow [3H]NMS binding kinetics, which are speeded up when the corresponding allosteric M2 amino acids are introduced (Buller et al., 2002; Prilla et al., 2006). The findings suggest that orthosteric ligands interact with epitopes of the extracellular allosteric site on their way to and from the orthosteric site.
The model of the NMS-bound M2 receptor (Figure 1A) is characterized by a narrow junction between the orthosteric and the allosteric area. Other conformations will have to occur allowing for sufficient free volume that is required for the passage of bulky inverse agonists such as N-methylscopolamine. In addition, with respect to the receptor conformation engaged in the binding of orthosteric agonists, there is strong evidence for a major conformational change of the allosteric/orthosteric junction in the active agonist-bound receptor protein (Grossmüller et al., 2006; Jäger et al., 2007).
Allosteric binding of a receptor ligand is experimentally obvious, when the equilibrium binding of an orthosteric probe is increased. If orthosteric radioligand binding is decreased by a test compound, allosteric binding is typically proven experimentally by an allosteric modulation of orthosteric radioligand dissociation. As can be taken intuitively from Figure 1A, muscarinic allosteric agents typically inhibit the dissociation of orthosteric radio-antagonists and radio-agonists (Stockton et al., 1983; Nedoma et al., 1986; Jepsen et al., 1988; Gnagey and Ellis, 1996; Kostenis and Mohr, 1996; Grossmüller et al., 2006).
Noteworthy, if a test compound has revealed an effect on orthosteric radioligand dissociation, this finding does not necessarily indicate that the test compound adopts the same binding topography in the orthosterically free receptor. Under this condition the ligand might slip down into the orthosteric orientation. For instance, even the classical orthosteric antagonist atropine is found to induce an allosteric inhibition of [3H]NMS-dissociation but only at excessive concentrations (Waelbroeck et al., 1992; Tränkle et al., 1996). Regarding the interaction of allosteric agents with the free receptor, an orthosteric radioligand association binding assay can hardly discriminate between an allosteric or an orthosteric binding topography of the test compound under investigation; both topographies would result in an inhibition of radioligand association. Additional features of allosteric agent binding such as the pronounced sensitivity of typical muscarinic allosteric agents on the ionic composition of the assay buffer (Schröter et al., 2000) or the dependence of allosteric binding affinity on allosterically versus orthosterically point mutated receptors may help to clarify the issue (Antony et al., 2009).
For archetypal allosteric agents such as alcuronium, gallamine and the alkane-bis-ammonio (ABA)-type compounds, there is no evidence that they move down into the orthosteric location in orthosterically free receptors (Schröter et al., 2000). Binding affinities of the ABA-type compounds W84 (Figure 1B) and naphmethonium do not respond to mutation of the orthosteric M2104Tyr (3.33) which is located next to M2103Asp (3.32) which is an orthosteric epitope of key importance for the binding of acetylcholine and other conventional orthosteric agonists and antagonists (Antony et al., 2009). Another experimental evidence for purely allosteric binding in free receptors comes from the ABA-type allosteric antagonist radioligand [3H]dimethyl-W84. Compared with orthosteric antagonist ligands such as [3H]N-methylscopolamine, the allosteric radioligand has dramatically faster receptor dissociation kinetics (Tränkle et al., 2003). This observation is in line with a superficial location in the allosteric binding site (Figure 1A).
Dualsteric binding is defined as simultaneous orthosteric/allosteric overlap binding of a receptor ligand. Other terms having been used to address such binding topography are ‘bitopic orthosteric/allosteric’ or ‘multivalent orthosteric/allosteric’. This binding mode has to be distinguished from ternary complex formation which means simultaneous binding of two independent molecules (different or identical) to the allosteric and the orthosteric site respectively. In addition, dualsteric binding has to be distinguished from a multiple binding mode, i.e. ligand binding that switches in a ‘flip-flop’-like fashion between two or more receptor topographies, in our case for instance between a purely allosteric and a purely orthosteric binding. Such ‘flip-flop’-binding might be worthwhile to consider in the case of small ligand molecules with comparably low binding affinity (i.e. ligands that have a poor fit to the receptor protein) – in contrast to voluminous high affinity ligands that are likely to have a fixed binding topography.
Muscarinic ligands heralding an allosteric/orthosteric binding topography
Initial experimental evidence that suggested an allosteric/orthosteric overlap binding emerged from a deeper mechanistic study of compounds that were developed in academic and industrial laboratories as subtype-selective muscarinic antagonists. With the advent of the antiulcer drug and M1-preferring muscarinic antagonist pirenzepine it became clear that the population of muscarinic receptors is heterogeneous (Hammer et al., 1980). Subsequently five subtypes of the muscarinic receptors were identified (Kubo et al., 1986; Bonner et al., 1987).
Methoctramine was introduced by Melchiorre and coworkers who developed polymethylene tetraamines as subtype-selective muscarinic receptor probes. Methoctramine (Figure 2) is an antagonist with pronounced preference for the M2 subtype (Melchiorre et al., 1987a; b;). Studies in living tissue preparations point to a competitive type of receptor interaction with orthosteric agonists over a wide concentration range of methoctramine. In addition, at very high concentrations (100-fold higher than its affinity constant) methoctramine was found to inhibit orthosteric radioantagonist dissociation – a hallmark of allosterism (Giraldo et al., 1988; Roffel et al., 1989). Melchiorre et al. (1989) proposed that methoctramine binds to both the orthosteric and allosteric site of the M2 receptor, capturing affinity from a non-conserved cluster of negatively charged allosteric amino acids (EDGE-sequence, 172Glu-173Asp-174Gly-175Glu). When the orthosteric site is blocked by a radioligand, the allosteric interaction is still possible, but with a lower affinity. In other words, methoctramine may switch from an allosteric/orthosteric high affinity binding to a purely allosteric low affinity binding. Receptor mutagenesis findings did not allow to decide whether methoctramine binds exclusively to the allosteric site or to both the orthosteric and the allosteric site (Matsui et al., 1995; Ellis and Seidenberg, 2000).
Figure 2.
