

Abstract
Background and purpose:
Human and rat 5-HT7 receptors were studied with a particular emphasis on the molecular interactions involved in ligand binding, searching for an explanation to the interspecies selectivity observed for a set of compounds. We performed affinity studies, molecular modelling and site-directed mutagenesis, with special focus on residue Phe(7.38) of the human 5-HT7 receptor [Cys(7.38) in rat].
Experimental approach:
Competition binding studies were performed for seven 5-HT7 receptor ligands at three different 5-HT7 receptors. The functional behaviour was evaluated by measuring 5-carboxytryptamine-stimulated cAMP production. Computational simulations were carried out to explore the structural bases in ligand binding observed for these compounds.
Key results:
Competition experiments showed a remarkable selectivity for the human receptor when compared with the rat receptor. These results indicate that mutating Cys to Phe at position 7.38 profoundly affects the binding affinities at the 5-HT7 receptor. Computational simulations provide a structural interpretation for this key finding. Pharmacological characterization of compounds mr25020, mr25040 and mr25053 revealed a competitive antagonistic behaviour. Compounds mr22423, mr22433, mr23284 and mr25052 behaved as partial agonists.
Conclusions and implications:
We propose that the interspecies difference in binding affinities observed for the compounds at human and rat 5-HT7 receptors is due to the nature of the residue at position 7.38. Our molecular modelling simulations suggest that Phe(7.38) in the human receptor is integrated in the hydrophobic pocket in the central part of the binding site [Phe(6.51)-Phe(6.52)] and allows a tighter binding of the ligands when compared with the rat receptor.
Keywords: GPCRs, interspecies selectivity, binding affinity, molecular modelling, site-directed mutagenesis
Introduction
5-Hydroxytryptamine (5-HT) is a biogenic amine mediating a wide range of physiological processes, with 5-HT and its receptors being distributed throughout the body. Dysfunction of this system has been implicated in cardiovascular and digestive as well as numerous psychiatric disorders (Hoyer et al., 2002). Seven distinct families of 5-HT receptors exist (5-HT1–7; nomenclature follows Alexander et al., 2008), and there is molecular and functional evidence for the existence of at least 14 different mammalian subtypes (Barnes and Sharp, 1999). With the exception of the 5-HT3 receptor, a ligand-gated ion channel, all 5-HT receptors are G protein-coupled receptors (GPCRs) (Hoyer et al., 1994; Barnes and Sharp, 1999) and constitute a large superfamily of very heterogeneous membrane receptors characterized by a typical heptahelical membrane-spanning fold, usually described as a seven-transmembrane (TM) domain (Horn et al., 2003; Kristiansen, 2004).
The 5-HT7 receptor has been identified in several species, including human, mouse, rat, guinea pig and pig. High sequence identity (∼90%) has been observed between 5-HT7 receptors from various species, whereas a low degree of identity (∼40%) is shared between 5-HT7 receptor and the other G protein-coupled 5-HT receptor subtypes. Although only a limited amount of work has been done on the 5-HT7 receptor so far, due to its relatively recent addition to the 5-HT receptor family, the current data have already provided some interesting insights (Hedlund and Sutcliffe, 2004; Thomas and Hagan, 2004). Expression of this GPCR has been demonstrated in human and rat hippocampus using radioligand binding, immunolabelling and in situ hybridization studies (Hedlund and Sutcliffe, 2004; Martin-Cora and Pazos, 2004). Several studies have implicated this receptor in the neurophysiology of depression, suggesting that modulation of 5-HT7 receptors might produce antidepressant-like effects (Bonaventure et al., 2007). The recent development of a 5-HT7 knockout mouse (Guscott et al., 2005) and the availability of specific 5-HT7 antagonists (Czeh et al., 2001) have facilitated behavioural studies into the role of this receptor in depression. The ability to correlate the behavioural effects of both genetic and pharmacological 5-HT7 modulation has also allowed a high degree of receptor specificity to be assigned to experimental findings. This may have contributed to the growing interest in 5-HT7 receptors. It has been proposed that 5-HT7 antagonism is capable of producing diverse antidepressant-like behavioural effects, alters hippocampal neuronal morphology and synergistically regulates hippocampal neurogenesis (Nandam et al., 2007).
Hence, the 5-HT7 receptor has been implicated in the sleep disturbances frequently seen in depression. Further studies have now confirmed that 5-HT7 receptors are directly involved in the modulation of REM sleep. Using SB656104 A (a 5-HT7 antagonist), 5-HT7 blockade in rodents leads to a decrease in the amount of time spent in REM sleep (Thomas et al., 2003). Decreases in REM sleep have also been produced in mice using SB269970, a pharmacologically distinct 5-HT7 antagonist (Hedlund et al., 2005). These REM effects are the opposite of the changes observed in depression, such that 5-HT7 blockade is producing behaviour that is being cautiously designated as ‘antidepressant like’ (Thomas et al., 2003; Hedlund et al., 2005). Regarding the potential therapeutic use of 5-HT7 receptor agonists, activation of 5-HT7 has been recently demonstrated to exert analgesic effects (Brenchat et al., 2009)
Homology modelling is a wide-used technique that allows the mapping of binding sites and the subsequent identification of the main molecular determinants for explaining the selectivity and affinity of different compounds. A community-wide assessment of GPCR modelling and ligand docking was recently conducted in coordination with the publication of the last GPCR experimental structure (the A2A adenosine receptor). The study concluded that, even if the field needs further development, the state-of-the-art modelling techniques perform reasonably well for the prediction of a GPCR structure and detection of the binding mode of antagonists (Michino et al., 2009). The lack of a crystallographic structure of any 5-HT receptors has been overcome by our groups and others by the generation of homology models of these receptors (Huang et al., 1999; Mialet et al., 2000; Brea et al., 2002; Rezmann-Vitti et al., 2004; Deng et al., 2007). These models were developed on the basis of any of the two GPCR existing templates: the historical α-carbon template for the TM helices from Baldwin (Baldwin et al., 1997) and lately, the X-ray crystal structure of bovine rhodopsin (Rh). More recently, the new high-resolution X-ray structure of the β2-adrenoceptor (Cherezov et al., 2007; Rasmussen et al., 2007; Rosenbaum et al., 2007) has allowed the generation of more confident homology models, based on the highest similarity with the template (de Graaf and Rognan, 2008; Selent et al., 2008; Wolf et al., 2008; Medina et al., 2009). Finally, other ligand-based approaches, such as the modelling of 3D pharmacophores, have contributed significantly to our knowledge of the molecular requirements of ligand binding to the 5-HT7 receptor. (Lopez-Rodriguez et al., 2000; Wilcox et al., 2001; Lopez-Rodriguez et al., 2003; Vermeulen et al., 2003).