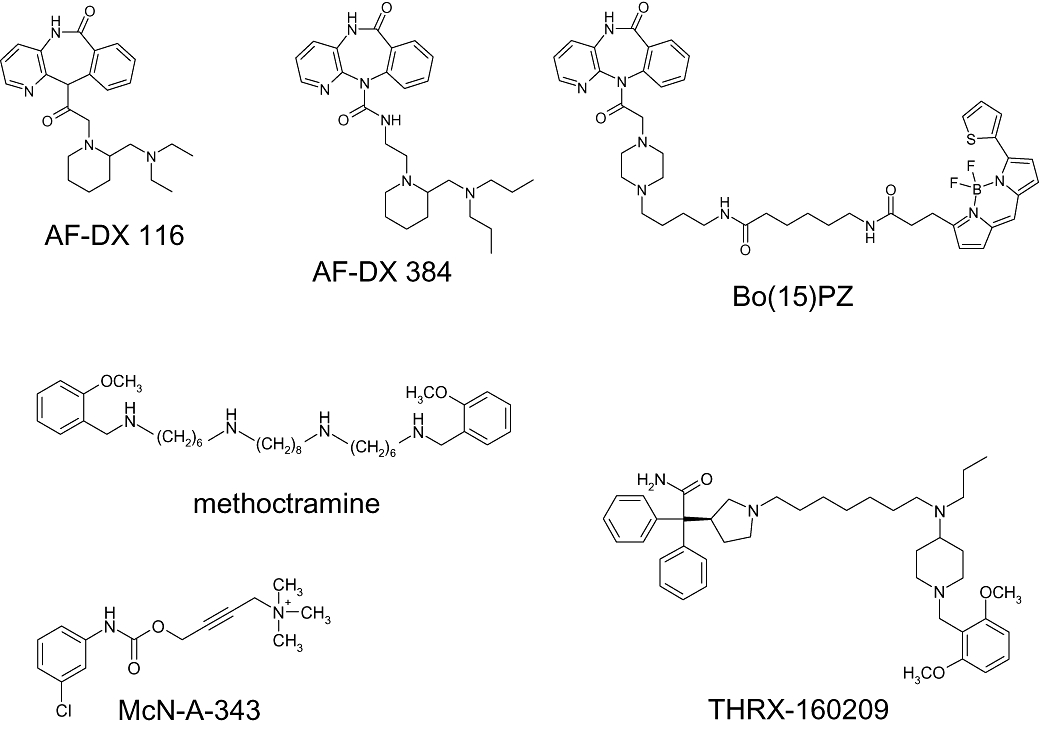
Structures of ligands with a suggested allosteric/orthosteric binding mode at muscarinic receptors.
AF-DX 116 and AF-DX 384 (Figure 2) are M2-preferring antagonists with a selectivity profile of M2 greater than M1 greater than M3 (Eberlein et al., 1989; Engel et al., 1989). Displacement of radiolabelled [3H]AF-DX 384 by atropine and AF-DX 116 was compatible with a competitive interaction (Entzeroth and Mayer, 1990). For AF-DX 116 (otenzepad) evidence for an allosteric interaction was found by Lee and El-Fakahany (1991) who applied radioligand binding in cloned muscarinic receptor subtypes and showed that M2 preferring antagonists including AF-DX 116 allosterically inhibit dissociation of the orthosteric [3H]N-methylscopolamine. Yet, ternary complex formation with [3H]NMS-occupied receptors requires high concentrations that by far exceed those that inhibit equilibrium binding of [3H]MNS. Noteworthy, both actions reveal the same order of subtype selectivity M2 > M1 > M3. The authors conclude that competitive displacement may be mediated via the primary (orthosteric) receptor binding site, whereas the inhibition of radioligand dissociation is the consequence of interaction with the secondary (allosteric) site of the receptor. Evidence for an allosteric component of AF-DX 116 was also provided by Lanzafame et al. (2001) who studied its interaction with allosteric and orthosteric antagonists in guinea pig atria and in radioligand binding assays.
For AF-DX 384, combination experiments in paced guinea pig atria suggested that the ABA-type allosteric antagonist modulator W84 binds with considerably lower affinity to M2 receptors that are liganded with AF-DX 384 compared with the conventional orthosteric antagonist N-methylscopolamine (NMS). In line with this, allosteric inhibition by W84 of the dissociation of [3H]NMS was much more pronounced than was dissociation of [3H]AF-DX 384. In addition, at high concentrations, purely allosteric binding of AF-DX 384 to [3H]NMS-liganded M2 receptors was shown. It was concluded that M2 receptor binding of AF-DX 384 extends beyond the orthosteric site and partially includes the allosteric site of the M2 receptor protein, thereby utilizing allosteric epitopes not being conserved among the muscarinic receptor subtypes (Tränkle et al., 1998). Use of stepwise shortened derivatives of W84 as probes suggested that the overlap of AF-DX 384 with the M2 allosteric site extends to about half of the area used by W84 (Mohr et al., 2004), i.e. part of AF-DX 384 would point from the orthosteric site into the allosteric core region (cf. Figure 1). A W84-derived building block was fused with AF-DX 384 hoping that this might further increase the allosteric overlap area of AF-DX 384. However, the resulting compound appeared to bind purely in the allosteric mode instead of the intended orthosteric/allosteric location. It was concluded that the AF-DX 384/W84-hybrid was hindered by its W84-building block from properly diving into the orthosteric binding pocket. Alternatively, the ABA-building block was introduced into the AF-DX 384 molecule at an unfavourable position not facing the allosteric site.
Taken together, the binding topography of the M2-preferring antagonists of the methoctramine- and AF-DX-type has not been fully elucidated until now. When the above-mentioned studies were carried out, allosteric key amino acids had not been identified. Now, that the close spatial vicinity between the orthosteric binding pocket and the allosteric core region of the M2 receptor has emerged (Figure 1A), the hypothesis of an orthosteric/allosteric overlap is revitalized with the orthosteric binding providing high affinity and the allosteric binding providing M2-selectivity.
Fluorescent pirenzepine derivatives
Ilien and coworkers (Tahtaoui et al., 2004) prepared fluorescent derivatives of the M1 antagonist pirenzepine. Fluorophores such as bodipy were linked with the antagonist through linkers of varying chain length. To check for simultaneous orthosteric/allosteric binding, dissociation from M1 receptors of the derivatives was measured in the absence and presence of brucine which is an allosteric ligand (Jakubík et al., 1997; Lazareno et al., 1998). Brucine inhibited the dissociation of fluorescent derivatives with a linker shorter than 12 atoms, indicating additional binding of brucine to the ligand receptor complex. Fluorescent derivatives with a chain length of 15 (Bo(15)PZ, Figure 2) and longer, however, were resistant to the allosteric action of brucine. This finding suggests that the area of receptor binding of these derivatives includes the allosteric domain where brucine binds. Taken that pirenzepine binds to the orthosteric site, the brucine-resistant derivatives were classified as ‘potential bitopic ligands’.
M2-preferring antagonist THRX-160209
Steinfeld and coworkers (2007) introduced a high-affinity, M2 subtype-selective muscarinic antagonist, THRX-160209 (Figure 2), and provided experimental evidence for a ‘multivalent’ orthosteric/allosteric receptor binding. This compound displays high affinity for the M2 subtype (pKI = 9.5). This affinity is far higher than that of the purely allosteric binding mode (pEC50,diss = 6.5) which was measured in M2 receptors the orthosteric site of which was blocked by [3H]NMS. The THRX-compound is composed of a 3-benzyhydryl pyrrolidine moiety (putative orthosteric building block, non-selective) that is linked by a seven carbon-chain with a 4-aminobenzylpiperidine motif (putative allosteric building block, M2-preferring). Three lines of evidence suggest a simultaneous orthosteric/allosteric binding. First, the binding affinity of the THRX-compound (pKI = 9.5) is higher than the binding affinity of each of the individual building blocks (pKI-values of 5.4 and 5.7 respectively). However, the gain in affinity cannot exclusively be related to the building blocks as the heptamethylene linker appears to contribute to THRX-binding. Second, the rank order of affinities is typical for an allosteric interaction (M2 > M4 > M1 > M3 > M5). Third, the receptor binding is sensitive to orthosteric and allosteric probes. This check included use of a [3H]THRX-radioligand, which allowed to directly follow receptor dissociation kinetics by an ‘infinite dilution assay’. A second, slow component of [3H]THRX-dissociation which is thought to represent bivalent orthosteric/allosteric binding was found to be sensitive to the individual building blocks and to other orthosteric and allosteric receptor ligands.
M2 partial agonist McN-A-343
Christopoulos and coworkers provided evidence for an allosteric/orthosteric overlap binding of the partial agonist McN-A-343 in M2 receptors (Valant et al., 2008). The mechanism of action of McN-A-343 has been studied for decades (Roszkowski, 1961; Fozard and Muscholl, 1971; Birdsall et al., 1983; Lambrecht et al., 1993; May et al., 2007b). There is evidence compatible with an orthosteric receptor interaction but an allosteric agonism has also been proposed. The ingenuity of the most recent experimental approach was to consider McN-A-343 (Figure 2) as a molecule containing two building blocks, i.e. tetramethylammonium (TMA) bromide, which is a full muscarinic agonist, and a chlorophenylcarbamate-type building block (Valant et al., 2008). The latter type of agent was shown, first, to act as (weakly potent) positive allosteric modulator on [3H]NMS-binding at the M2 receptor and, second, to reduce the intrinsic efficacies and the affinities of TMA and acetylcholine in an EKR1/2-phosphorylation assay. Mutation of M2177Tyr, an allosteric core amino acid (Voigtländer et al., 2003), was shown to increase the efficacy of McN-A-343 and to weaken the action of the chlorophenylcarbamate-type building blocks with respect to both allosteric inhibition of [3H]NMS-dissociation and diminution of TMA- and acetylcholine-intrinsic efficacy. Docking of McN-A-343 in a M2 receptor model was compatible with an allosteric location of the chlorophenylcarbamate-type building block and with an orthosteric interaction of the TMA building block with the orthosteric M2103Asp. Taken together the findings of Valant et al. (2008) suggest that the chlorophenylcarbamate moiety is a built-in allosteric quencher of an orthosterically mediated intrinsic efficacy of McN-A-343.
This hypothesis is in line with findings that were obtained by Disingrini et al. (2006) who used the concept displayed in Figure 1B for the design of allosteric/orthosteric ligands and who encountered a loss of orthosteric full agonist efficacy upon fusion with allosteric inverse agonistic building blocks (see below).
Design of dualsteric agonists
For most of the compounds being introduced in the preceding chapter, evidence for the proposed allosteric/orthosteric receptor binding topography came by serendipity, none of these compounds was designed from pre-existing orthosteric and allosteric building blocks. In addition, none of these compounds has efficacy for appreciable M2 receptor activation. The dualsteric compounds described in this review are the first to emerge from a rational design concept aiming at ligands that encompass orthosteric and allosteric functionalities, i.e. high affinity orthosteric receptor activation and allosteric M2 subtype-selectivity (Figure 1B). In these ligands, oxotremorine M-related receptor activators serve as orthosteric building blocks. On their own these agonists are devoid of receptor subtype selectivity (Figure 3, compounds a to d upper panel). These orthosteric building blocks are connected, by fusing the common TMA head, with fragments of ABA-type compounds such as the truncated W84 derivative (1 in Figure 3). W84 and its shortened fragments are subtype-selective for M2 (Figure 3; Antony et al., 2009). In isolated tissue preparations that serve as M1, M2 and M3 models (rabbit vas deferens, guinea pig atrium, guinea pig ileum), all hybrid compounds lose over-all potency, but gain subtype-selectivity. However, compounds 1a, 1b and 1c have almost no intrinsic efficacy. These allosteric/orthosteric hybrids behave as antagonists in the M1 and M2 model and, at best, as very weak partial agonists in the M3 model (Figure 3, lower panel; Disingrini et al., 2006). In membranes from CHO-cells overexpressing human M2 receptors, however, partial agonist-like G protein activation is observed with some of the hybrid compounds (Disingrini et al., 2006). Therefore, the latter already combine receptor subtype selectivity with weak (but physiologically irrelevant) partial agonism. Compound 1d, which includes the extremely potent agonist building block iperoxo d, acts as a full agonist in the M1, the M2 and the M3 model. With this and a closely related compound, which contains an allosteric building block from the ABA-type compound naphmethonium (cf. Antony et al., 2009), the intended M2-preferring agonism is achieved. A true dualsteric binding topography is to be validated by various experimental approaches. In particular it is necessary to exclude that the compounds bind in a purely allosteric topography, as activation of muscarinic receptors from the allosteric site has been shown for various subtypes (for review De Amici et al., in press). The M2 subtype, for instance, is activated by autoantibodies that bind to the second outer loop (e.g. Sterin-Borda et al., 1991; Goin et al., 1997; Hernandez et al., 2008). Therefore, the proposed allosteric/orthosteric binding topography has been verified (Figure 4; for details see Antony et al., 2009). In brief, binding experiments using an orthosteric antagonist radioligand prove that binding of hybrid 1d is sensitive to both, allosteric and orthosteric M2 receptor mutations. In addition, the mutation-induced changes of cooperativity between the hybrids and an orthosteric radioligand are in line with the predictions of a model for dualsteric ligand binding introduced by May et al. (2007a). Furthermore, the hybrid agonist is sensitive to the orthosteric antagonist atropine which eliminates the hybrid's intrinsic efficacy, while still allowing for hybrid binding in the purely allosteric mode. The allosteric antagonist W84, however, interacts with the hybrid in a competitive-type antagonism (Antony et al., 2009). A docking simulation shows that the receptor may well accommodate the hybrid in the orthosteric/allosteric binding topography, with the orthosteric agonist moiety being positioned in the orthosteric site and the allosteric building block in the allosteric core region (Antony et al., 2009). Taken together, the allosteric/orthosteric ligand design translates into a dualsteric receptor binding. In addition, the dualsteric binding mode goes along with biased signalling. While common M2 receptor activating agonists may trigger the Gs pathway in addition to the Gi pathway, the hybrid compound is devoid of Gs activating properties (Antony et al., 2009; Kebig et al., 2009).
Figure 3.