The present work describes a multidisciplinary effort combining pharmacological evaluation, molecular modelling and site-directed mutagenesis, in order to explore the molecular basis for interspecies selectivity between human and rat 5-HT7 receptors. The exploration, at the molecular level, of interspecies differences between human and rat receptors is a key point in molecular pharmacology of GPCRs (Tucker et al., 1994; Laurila et al., 2007; Michel et al., 2008). The proposed 3D models show a high correlation with the interspecies selectivity behaviour, experimentally assessed here for the set of compounds under consideration, and have been successfully challenged by site-directed mutagenesis. Our work highlights the contribution of a single residue in the receptor (position 7.38) to the binding properties of some compounds exhibiting a selectivity profile for human versus rat 5-HT7 receptors.
Methods
Residue numbering and nomenclature
The residue numbering for GPCRs proposed by Ballesteros and Weinstein (1994) was adopted throughout this manuscript, and is always indicated with brackets. Whenever a residue number is displayed without brackets, it accounts for human/rat-specific sequence numbers.
Alignment of amino acid sequences
The amino acid sequences of human and rat 5-HT7 receptor subtypes were retrieved from the Swiss-Prot database (accession numbers: human 5-HT7 receptor, P34969; rat receptor, P32305) and aligned to the sequence of the β2-adrenoceptor (accession number: P07550) (Figure 1). The alignment was carried out using ClustalX (Thompson et al., 1997). A slow pairwise alignment using BLOSUM matrix series (Henikoff and Henikoff, 1992) and a gap opening penalty of 15.0 werechosen for aligning the amino acid sequences to the sequence of the β2-adrenoceptor.
Figure 1.
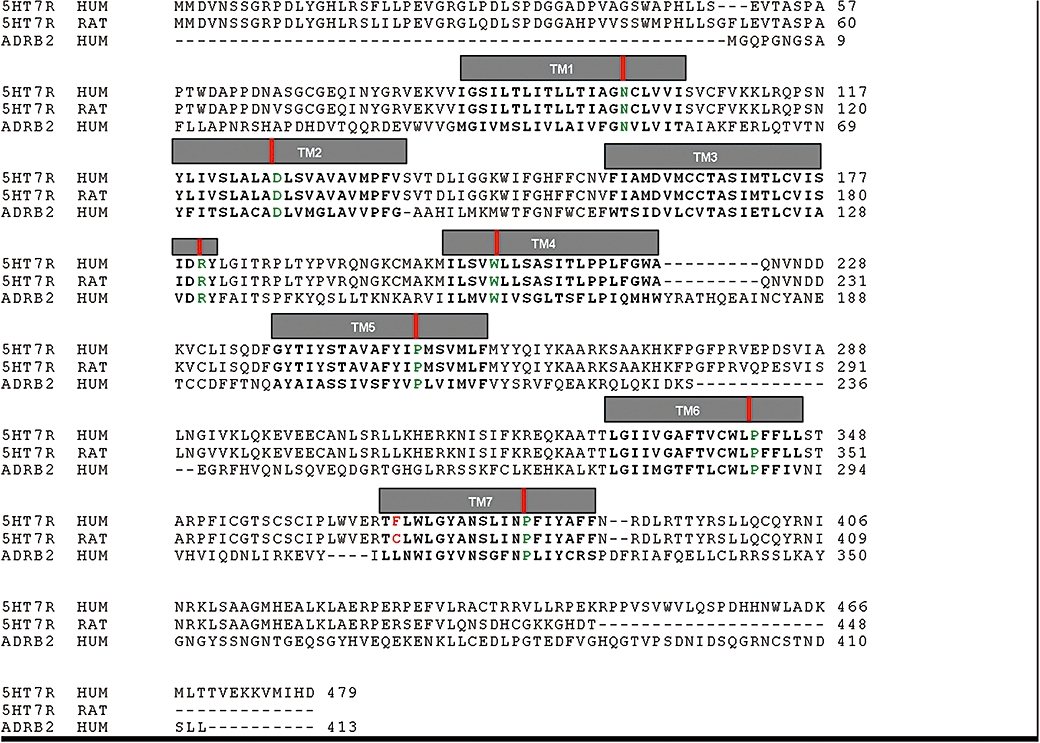
Amino acid sequence alignment of human and rat 5-HT7 receptors (5-HT7R HUM: human 5-HT7 receptor, 5-HT7R RAT: rat 5-HT7 receptor) to the human β2-adrenoceptor (ADRB2 HUM). Transmembrane helices (TMI to TMVII) are delimited by grey boxes. The amino acid residue mutated in this work is marked in red. Position 50 (Ballesteros numbering) of each TM is marked by a red vertical bar and coloured in green (Asn for TMI, Asp for TMII, Arg for TMIII, Trp for TMIV, Pro for TMs V–VII).
Modelling the antagonist-bound conformations of human and rat 5-HT7 receptors
The models for human and rat 5-HT7 receptors were built using the module Modeler (Marti-Renom et al., 2000) of Discover Studio 2.0 suite of programs using the structure of the β2-adrenoceptor as a template (PDB code 2RH1). Three models were initially built with the default Modeler parameters. Thereafter, for each of these three structures we generated three refined models with the LoopSearch routine implemented in modeller, thus improving the modelling of the loop regions. With this strategy we ended up with nine models for each of the two (human and rat) 5-HT7 receptors under consideration. A single structure was selected for each receptor taking into consideration the energetic scoring provided by Modeler. In this respect, Modeler considers the best model as the one with the lowest PDF Total Energy (fitness function use during the optimization process is derived from the PDF Total Energy). Then, each receptor was minimized with AMBER8.0 (Case et al., 2004) using the AMBER03 force field to relax the structure and to remove steric bumps. The minimizations were carried out by 1000 steps of steepest descent followed by conjugate gradient minimization until the rms gradient of the potential energy was lower than 0.05 kcal·molÅ−1. A twin cut-off (10.0, 15.0 Å) was used to calculate non-bonded electrostatic interactions at every minimization step, and the non-bonded pair list was updated every 25 steps. A distance-dependent (ε = 4r) dielectric function was used. The stereochemistry parameters of quality (Ramachandran plots) were also checked after minimization.