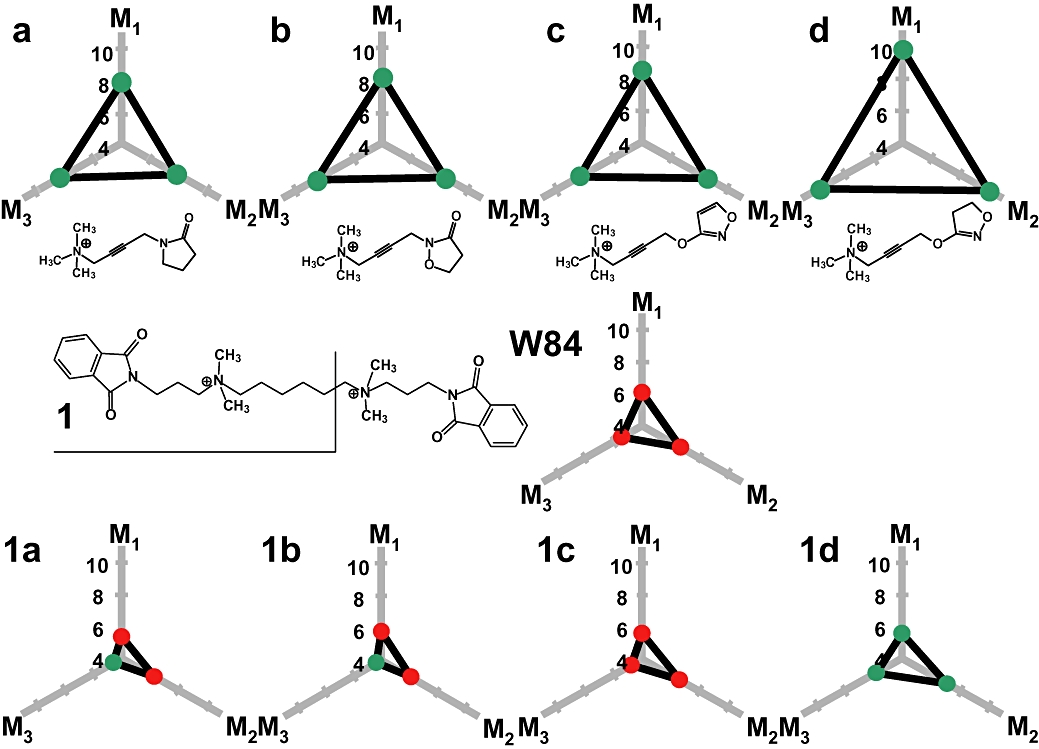
Potency, efficacy and muscarinic subtype-selectivity of building blocks and resulting allosteric/orthosteric hybrid compounds. Potency is indicated as minus log EC50 and minus log KB in the case of agonism and antagonism respectively, for the action of orthosteric agonists a–d (upper panel), allosteric inverse agonist W84 (middle panel) and hybrids 1a–1d (lower panel) in isolated organ models for M1 (rabbit vas deferens), M2 (guinea pig left atrium), M3 (guinea pig ileum). Hybrids consist of the fragment 1 from W84 and the respective agonist molecules a–d. Efficacy is indicated by dot colour – green: agonist activity, red: antagonist activity. Values for a–d are taken from Dallanoce et al. (1999), for W84 from Tränkle et al., 1998, for 1a–1c from Disingrini et al. (2006) and for 1d from Antony et al. (2009).
Figure 4.
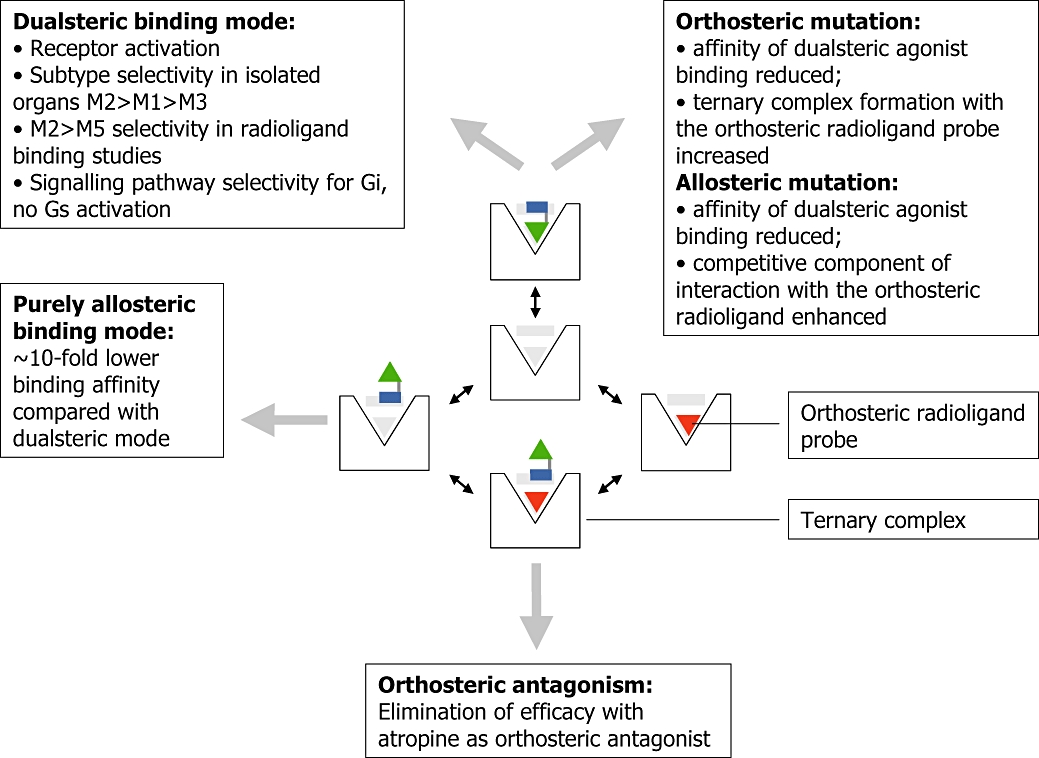
Binding modes of muscarinic dualsteric agonists in the absence and presence of an orthosteric radioligand probe – related experimental evidence and functional consequences. Centre: a symbolized muscarinic receptor contains in its binding cavity the inner orthosteric and the outer allosteric binding region (grey shaded). The dualsteric ligand such as compound 1d (Figure 3) consists of an orthosteric agonist building block (green triangle) which is connected by a linker with an allosteric building block (blue rectangle); orthosteric probe: red triangle. Boxes, right and bottom: experimental evidence for dualsteric binding arising from radioligand binding assays in wild-type and point-mutated human M2 muscarinic receptors (right box) and from G protein-activation experiments ([35S]GTPγS-assay, bottom box). Boxes, left: functional consequences related to dualsteric binding arising from radioligand binding assays, G protein-activation assays, label-free signalling pathway analysis by dynamic mass redistribution and organ-bath experiments. For details see Antony et al. (2009) and Kebig et al. (2009).
Thermodynamic aspects of dualsteric receptor interactions
Here we consider dualsteric binding as a special case of polyvalent interaction (Mammen et al., 1998) in order to predict which affinity can be expected for the dualsteric interaction relative to the orthosteric and allosteric binding component. In the simplest case, the total free energy change ΔGdual should be approximately the sum of the free energy changes of the individual components ΔGortho and ΔGallo together with an entropic correction. The free energy change corresponds to receptor binding affinity according to ΔG = − RTln(K), where K is the association constant of ligand binding, R is the gas constant and T is temperature in Kelvin. Therefore, the expected affinity of dualsteric binding Kdual should be near the product of the component affinities Kortho and Kallo. However, additional aspects have to be considered which may contribute to a less than expected binding affinity. These aspects shall be discussed in the light of the enthalpic and the entropic component of the free energy change ΔG = ΔH − TΔS.
Negative cooperativity: The enthalpy change ΔHdual of the dualsteric interaction will be smaller than the sum of ΔHortho and ΔHallo, if binding of one of the building blocks imprints a conformation on the receptor protein that impairs binding of the other building block. As will be discussed below, negative cooperativity is likely to play a role for the receptor interaction of the dualsteric ligands shown in Figure 3.