Finally, the selected model for each protein was prepared for molecular dynamics (MD) simulations following the procedure of Ander et al. (2008). A ∼30-Å-thick layer of octane molecules was built around the model, using Packmol (Martinez and Martinez, 2003), to emulate the membrane in which the receptor is situated. The octane layer/5-HT7 assembly was then solvated with TIP3P water molecules (Jorgensen et al., 1996) in a cubic periodic box with dimensions 100 Å × 100 Å × 86 Å, neutralized with necessary counterions and equilibrated for 500 ps in an MD simulation at 300 K. During this relaxation all heavy atoms of the protein were constrained with a 10 kcal/molÅ2 force constant, while allowing the solvent and octane molecules to equilibrate around the receptor assembly. Software NAMD (Phillips et al., 2005) with the OPLS force-field was used in this step.
In order to check the reliability of the models generated, and the set-up of the docking protocol, we carried out preliminary docking of the 5-HT7 antagonist SB656104 A (Thomas et al., 2003) (Figure 2) with GOLD4.0. (Jones et al., 1997). The location of the binding pocket is indicated by the position of the Asp162(3.32), which is known to establish a charge-reinforced hydrogen bond with the protonated nitrogen present in amine ligands. The centre of the binding site was placed on this residue and expanded by an area of 12 Å. Default GOLD parameters were used, with additional flexibility for the ligand (flip ring corners and flip pyramidal nitrogens), and 20 independent genetic algorithm runs were generated. The docking of our set of seven compounds was conducted under identical conditions, with the exception that we expanded the search area by adding a 12 Å patch around the selected pose of SB656104 A. An H-bond constraint between the charged amino group of each ligand and Asp(3.32) was imposed in order to avoid spurious results. Ligand preparation was done in Sybyl 8.0 (Tripos International, S.L., Missouri, USA) each molecule was built in 3D, treated as charged and energy minimized, saved in mol2 format ensuring the correct atom typing for GOLD.
Figure 2.
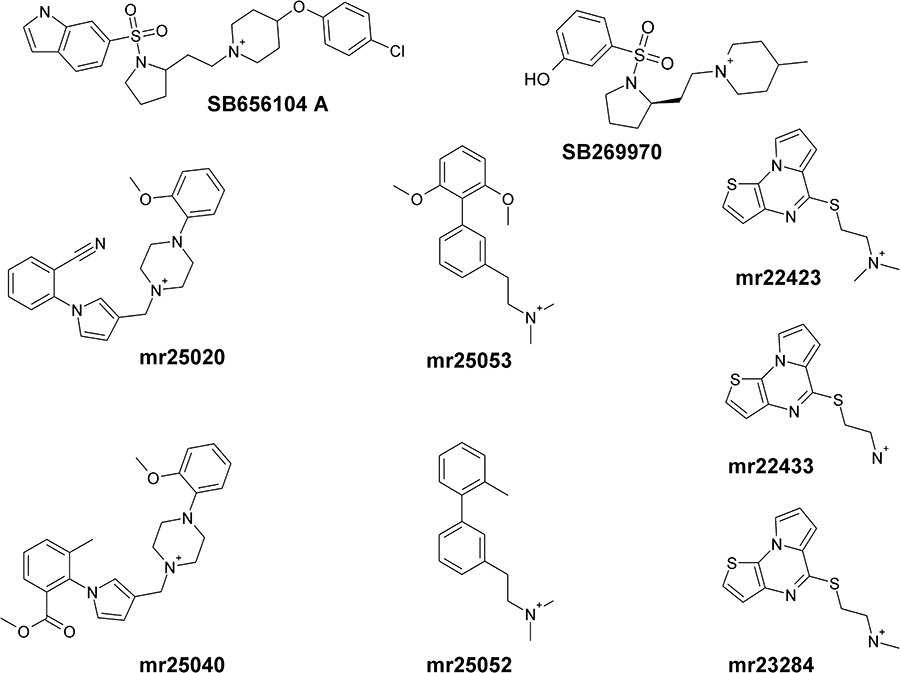
Chemical structures of 5-HT7 receptor ligands involved in these studies. Reference ligands for 5-HT7 receptors SB656104 A and SB269970 are also shown.
MD and binding free energy calculations
Relative binding affinities were calculated using the linear interaction energy (LIE) method, described in detail elsewhere.(Åqvist et al., 1994; Marelius et al., 1998) Originally, this approach estimates the absolute ligand free energy of binding from the difference in the ligand − surrounding interaction energies in both its bound and free states (e.g. water-solvated ligand). This method can be easily adapted to obtain relative binding affinities of a given compound between different proteins as a measure of ligand selectivities (Gutiérrez-de-Terán et al., 2006). Following this approach, for the case of human and rat 5-HT7 receptors, we defined the bound state 1 as the human 5-HT7 receptor bound conformation, and bound state 2 as the rat 5-HT7 receptor bound conformation. The relationship between the ligand intermolecular interaction energies and the relative free energy of binding is therefore given by the equation:
where 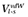 and
and 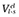 denote, respectively, the Lennard–Jones and electrostatic interactions between the ligand and its surroundings (l-s). These interactions are evaluated as energy averages (denoted by the broken brackets) from separate MD simulations of the two bound states to be compared (h for human, r for rat). The difference between such averages for each type of potential is scaled by different coefficients (Marelius et al., 1998) giving the polar and non-polar contributions to the binding free energy. For the non-polar contribution, we used the empirical value of α = 0.181, while for the polar contribution, the scaling factor follows the linear response approximation (β = 0.5) since the ligands hold a net positive charge. An offset factor (denoted by γ), which is frequently introduced for the calculation of absolute energies, is assumed to cancel out for the calculation of relative energies between highly similar proteins, and therefore it is not considered in the present implementation of the LIE methodology.
denote, respectively, the Lennard–Jones and electrostatic interactions between the ligand and its surroundings (l-s). These interactions are evaluated as energy averages (denoted by the broken brackets) from separate MD simulations of the two bound states to be compared (h for human, r for rat). The difference between such averages for each type of potential is scaled by different coefficients (Marelius et al., 1998) giving the polar and non-polar contributions to the binding free energy. For the non-polar contribution, we used the empirical value of α = 0.181, while for the polar contribution, the scaling factor follows the linear response approximation (β = 0.5) since the ligands hold a net positive charge. An offset factor (denoted by γ), which is frequently introduced for the calculation of absolute energies, is assumed to cancel out for the calculation of relative energies between highly similar proteins, and therefore it is not considered in the present implementation of the LIE methodology.
MD simulations were done using the program Q (Marelius et al., 1998) and the OPLS force field there implemented (Jorgensen et al., 1996). Each system was solvated with a simulation sphere of TIP3P waters (Jorgensen et al., 1996) of radius 25 Å, centred on the central atom of the ligand. The water surface of this sphere was subjected to radial and polarization restraints (King and Warshel, 1989) in order to mimic bulk water at the sphere boundary. Ionizable residues in the inner solvation sphere were modelled as charged, while residues close to the boundary of this sphere were considered in their neutral form, except if they form salt bridges. The resulting total charge of the sphere was in all cases equal to (−1). Non-bonded interaction energies were calculated up to a 10 Å cut-off, except for the ligand atoms for which no cut-off was applied. Beyond the cut-off, long-range electrostatics were treated with the local reaction field multi-pole expansion method (Li et al., 2005). Protein atoms outside the simulation sphere were restrained to their initial positions, and only interacted with the system through bonds, angles and torsions. A heating and equilibration procedure was applied before the data collection phase. The equilibration protocol started with 1000 steps of MD using very short-time step (0.2 fs) at 1 K temperature, coupled to a strong bath (0.2 fs bath coupling) with positional restraints on heavy atoms. Then the system was gradually heated up to 300 K, relaxing the bath coupling to 100 fs and increasing the time step to 1.5 fs, while the positional restraints were smoothly released. This equilibration phase was followed by a collection period consisting on 750 ps of unrestrained MD, with energies collected at regular intervals of 15 fs. Energy averaging was performed on the energetically stable phase of the collection period, which was never shorter than 450 ps, where stability was addressed by comparing the average binding free energy values of the first and second halves of the data collection period.
Site-directed mutagenesis
Rat 5-HT7 cDNA was obtained from total RNA from Sprague–Dawley rat brain by RT-PCR using specific primers homologous to the reported sequence of rat 5-HT7 receptor (EMBL accession number L19654). Rat 5-HT7-Cys372Phe receptor mutant cDNA was generated by PCR using cDNA of the wild-type rat 5-HT7 receptor as template and with the primers 5′-gagaggacatttctgtggctgggc and 5′-cagccacagaaatgtcctctccac. The full-length cDNAs were cloned into the pcDNA3 vector (Invitrogen) by introducing BamHI and EcoRI restriction sites. The constructs were transformed and amplified in the Escherichia coli XL1_blue strain (Stratagene, La Jolla, CA), and then purified with the QIAfilter Plasmid Midi Kit (Qiagen Iberia, S.L., Madrid). All the cDNA constructs were verified by sequencing.
Expression of wild-type and mutant 5-HT7 receptors and cell culture
HEK293 cells stably expressing human 5-HT7A receptors were kindly provided by Laboratorios Esteve (Spain). The wild type and mutant Cys372Phe rat 5-HT7 receptors were stably expressed in HEK293 cells after transfection following the calcium phosphate method (Cullen, 1987) and selection with 400 µg·mL−1 geneticin. Stable clones expressing the receptors were selected by binding of [3H]SB269970 in cell membranes prepared as following described. All the stable HEK293 cell lines were maintained in culture in Eagle's minimum essential medium (MEM), supplemented with 10% (v/v) foetal calf serum, 100 U mL−1 penicillin, 100 µg·mL−1 streptomycin, 1% (v/v) MEM non-essential amino acid solution and 1 mM MEM sodium pyruvate. Cells were grown at 37°C in a 5% CO2 humidified atmosphere and in 150 mm Petri dishes.
Membrane preparation
HEK293 cells stably expressing the wild type or mutant receptors were washed twice in buffer A (10 mM EDTA, 10 mM EGTA, 10 NaHCO3, protease inhibitor cocktail 1×, pH = 7.4), harvested in the same buffer and homogenized using a Teflon-glass homogenizer. Cell suspension was centrifuged at 500× g for 5 min at 4°C and supernatant was recovered and centrifuged at 25 000× g for 20 min at 4°C. Pellets were washed in buffer A and centrifuged at 25 000× g for 20 min at 4°C. Membranes were suspended in a small volume of buffer B (25 mM Tris-HCl pH = 7.4, 5 mM MgCl2 and 1 mM EDTA). Protein concentration was determined by the method of Bradford with the Bio-Rad protein assay kit and using bovine serum albumin as a standard. Membranes were stored at −80°C before use.
Binding assays
Membrane binding assays were performed using [3H]SB269970 as radioligand. Membranes (5 µg protein) were incubated for 60 min at 37°C in a medium containing: 50 mM Tris-HCl pH = 7.4, 4 mM MgCl2, 1 mM ascorbic acid and 0.1 mM pargyline. Expression of receptors and affinity of the radioligand for the wild type and the mutated 5-HT7 receptors was determined by saturation experiments using concentrations of labelled [3H]SB269970 ranging from 0.1 to 30 nM. For each concentration of radioligand, total and non-specific binding were determined in absence or presence of 25 µM of clozapine respectively. The affinities (Ki) of the unlabelled compounds were determined by competition experiments. Briefly, 2 nM of [3H]SB269970 was added in the incubation medium with increasing concentrations of the unlabelled compounds to be tested. Non-specific binding was evaluated using 25 µM clozapine. After 60 min at 37°C, the incubation was stopped by filtration through GF/B filters. Specific binding was calculated and expressed as percentage ofthe maximal binding capacity determined in the absence of unlabelled compound.