Non-complementary spatial geometry between receptor and ligand: enthalpies ΔHortho and ΔHallo will be less than additive, if the linker between the building blocks of the ligand does not fit the distance between the orthosteric and the allosteric receptor site. Structure-activity-relationships addressing the linker chain length of the dualsteric ligands would help to clarify this issue.
Entropic cost: With respect to translational and rotational entropy (movement through space and freedom for rotation), linkage of two independent ligands will reduce the entropic cost of receptor binding. With respect to the total entropic cost of binding, however, conformational entropy is important, in particular with respect to the linker (Mammen et al., 1998). Bivalent ligands containing a flexible linker will cause a considerable entropic cost when the molecule is fixed on the receptor surface. Therefore, the highly flexible hexamethylene linker of the dualsteric agonists may diminish binding affinity because of entropic reasons. On the other hand, linker flexibility may be beneficial for spatial geometry reasons (see above). Worth mentioning, the linker itself may have a ligand function and thereby contribute to binding enthalpy.
An unsettled quest: rational tailoring of dualsteric ligands for active receptors
There are two issues in the pharmacological characteristics of the ABA-/iperoxo-type hybrid ligands that await clarification on the receptor level before a fully rational design can be achieved. First, when an additional anchor for receptor binding is introduced into a receptor ligand, one would expect receptor binding affinity to be considerably increased (see above). However, relative to their respective parent agonists, all hybrids lose potency and affinity (Figure 3). For instance, in radioligand binding experiments the M2-receptor affinity of the orthosteric building block iperoxo was pK ∼9, the affinity of one half of the ABA-type ligand W84 was pK ∼4.5, the measured affinity of the resulting dualsteric ‘1d’ (Figure 3) was only pK ∼8 (Antony et al., 2009; table 1 and Supplemental figure 1 therein), whereas the expected affinity would amount to pK ∼13.5. Second, hybrid formation results in a dramatic loss of intrinsic efficacy relative to the agonist building block (Figure 3 – agents 1a, 1b and 1c), except for 1d, which is derived from the ‘superagonist’ iperoxo (compound d).
As delineated above, possible reasons for a less than expected level of affinity are negative cooperativity, non-complementary spatial geometry and entropic cost. The concept of a ‘functional misfit of building blocks’ addresses negative cooperativity (Figure 5). This hypothesis is based on the evidence that GPCR activation involves a global conformational change of the receptor protein including the allosteric site. The rearrangement of the intracellular loop region required for G protein-activation is accompanied by a corresponding conformational transition of the extracellular loop region (for review e.g. Schwartz et al., 2006; Hoffmann et al., 2008; Wess et al., 2008). In fact, there is experimental evidence that the allosteric site of muscarinic receptors undergoes a pronounced conformational rearrangement, depending on whether the orthosteric site is liganded by either a receptor activator or an inactivator (Grossmüller et al., 2006). For instance, allosteric ligand binding which is promoted by bound orthosteric antagonists (positive cooperative interaction) is strongly reduced when the receptor is bound by orthosteric agonists (negative cooperative interaction; Jäger et al., 2007). In line with this it has been shown that the inactive-to-active transition involves a functional and most probably spatial reorientation of the allosteric core epitope M2422Trp (7.35). In a ‘Janus-like’ fashion this amino acid provides affinity for allosteric antagonist binding in the inactive receptor and for orthosteric agonist binding in the active receptor (Jäger et al., 2007). The dualsteric agents described here are composed of building blocks that stabilize functionally opposite receptor conformations: the oxotremorine M-like compounds are orthosteric agonists, the ABA-ligands are allosteric inverse agonists (Figure 5). As individual compounds they would diminish each other's binding affinity in a negative cooperative fashion. Therefore, this ‘built-in’ functional misfit of building blocks is likely to contribute to the loss of affinity observed with the dualsteric agonists compared with their respective agonist building blocks.
Figure 5.
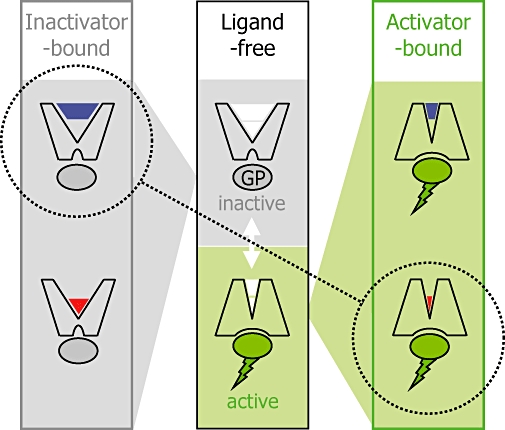
‘Functional misfit’ of dualsteric ligands which consist of an orthosteric agonist building block and an allosteric inverse agonist building block. Middle panel: the ligand-free receptor may switch spontaneously from the inactive (top) into the active (conformation) which stimulates a G protein (GP); the inactive-to-active conformational transition includes the orthosteric and the allosteric binding site (symbolized as white areas in the ligand binding pocket of the receptor protein). The inactive conformation (left panel) is stabilized by inverse agonist ligands that may bind either to the allosteric (left panel, top) or to the orthosteric site (left panel, bottom). Likewise, the active conformation (right panel) can be stabilized by appropriate allosteric and orthosteric ligands. The building blocks of the hybrid compounds shown in Figure 3 prefer functionally opposing receptor conformations (stippled line).
In contrast to the loss of affinity, the clear loss of intrinsic efficacy seen with hybrids 1a, 1b and 1c is unlikely to be solely explained by the concept of functional misfit of building blocks. There is ample evidence that allosteric inverse agonist binding does not diminish maximum receptor activation by orthosteric full agonists (Zahn et al., 2002; Jäger et al., 2007). Worth mentioning, however, allosteric inverse agonists may reduce partial agonist efficacy at muscarinic M2 receptors (Zahn et al., 2002; Jäger et al., 2007). In any case, the agonist building blocks of the dualsteric ligands are derived from oxotremorine M which is a full muscarinic agonist. Therefore, the loss of intrinsic efficacy observed upon hybrid formation with oxotremorine M-like building blocks can hardly be explained by a ‘built-in’ negative activation cooperativity between the dualsteric building blocks. Instead, non-complementary spatial geometry (spatial misfit) may play a role. Intriguingly, only the ‘super-high-affinity’ iperoxo building block endowed the hybrids with intrinsic efficacy. Its extremely high affinity probably reflects an optimum fit to the orthosteric site of the active receptor. Studies carried out by Kobilka and coworkers in β-adrenergic receptors suggest that agonist-induced receptor activation is a multi-step process that involves more than one receptor conformation (Kobilka and Deupi, 2007). Instead a spectrum of agonist-bound conformations might occur which differ in efficacy reaching from fully active over partially active to inactive. In the light of this concept it can be imagined that an allosteric anchor which is not at an optimal distance from the orthosteric building block might put a strain on the orthosteric agonist building block. In the case of a less tightly fitting agonist building block the strain might move it out of the optimal location within the orthosteric site, thereby favouring less active or even inactive receptor conformations. Due to its tight fit, iperoxo seems to be a ‘high fidelity’ receptor activator that is resistant to strain imposed by an allosteric anchor. This issue requires more insight into how the inactive-to-active transition of the receptor affects the spatial orientation of allosteric key epitopes relative to the orthosteric site. With respect to dualsteric ligands this implies that the length of the linker chain may have a function for intrinsic efficacy. In addition, a relaxation of strain by optimizinglinker length would increase affinity of dualsteric agent binding.