Functional assays
Cells were seeded (15000 cells/well) in 96-well microplates in MEM Eagle, supplemented with 10% (v/v) foetal calf serum, 100 U mL−1 penicillin, 100 µg·mL−1 streptomycin, 1% (v/v) MEM non-essential amino acid solution and 1 mM MEM sodium pyruvate and maintained for 24 h at 37°C in at atmosphere with 5% CO2. At the time of the experiments, cells were washed with assay buffer (MEM Eagle containing 25 mM HEPES pH = 7.4), 30 µM rolipram was added and cells were pre-incubated for 15 min at 37°C. Concentration–response curves for the agonist 5-carboxytryptamine (5-CT), as well as for the partial agonist test compounds, were performed by adding the agonists after this pre-incubation time and incubating the cells for additional 15 min. For the characterization of the effect of test compounds on the 5-CT-stimulated production of cAMP, test compounds were added to the cells at the time of rolipram and 15 min latter, 5-CT was added for additional 15 min. After this time, the medium was removed from the microplates and the intracellular cAMP formation was measured using an enzymoimmunoassay kit from GE Healthcare (Barcelona, Spain).
Data analysis
The data were analysed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA). The dissociation constant (KD) of the radioligand was determined by non-linear regression curve fitting. The inhibitory dissociation constants (Ki) for the compounds under study were calculated from binding competition experiments according to the Cheng & Prusoff equation: Ki = IC50/(1 + [L]/KD), where IC50 is the concentration of the unlabelled compounds leading to half-maximal inhibition of specific binding, [L] the concentration of the radioligand present in the assay and KD its affinity for the receptor studied. Results are expressed as the mean ± SEM of the number of distinct experiments indicated. Agonist potency (EC50) and efficacy (Emax) were calculated by fitting the data to a sigmoidal dose–response curve using GraphPad Prism. Antagonist potency was expressed as KB and was calculated with the equation KB = IC50/(1 + [L]/EC50), where IC50 is the concentration of compound leading to half-maximal inhibition of cAMP formation, [L] the concentration of the agonist used for eliciting cAMP accumulation and EC50 is the concentration of the agonist that elicits a half-maximal response.
Materials
All reagents used were of analytical grade. Most standard chemicals were purchased from Sigma (Madrid, Spain), Roche Molecular Biochemicals (Mannheim, Germany) or Merck & Co., Inc. (Darmstadt, Germany), unless otherwise indicated. The set of seven original compounds under study were synthesized in our laboratory, following previously described protocols (Lepailleur et al., 2004; 2005; Paillet-Loilier et al., 2005; 2007;). [Correction added after online publication 25 February 2010: the sitations of Lepailleur et al. 2004 and 2005 were added to the preceding sentence.] DMEM, penicillin-streptomycin, L-glutamine and non-essential amino acids were purchased from Sigma (Madrid, Spain).
Results
Pharmacological characterization of compounds at human and rat 5-HT7 receptors
Affinities of seven compounds, showing different scaffolds, from our chemical library were characterized at human 5-HT7 receptors. All the compounds tested showed a concentration-dependent inhibition of specific [3H]SB269970 binding with Ki values ranging from 31 to 88 nM (Table 1, Figure 3).
Table 1.
Binding properties of human and rat wild type and rat Cys372Phe mutant 5-HT7 receptors for the different compounds included in this study
| Compound | Ki (nM) human 5-HT7 | Ki (nM) rat 5-HT7 | Ki (nM) rat Cys372Phe mutant 5-HT7 |
|---|---|---|---|
| mr22423 | 74.46 ± 16.51 | 215.90 ± 73.98 | 55.78 ± 21.02 |
| mr22433 | 58.78 ± 8.53 | 409.17 ± 90.75 | 42.68 ± 11.06 |
| mr23284 | 31.25 ± 3.31 | 93.41 ± 30.92 | 26.48 ± 4.53 |
| mr25020 | 42.94 ± 7.34 | 343.96 ± 126.91 | 42.60 ± 8.91 |
| mr25040 | 88.09 ± 16.19 | 192.20 ± 17.83 | 68.66 ± 21.92 |
| mr25052 | 34.03 ± 2.26 | 50.88 ± 19.17 | 17.27 ± 4.26 |
| mr25053 | 68.38 ± 13.42 | 399.86 ± 88.68 | 63.22 ± 24.53 |
Figure 3.
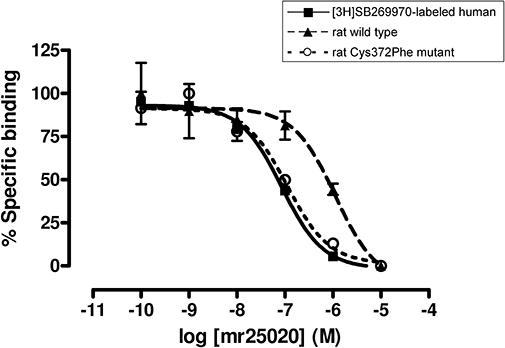
Different affinity of compounds at human, rat wild type and rat Cys372Phe mutant 5-HT7 receptors. An example is shown with competition binding curves at [3H]SB269970-labelled human, rat wild type and rat Cys372Phe mutant 5-HT7 receptors with mr25020. Points represent the mean ± SEM (vertical bars) of two (n = 2) independent assays performed in duplicate.
The functional behaviour of the compounds at human 5-HT7 receptors was studied by making cAMP measurements. Compounds mr25020, mr25040 and mr25053 showed a competitive antagonistic behaviour by inhibiting the cAMP formation elicited by 0.1 µM 5-CT (Figure 4A) with KB values very close to the Ki values obtained in binding assays (Table 2). On the other hand, compounds mr22423, mr22433, mr23284 and mr25052 behaved as partial agonists at human 5-HT7 receptors when compared with 5-CT (Figure 4B; Figure S1), eliciting concentration-dependent increases in cAMP accumulation with EC50 values higher than the Ki values obtained in radioligand binding assays (Table 2). Discrepancies between binding affinity and potency of 5-HT7 receptor agonists have been described and it has been proposed to be due to low efficiency in receptor-effector coupling that is inherent to this receptor subtype (Adham et al., 1998). While all these four compounds increased the cAMP accumulation when co-incubated with low concentrations of 5-CT, a rightward shift in the EC50 of the agonist 5-CT was observed only in the presence of three of them (mr22423, mr22433, mr23284), as would be expected for partial agonists (Figure S1). However, compound mr25052 showed a more striking behaviour as it did not elicited the rightward shift in the concentration–response curve of 5-CT. This result prevents us from discarding the possibility of a mixed allosteric profile for this particular compound.
Figure 4.
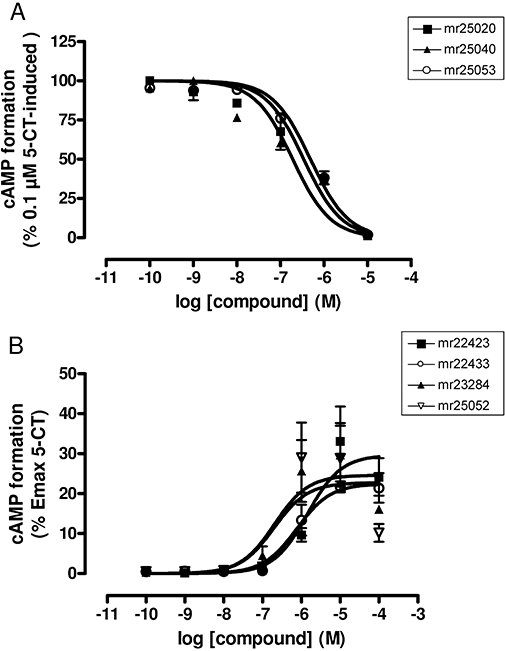
Functional characterization of selected compounds at human 5-HT7 receptors. (A) Concentration–response curves of compounds mr25020, mr25040 and mr25053 inhibiting the cAMP formation elicited by 0.1 µM 5-CT. Data represent the mean ± SEM (vertical bars) of two independent (n = 2) experiments performed in duplicate. (B) Concentration–response curves of compounds mr22423, mr22433, mr23284 and mr25052 measuring the formation of intracellular cAMP. Data are expressed as the % of the maximal cAMP formation induced by 5-CT and represent the mean ± SEM (vertical bars) of two independent (n = 2) experiments performed in duplicate.
Table 2.
Functional characterization of different compounds included in this study at human 5-HT7 receptors by measuring cAMP formation
| Compound | Antagonists |
Agonists |
|
|---|---|---|---|
| KB (nM) | EC50 (nM) | Emax (% of 5-CT) | |
| Mr22423 | 1402.23 ± 582.62 | 33.06 ± 3.12 | |
| mr22433 | 764.74 ± 54.46 | 21.69 ± 0.35 | |
| mr23284 | 302.67 ± 93.41 | 38.68 ± 3.51 | |
| mr25020 | 57.96 ± 0.18 | ||
| mr25040 | 82.00 ± 33.36 | ||
| mr25052 | 209.12 ± 105.23 | 28.99 ± 5.42 | |
| mr25053 | 37.48 ± 10.32 | ||
When the affinity of these compounds was evaluated over rat 5-HT7 receptors, a decrease in their affinity was observed. All the compounds showed higher affinity at human than at rat 5-HT7 receptors, showing – with the exception of mr25052 – Ki values more than three times lower at human than at rat 5-HT7 receptors (Table 1, Figure 3).
Taking all these data into account, we performed a computational study in order to evaluate the binding mode and specific molecular interactions of our series of 5-HT7 ligands at both human and rat 5-HT7 receptors.
Three-dimensional molecular modelling
Until very recently (November 2007) the best starting material for the homology building of GPCR models was the crystal structure of rhodopsin, even if the low-sequence homology (≈20% identity) between the 5-HT7 receptors and rhodopsin made the application of standard homology modelling methods difficult. However, the new X-ray structure of the β2-adrenoceptor, a receptor belonging to the same subfamily of GPCRs (the biogenic amine subfamily), opens the door to obtain more accurate models of amine receptors. The higher sequence homology between 5-HT7 receptors and the β2-adrenoceptor (showing a sequence identity of around 35% and sequence similarity of around 60%) lead us to expect an improvement in the quality of the models derived herein with respect to previous works exploring this receptor (Lopez-Rodriguez et al., 2003).
Alignment of the amino acid sequences of human and rat 5-HT7 receptors with that of the β2-adrenoceptor is rather straightforward as all rhodopsin-like fingerprints (Attwood, 2001; Bissantz et al., 2004) are common to all entries (Figure 1).
The only difference within the TM regions between human and rat receptors was located at TMVII, position 7.38 (Phe369 in human and a Cys372 in the rat 5-HT7 receptor). In fact, this is the TM position with the highest variability according to a multiple sequence alignment of all 5-HT7 receptor species available at the GPCR Database (http://www.gpcr.org/7tm/classes/serotonin-type-7/). Our molecular models show that the residue at position 7.38 is located close to the highly conserved Phe(6.52), but available to the binding crevice.
The antagonist binding site has been defined by automated docking with SB656104 A in both the human and rat receptors. This reference ligand for 5-HT7 receptors is bound to our receptor models in an L-shape with the longest axis perpendicular to the TM helices (see Figure S2). The SB656104 A/5-HT7 receptor complex is in good agreement with site-directed mutagenesis data available from the literature, as the ligand is in close contact with residues previously identified as important for the ligand binding (Figure S2). Briefly, the side chain of Asp162(3.32) and the charged nitrogen of SB656104 A interact making a charge-reinforced hydrogen bond. The indole moiety faces helix 7, at hydrogen-bond distance with Ser381(7.46) and is also stabilized with hydrophobic interactions with Trp340(6.48). Additional polar contacts are observed between the sulphonamide group and the Tyr374(7.43) phenolic hydroxyl group. The p-fluorobenzyl is immersed in a hydrophobic pocket, delimited by Phe343(6.51), Phe344(6.52). These residues, together with Trp340(6.48), constitute the so-called hydrophobic toggle switch, which have been suggested to participate in a coordinated conformational change in GPCRs, potentially important for the activation process (Ballesteros et al., 2001; Sylte et al., 2001; Shapiro et al., 2002; Bissantz, 2003). To conclude, the fluorine substituent of the aromatic ring is located close to the Ser243(5.43) and Thr244(5.44), at hydrogen-bond distance with the last residue. Ser(5.43) and Ser(5.46) in several 5-HT receptors are residues that have been postulated to be relevant for 5-HT binding (Braden and Nichols, 2007). In human and rat 5-HT7 receptors, position 5.46 is substituted by a valine. Probably, Thr244(5.44) plays the role of Ser(5.46) in other subfamilies of 5-HT receptors.