Taken together, the suspected functional and spatial misfit in the dualsteric binding mode might contribute to the finding that dualsteric binding affinity is only about 10-fold higher than the affinity of purely allosteric binding (Figure 4).
Conclusion
Rationally designed dualsteric GPCR agonists allow to exploit favourable characteristics of orthosteric and allosteric receptor sites simultaneously. In a single molecular entity, orthosteric receptor activation may be linked with allosteric subtype-selectivity and intracellular signalling pathway selectivity. The choice of building blocks allows to fine-tune intrinsic efficacy and, possibly, routes of signalling pathway activation. In addition to a potential therapeutic use, systematically varied dualsteric molecules may serve as valuable research tools for a label-free approach to gain deeper insight into the geometry of activation-related conformational changes of GPCRs.
Acknowledgments
U.H., K.M. and E.K. gratefully acknowledge grants of the Deutsche Forschungsgemeinschaft (HO 1368/7-1 to 7-4 and HO 1368/12-1; MO 821/1-1 to 1-4 and MO 821/2-1; KO 1582/3-1). MDA has been financially supported by the University of Milano.
Glossary
Abbreviations:
- ABA
alkane-bis-ammonio
- GPCR
G protein-coupled receptor
- M-receptor
muscarinic acetylcholine receptor
- TMA
tetramethylammonium
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony J, Kellershohn K, Mohr-Andrä M, Kebig A, Prilla S, Muth M, et al. Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 2009;23:442–450. doi: 10.1096/fj.08-114751. [DOI] [PubMed] [Google Scholar]
- Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, Christopoulos A. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem. 2007;282:25677–25686. doi: 10.1074/jbc.M702311200. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Birdsall NJ, Lazareno S. Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev Med Chem. 2005;5:523–543. doi: 10.2174/1389557054023251. [DOI] [PubMed] [Google Scholar]
- Birdsall NJ, Burgen AS, Hulme EC, Stockton JM, Zigmond MJ. The effect of McN-A-343 on muscarinic receptors in the cerebral cortex and heart. Br J Pharmacol. 1983;78:257–259. doi: 10.1111/j.1476-5381.1983.tb09388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a family of muscarinic acetylcholine receptor genes. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- Buller S, Zlotos DP, Mohr K, Ellis J. Allosteric site on muscarinic acetylcholine receptors: a single amino acid in transmembrane region 7 is critical to the subtype selectivities of caracurine V derivatives and alkane-bisammonium ligands. Mol Pharmacol. 2002;61:160–168. doi: 10.1124/mol.61.1.160. [DOI] [PubMed] [Google Scholar]
- Chung S, Funakoshi T, Civelli O. Orphan GPCR research. Br J Pharmacol. 2008;153(Suppl. 1):S339–346. doi: 10.1038/sj.bjp.0707606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AL, Mitchelson F. The inhibitory effect of gallamine on muscarinic receptors. Br J Pharmacol. 1976;58:323–331. doi: 10.1111/j.1476-5381.1976.tb07708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallanoce C, Conti P, De Amici M, De Micheli C, Barocelli E, Chiavarini M, et al. Synthesis and functional characterization of novel derivatives related to oxotremorine and oxotremorine-M. Bioorg Med Chem. 1999;7:1539–1547. doi: 10.1016/s0968-0896(99)00107-8. [DOI] [PubMed] [Google Scholar]
- De Amici M, Dallanoce C, Tränkle C, Holzgrabe U, Mohr K. Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med Res Rev. 2009 doi: 10.1002/med.20166. in press (DOI 10.1002/Med. [DOI] [PubMed] [Google Scholar]
- Disingrini T, Muth M, Dallanoce C, Barocelli E, Bertoni S, Kellershohn K, et al. Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J Med Chem. 2006;49:366–372. doi: 10.1021/jm050769s. [DOI] [PubMed] [Google Scholar]
- Eberlein WG, Engel W, Mihm G, Rudolf K, Wetzel B, Entzeroth M, et al. Structure-activity relationships and pharmacological profile of selective tricyclic antimuscarinics. Trends Pharmacol Sci. 1989;(Suppl.):50–54. [PubMed] [Google Scholar]
- Ellis J, Seidenberg M, Brann MR. Use of chimeric muscarinic receptors to investigate epitopes involved in allosteric interactions. Mol Pharmacol. 1993;44:583–588. [PubMed] [Google Scholar]
- Ellis J, Huyler J, Brann MR. Allosteric regulation of cloned m1-m5 muscarinic receptor subtypes. Biochem Pharmacol. 1991;42:1927–1932. doi: 10.1016/0006-2952(91)90591-r. [DOI] [PubMed] [Google Scholar]
- Ellis J, Seidenberg M. Interactions of alcuronium, TMB-8, and other allosteric ligands with muscarinic acetylcholine receptors: studies with chimeric receptors. Mol Pharmacol. 2000;58:1451–1460. doi: 10.1124/mol.58.6.1451. [DOI] [PubMed] [Google Scholar]
- Engel WW, Eberlein WG, Mihm G, Hammer R, Trummlitz G. Tricyclic compounds as selective muscarinic receptor antagonists. 3. Structure-selectivity relationships in a series of cardioselective (M2) antimuscarinics. J Med Chem. 1989;32:1718–1724. doi: 10.1021/jm00128a008. [DOI] [PubMed] [Google Scholar]
- Entzeroth M, Mayer N. Labeling of rat heart muscarinic receptors using the new M2 selective antagonist [3H]AF-DX 384. Biochem Pharmacol. 1990;40:1674–1676. doi: 10.1016/0006-2952(90)90473-x. [DOI] [PubMed] [Google Scholar]
- Fozard JR, Muscholl E. Effects of several muscarinic agonists on cardiac performance and the release of noradrenaline from the sympathetic nerves of the rabbit heart. Br J Pharmacol. 1971;43:454P–455P. [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Jacobson KA. Allosterism in membrane receptors. Drug Discov and Today. 2006;11:191–202. doi: 10.1016/S1359-6446(05)03689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo E, Micheletti R, Montagna E, Giachetti A, Viganò MA, Ladinsky H, et al. Binding and functional characterization of the cardioselective muscarinic antagonist methoctramine. J Pharmacol Exp Ther. 1988;244:1016–1020. [PubMed] [Google Scholar]
- Gnagey A, Ellis J. Allosteric regulation of the binding of [3H]acetylcholine to m2 muscarinic receptors. Biochem Pharmacol. 1996;52:1767–1775. doi: 10.1016/s0006-2952(96)00598-9. [DOI] [PubMed] [Google Scholar]
- Gnagey AL, Seidenberg M, Ellis J. Site-directed mutagenesis reveals two epitopes involved in the subtype selectivity of the allosteric interactions of gallamine at muscarinic acetylcholine receptors. Mol Pharmacol. 1999;56:1245–1253. doi: 10.1124/mol.56.6.1245. [DOI] [PubMed] [Google Scholar]