Once the binding site was defined with the docking of a reference 5-HT7 ligand, this was followed by an automated docking exploration, MD and binding free energy estimation for the seven 5-HT7 ligands extracted from our chemical library (Paillet-Loilier et al., 2005; 2007;) in both human and rat receptors. The antagonists mr25020 and mr25040 adopt a rather similar binding pose to SB656104 A in both human and rat 5-HT7 receptors (Figure 5), which recalls earlier binding modes described by other authors for ketanserin-like compounds. (Lopez-Rodriguez et al., 2003; Dezi et al., 2007). On the other hand, the binding mode of tricyclic compounds, which have been characterized as partial agonists in the functional assays (mr22423, mr22433 and mr23284, Figure S3E–J of the Supporting information) shows a good overlay with the binding pose of clozapine in 5-HT2A receptors, as recently published by us and other groups (Selent et al., 2008; Gutiérrez-de-Terán et al., 2009). Biphenyl compounds (mr25052 and mr25053, Figure S3K–N) also occupy the binding site defined between helices 3, 5 and 6. In all the 14 complexes simulated (7 compounds in 2 receptors), the interaction between the protonated amino group of the ligand and Asp162(3.32) is the major contribution to the ligand-receptor polar interactions, as is commonly accepted for the biogenic amine subfamily of GPCRs (Kristiansen et al., 2000). Notably, every compound shows a strong π aromatic interaction with the side chains of Phe343(6.51) and Phe344(6.52), belonging to the hydrophobic toggle switch. In the case of the human receptor, the Phe at position 7.38 represents an expansion of this aromatic cluster, and in most cases a direct interaction with the ligand is frequently observed along the MD simulations. On the contrary, the equivalent residue in the rat receptor [Cys(7.38)], possesses a smaller side-chain with different physicochemical properties, which is frequently pointing outside the binding site (see Figure S3). The estimation of relative ligand affinities between the two receptors is in excellent agreement with the experimental data on interspecies selectivity here reported, as we observed a stronger binding to the human receptor for every compound under consideration (see Table 3). The presence of a bulkier Phe at 7.38 contributes to keeping the ligand in place, and this is reflected in stronger interactions with the human receptor. In general, the electrostatic component of binding is stronger in the human receptor, although in two cases (mr25020 and mr22423) a direct increase of the non-polar contribution to ligand binding was also observed.
Figure 5.
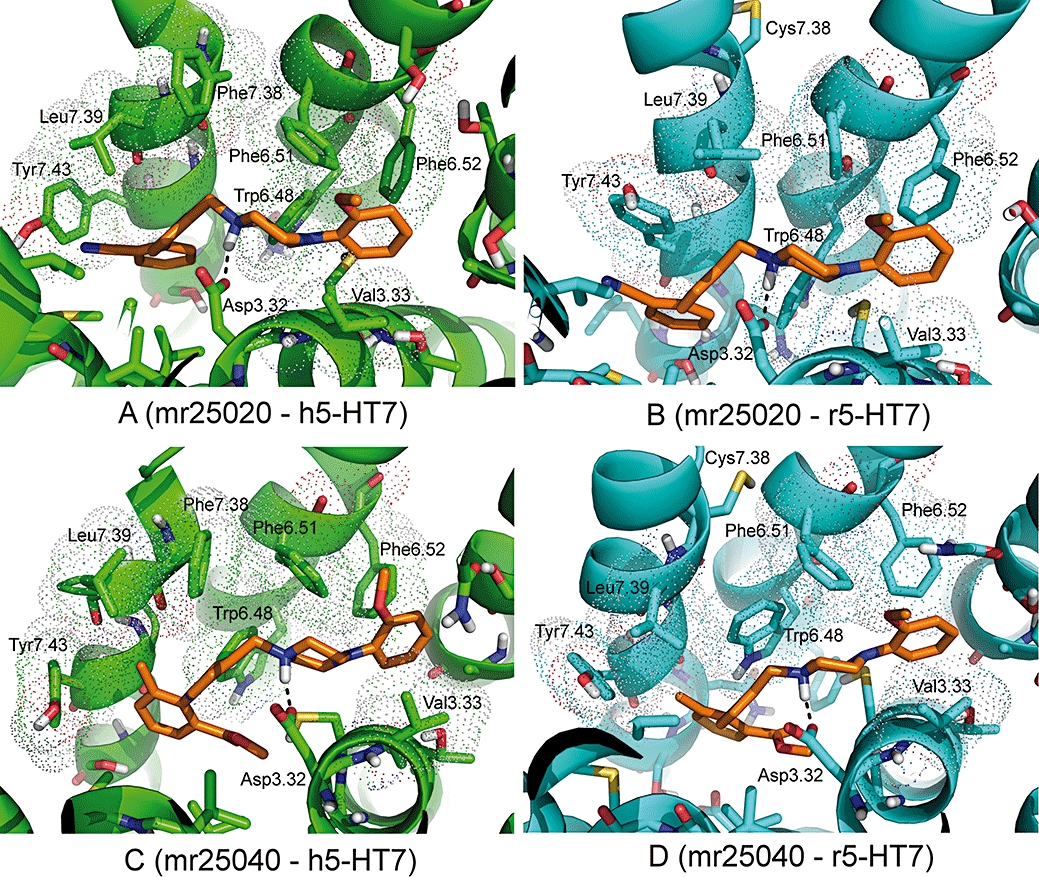
Proposed binding mode of mr25020 (top) and mr25040 (bottom) to human (left, green) and rat (right, cyan) 5-HT7 receptors. The TM helices of each receptor are displayed by green (human) or cyan (rat) cylinders. The ligands and selected side chains interacting with them are represented as sticks. Residues making non-polar contacts with the ligand are indicated with dotted surface, while H-bonds are indicated by dashed lines.
Table 3.