- Goin JC, Leiros CP, Borda E, Sterin-Borda L. Interaction of human chagasic IgG with the second extracellular loop of the Hum heart muscarinic acetylcholine receptor. functional and pathological implications. FASEB J. 1997;11:77–83. doi: 10.1096/fasebj.11.1.9034169. [DOI] [PubMed] [Google Scholar]
- Grossmüller M, Antony J, Tränkle C, Holzgrabe U, Mohr K. Allosteric site in M2 acetylcholine receptors: evidence for a major conformational change upon binding of an orthosteric agonist instead of an antagonist. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:267–276. doi: 10.1007/s00210-005-0023-4. [DOI] [PubMed] [Google Scholar]
- Hammer R, Berrie CP, Birdsall NJ, Burgen AS, Hulme EC. Pirenzepine distinguishes between different subclasses of muscarinic receptors. Nature. 1980;283:90–92. doi: 10.1038/283090a0. [DOI] [PubMed] [Google Scholar]
- Heilker R, Wolff M, Tautermann CS, Bieler M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov and Today. 2009;14:231–240. doi: 10.1016/j.drudis.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Nascimento JH, Chaves EA, Costa PC, Masuda MO, Kurtenbach E, et al. Autoantibodies enhance agonist action and binding to cardiac muscarinic receptors in chronic Chagas' disease. J Recept Signal Transduct. 2008;28:375–401. doi: 10.1080/10799890802262319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Zürn A, Bünemann M, Lohse MJ. Conformational changes in G-protein-coupled receptors-the quest for functionally selective conformations is open. Br J Pharmacol. 2008;153(Suppl. 1):S358–366. doi: 10.1038/sj.bjp.0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XP, Ellis J. Mutational disruption of a conserved disulfide bond in muscarinic acetylcholine receptors attenuates positive homotropic cooperativity between multiple allosteric sites and has subtype-dependent effects on the affinities of muscarinic allosteric ligands. Mol Pharmacol. 2007;71:759–768. doi: 10.1124/mol.106.028944. [DOI] [PubMed] [Google Scholar]
- Huang XP, Prilla S, Mohr K, Ellis J. Critical amino acid residues of the common allosteric site on the M2 muscarinic acetylcholine receptor: more similarities than differences between the structurally divergent agents gallamine and bis(ammonio)alkane-type hexamethylene-bis-[dimethyl-(3-phthalimidopropyl)ammonium]dibromide. Mol Pharmacol. 2005;68:769–778. doi: 10.1124/mol.105.014043. [DOI] [PubMed] [Google Scholar]
- Jäger D, Schmalenbach C, Prilla S, Kebig A, Sennwitz M, Heller E, et al. Allosteric small molecules unveil a role of an extracellular E2/transmembrane helix 7 junction for G protein-coupled receptor activation. J Biol Chem. 2007;282:34968–34976. doi: 10.1074/jbc.M705563200. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Tuček S. Protection by alcuronium of muscarinic receptors against chemical inactivation and location of the allosteric binding site for alcuronium. J Neurochem. 1994;63:1932–1940. doi: 10.1046/j.1471-4159.1994.63051932.x. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Bacáková L, El-Fakahany EE, Tuček S. Positive cooperativity of acetylcholine and other agonists with allosteric ligands on muscarinic acetylcholine receptors. Mol Pharmacol. 1997;52:172–179. doi: 10.1124/mol.52.1.172. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Krejcí A, Dolezal V. Asparagine, valine, and threonine in the third extracellular loop of muscarinic receptor have essential roles in the positive cooperativity of strychnine-like allosteric modulators. J Pharmacol Exp Ther. 2005;313:688–696. doi: 10.1124/jpet.104.080358. [DOI] [PubMed] [Google Scholar]
- Jepsen K, Lüllmann H, Mohr K, Pfeffer J. Allosteric stabilization of [3H]N-methylscopolamine binding in guinea-pig myocardium by an antidote against organophosphate intoxication. Pharmacol Toxicol. 1988;63:163–168. doi: 10.1111/j.1600-0773.1988.tb00932.x. [DOI] [PubMed] [Google Scholar]
- Jöhren K, Höltje HD. A model of the human M2 muscarinic acetylcholine receptor. J Comput Aided Mol Des. 2002;16:795–801. doi: 10.1023/a:1023880611709. [DOI] [PubMed] [Google Scholar]
- Kebig A, Kostenis E, Mohr K, Mohr-Andrä M. An optical dynamic mass redistribution assay reveals biased signaling of dualsteric GPCR activators, J. Recept Signal Transduct. 2009;29:140–145. doi: 10.1080/10799890903047437. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kostenis E, Mohr K. Two-point kinetic experiments to quantify allosteric effects on radioligand dissociation. Trends Pharmacol Sci. 1996;17:280–283. doi: 10.1016/0165-6147(96)10034-1. [DOI] [PubMed] [Google Scholar]
- Krejcí A, Tuček S. Changes of cooperativity between N-methylscopolamine and allosteric modulators alcuronium and gallamine induced by mutations of external loops of muscarinic M(3) receptors. Mol Pharmacol. 2001;60:761–767. [PubMed] [Google Scholar]
- Kubo T, Fukuda K, Mikami A, Maeda A, Takahashi H, Mishina M, et al. Cloning. sequencing and expression of complementary DNA encoding the muscarinic acetylcholine receptor. Nature. 1986;323:411–416. doi: 10.1038/323411a0. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Moser U, Grimm U, Pfaff O, Hermanni U, Hildebrandt C, et al. New functionally selective muscarinic agonists. Life Sci. 1993;52:481–488. doi: 10.1016/0024-3205(93)90305-m. [DOI] [PubMed] [Google Scholar]
- Lanzafame A, Christopoulos A, Mitchelson F. The allosteric interaction of otenzepad (AF-DX 116) at muscarinic M2 receptors in guinea pig atria. Eur J Pharmacol. 2001;416:235–244. doi: 10.1016/s0014-2999(01)00827-5. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Gharagozloo P, Kuonen D, Popham A, Birdsall NJ. Subtype-selective positive cooperative interactions between brucine analogues and acetylcholine at muscarinic receptors: radioligand binding studies. Mol Pharmacol. 1998;53:573–589. doi: 10.1124/mol.53.3.573. [DOI] [PubMed] [Google Scholar]
- Lee NH, El-Fakahany EE. Allosteric interactions at the m1, m2 and m3 muscarinic receptor subtypes. J Pharmacol Exp Ther. 1991;256:468–479. [PubMed] [Google Scholar]
- Leppik RA, Miller RC, Eck M, Paquet JL. Role of acidic amino acids in the allosteric modulation by gallamine of antagonist binding at the m2 muscarinic acetylcholine receptor. Mol Pharmacol. 1994;45:983–990. [PubMed] [Google Scholar]
- Leppik RA, Lazareno S, Mynett A, Birdsall NJ. Characterization of the allosteric interactions between antagonists and amiloride analogues at the human α2A-adrenergic receptor. Mol Pharmacol. 1998;53:916–925. [PubMed] [Google Scholar]
- Lüllmann H, Ohnesorge FK, Schauwecker GC, Wassermann O. Inhibition of the actions of carbachol and DFP on guinea pig isolated atria by alkane-bis-ammonium compounds. Eur J Pharmacol. 1969;6:241–247. doi: 10.1016/0014-2999(69)90181-2. [DOI] [PubMed] [Google Scholar]
- Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angewandte Chemie Int Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Matsui H, Lazareno S, Birdsall NJ. Probing of the location of the allosteric site on m1 muscarinic receptors by site-directed mutagenesis. Mol Pharmacol. 1995;47:88–98. [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007a;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM, et al. Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007b;72:463–476. doi: 10.1124/mol.107.037630. [DOI] [PubMed] [Google Scholar]
- Melchiorre C, Cassinelli A, Quaglia W. Differential blockade of muscarinic receptor subtypes by polymethylene tetraamines. Novel class of selective antagonists of cardiac M-2 muscarinic receptors. J Med Chem. 1987a;30:201–204. doi: 10.1021/jm00384a034. [DOI] [PubMed] [Google Scholar]
- Melchiorre C, Angeli P, Lambrecht G, Mutschler E, Picchio MT, Wess J. Antimuscarinic action of methoctramine, a new cardioselective M-2 muscarinic receptor antagonist, alone and in combination with atropine and gallamine. Eur J Pharmacol. 1987b;144:117–124. doi: 10.1016/0014-2999(87)90509-7. [DOI] [PubMed] [Google Scholar]
- Melchiorre C, Minarini A, Angeli P, Giardinà D, Gulini U, Quaglia W. Polymethylene tetraamines as muscarinic receptor probes. Trends Pharmacol Sci. 1989;(Suppl.):55–59. [PubMed] [Google Scholar]
- Mohr K, Tränkle C, Holzgrabe U. Structure/activity relationships of M2 muscarinic allosteric modulators. Receptors Channels. 2003;9:229–240. [PubMed] [Google Scholar]
- Mohr M, Heller E, Ataie A, Mohr K, Holzgrabe U. Development of a new type of allosteric modulator of muscarinic receptors. hybrids of the antagonist AF-DX 384 and the hexamethonio derivative W84. J Med Chem. 2004;47:3324–3327. doi: 10.1021/jm031095t. [DOI] [PubMed] [Google Scholar]
- Nedoma J, Tuček S, Danilov AF, Shelkovnikov SA. Stabilization of antagonist binding to cardiac muscarinic acetylcholine receptors by gallamine and other neuromuscular blocking drugs. J Pharmacol Exp Ther. 1986;236:219–223. [PubMed] [Google Scholar]
- Potter LT, Ferrendelli CA, Hanchett HE, Hollifield MA, Lorenzi MV. Tetrahydroaminoacridine and other allosteric antagonists of hippocampal M1 muscarine receptors. Mol Pharmacol. 1989;35:652–660. [PubMed] [Google Scholar]
- Prilla S, Schrobang J, Ellis J, Höltje HD, Mohr K. Allosteric interactions with muscarinic acetylcholine receptors: complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol Pharmacol. 2006;70:181–193. doi: 10.1124/mol.106.023481. [DOI] [PubMed] [Google Scholar]
- Roffel AF, Elzinga CR, Meurs H, Zaagsma J. Allosteric interactions of three muscarine antagonists at bovine tracheal smooth muscle and cardiac M2 receptors. Eur J Pharmacol. 1989;172:61–70. doi: 10.1016/0922-4106(89)90045-x. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszkowski AP. An unusual type of sympathetic ganglionic stimulant. J Pharmacol Exp Ther. 1961;132:156–170. [PubMed] [Google Scholar]
- Schröter A, Tränkle C, Mohr K. Modes of allosteric interactions with free and [3H]N-methylscopolamine-occupied muscarinic M2 receptors as deduced from buffer-dependent potency shifts. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:512–519. doi: 10.1007/s002100000316. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation – a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Steinfeld T, Mammen M, Smith JA, Wilson RD, Jasper JR. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol Pharmacol. 2007;72:291–302. doi: 10.1124/mol.106.033746. [DOI] [PubMed] [Google Scholar]
- Sterin-Borda L, Gorelik G, Borda ES. Chagasic IgG binding with cardiac muscarinic cholinergic receptors modifies cholinergic-mediated cellular transmembrane signals. Clin Immunol Immunopathol. 1991;61:387–397. doi: 10.1016/s0090-1229(05)80010-8. [DOI] [PubMed] [Google Scholar]
- Stockton JM, Birdsall NJ, Burgen AS, Hulme EC. Modification of the binding properties of muscarinic receptors by gallamine. Mol Pharmacol. 1983;23:551–557. [PubMed] [Google Scholar]
- Tahtaoui C, Parrot I, Klotz P, Guillier F, Galzi JL, Hibert M, et al. Fluorescent pirenzepine derivatives as potential bitopic ligands of the human M1 muscarinic receptor. J Med Chem. 2004;47:4300–4315. doi: 10.1021/jm040800a. [DOI] [PubMed] [Google Scholar]
- Tränkle C, Kostenis E, Burgmer U, Mohr K. Search for lead structures to develop new allosteric modulators of muscarinic receptors. J Pharmacol Exp Ther. 1996;279:926–933. [PubMed] [Google Scholar]
- Tränkle C, Andresen I, Lambrecht G, Mohr K. M2 receptor binding of the selective antagonist AF-DX 384: possible involvement of the common allosteric site. Mol Pharmacol. 1998;53:304–312. doi: 10.1124/mol.53.2.304. [DOI] [PubMed] [Google Scholar]
- Tränkle C, Weyand O, Voigtländer U, Mynett A, Lazareno S, Birdsall NJ, et al. Interactions of orthosteric and allosteric ligands with [3H]dimethyl-W84 at the common allosteric site of muscarinic M2 receptors. Mol Pharmacol. 2003;64:180–190. doi: 10.1124/mol.64.1.180. [DOI] [PubMed] [Google Scholar]
- Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, et al. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigtländer U, Jöhren K, Mohr M, Raasch A, Tränkle C, Buller S, et al. Allosteric site on muscarinic acetylcholine receptors: identification of two amino acids in the muscarinic M2 receptor that account entirely for the M2/M5 subtype selectivities of some structurally diverse allosteric ligands in N-methylscopolamine-occupied receptors. Mol Pharmacol. 2003;64:21–31. doi: 10.1124/mol.64.1.21. [DOI] [PubMed] [Google Scholar]
- Waelbroeck M, Camus J, Tastenoy M, Christophe J. Binding properties of nine 4-diphenyl-acetoxy-N-methyl-piperidine (4-DAMP) analogues to M1, M2, M3 and putative M4 muscarinic receptor subtypes. Br J Pharmacol. 1992;105:97–102. doi: 10.1111/j.1476-5381.1992.tb14217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Wess J, Han SJ, Kim SK, Jacobson KA, Li JH. Conformational changes involved in G-protein-coupled-receptor activation. Trends Pharmacol Sci. 2008;29:616–625. doi: 10.1016/j.tips.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn K, Eckstein N, Tränkle C, Sadée W, Mohr K. Allosteric modulation of muscarinic receptor signaling: alcuronium-induced conversion of pilocarpine from an agonist into an antagonist. J Pharmacol Exp Ther. 2002;301:720–728. doi: 10.1124/jpet.301.2.720. [DOI] [PubMed] [Google Scholar]