Experimental and calculated energetics (kcal·mol−1) of 5-HT7 binding, expressed in terms of human/rat receptor selectivity (ΔΔGbind)
| Compound | ΔΔGbind, exp (kcal·mol−1)a,b | ΔΔGbind, calc (kcal·mol−1)a |
ligand-surrounding interactions (kcal·mol−1)c |
|||
|---|---|---|---|---|---|---|
 |
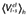 |
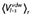 |
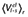 |
|||
| mr22423 | −0.6 | −2.0 ± 0.4 | −43.7 ± 0.2 | −148.5 ± 0.3 | −41.1 ± 0.0 | −145.5 ± 0.4 |
| mr22433 | −1.1 | −3.6 ± 0.7 | −32.9 ± 0.6 | −184.8 ± 0.5 | −32.6 ± 0.1 | −177.7 ± 0.6 |
| mr23284 | −0.7 | −3.2 ± 0.9 | −35.6 ± 0.7 | −166.1 ± 0.1 | −37.0 ± 0.1 | −159.3 ± 1.3 |
| mr25020 | −1.2 | −0.5 ± 0.8 | −58.3 ± 0.5 | −145.0 ± 0.9 | −56.4 ± 0.7 | −144.8 ± 0.2 |
| mr25040 | −0.5 | −2.4 ± 0.4 | −61.9 ± 0.1 | −148.2 ± 0.0 | −61.8 ± 0.6 | −143.5 ± 0.5 |
| mr25052 | −0.2 | −2.3 ± 0.5 | −39.5 ± 0.5 | −151.1 ± 0.3 | −39.5 ± 0.2 | −146.6 ± 0.4 |
| mr25053 | −1.1 | −0.5 ± 0.5 | −42.2 ± 0.4 | −150.7 ± 0.7 | −46.6 ± 0.5 | −148.2 ± 0.0 |
As a convention, selectivity for human 5-HT7 receptor is denoted by a negative sign for ΔΔGbind, while a positive sign would indicate selectivity for the rat receptor.
The binding free energies are calculated from experimentally determined Ki's using the relation 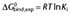 .
.
The calculated average electrostatic 〈Ve1〉 and non-polar 〈Vvdw〉energies for ligand surrounding (l-s) interactions. The subscripts h and r denote simulations of the ligand in complex with the human and rat 5-HT7 receptors respectively.
Phe369(7.38) is a key residue to explain the different selectivity in human versus rat 5-HT7 receptors
The molecular models we proposed were first tested by classical site-directed mutagenesis. The rat 5-HT7 receptor mutant (Cys372Phe) was engineered and tested for its binding properties for the seven compounds considered in this work. Competition experiments using [3H]SB269970 as radioligand with this set of selective human 5-HT7 ligands showed that mutating Cys to Phe profoundly affected the binding affinities of the rat receptor. As predicted, mutation of Cys372Phe increased the affinity of the rat 5-HT7 receptor for the different compounds by factors of between 2.79 and 9.58 (Table 1, Figure 3), depending on the compound, thus confirming its utmost importance for binding recognition.
Discussion and conclusions
In the present study we deal with the issue of interspecies selectivity in a given receptor, here the 5-HT7 receptor. Experimental binding affinity data reported in this work show a systematic drop in affinity when moving from the human to the rat receptors, for a series of compounds retrieved from our chemical library, (Paillet-Loilier et al., 2005; 2007;) all of them being in the low nanomolar range for the human receptor. As a result of our multidisciplinary approach, we report the identification of a single residue in helix 7, Phe/Cys(7.38), as responsible for the interspecies selectivity between human and rat 5-HT7 receptors for the series of compounds considered. Several pieces of published evidence have involved position 7.38 in GPCR-ligand binding. On the basis of previous homology models derived from rhodopsin and bacteriorhodopsin structures, Phe(7.38) has been identified as an important residue in the aromatic box forming the 5-HT binding site of 5-HT2B receptors (Manivet et al., 2002). However, even though several mutations were performed in that work, Phe(7.38) was not studied by site-directed mutagenesis. In any case, it is commonly accepted that the results obtained with one 5-HT receptor cannot necessarily be directly extrapolated to other 5-HT receptor subtypes. Analogously, in the mammalian type I and type II gonadotropin-releasing hormone (GnRH) receptors, Li et al. (2005) showed the importance of Phe(7.38) for differential ligand selectivity to GnRH-I and GnRH-II.
A precise mapping of molecular interactions between ligands and their receptors is a prerequisite for determining the functional role of such receptors and designing new agonists or antagonists. Up till now, a few 3D models of 5-HT7 receptors have been published (Lopez-Rodriguez et al., 2003; Nordling and Homan, 2004; Vermeulen et al., 2004) and all of them are based on the X-ray structure of bovine rhodopsin, with the exception of recent work that appeared during the preparation of this manuscript (Medina et al., 2009). Moreover, very few studies of the pharmacophores of 5-HT7 ligands are present in the literature, one of them performed by our group (Lopez-Rodriguez et al., 2000; 2003; Nordling and Homan, 2004; Vermeulen et al., 2004; Lepailleur et al., 2005). Thus, an important issue of the molecular modelling approach undertaken in this work is to take advantage of the recent crystal structure of the first GPCR belonging to the amine family, the β2-adrenoceptor. The proposed binding site, as defined with the preliminary docking exploration with the specific 5-HT7 antagonist SB656104 A, is in excellent agreement with previously described site-directed mutagenesis-guided mapping of the 5-HT family binding site. This binding site was exhaustively explored by automated docking of our series of compounds, proposing a single binding mode for the three families of compounds under study (ketanserin-like, tricyclic compounds and biphenyl derivatives). All molecules display quite similar binding modes in the human and rat 5-HT7 receptors, with ligand average RMSDs between the corresponding human and rat complexes being below 2 Å in all cases. On the basis of these binding modes, we have undertaken an MD and binding free energy analysis with the well-established LIE methodology. The adaptation of this approach for the estimation of selectivity between related proteins has been described previously, and it basically consists of a direct comparison of the ligand-surrounding energies in the two binding sites (Gutiérrez-de-Terán et al., 2006). In this case the MD simulations of the free (water solvated) state of the ligand, which often can be a major source of variance in the computed average energies (Marelius et al., 1998), are not necessary. As the experimental differences in binding affinity between human and rat receptors are approximately of the same magnitude as the errors usually associated with LIE calculations (typically around 1 kcal·mol−1), one can perhaps not expect a quantitative correlation between experimental and calculated interspecies selectivity for the 5-HT7 receptors. However, an excellent qualitative agreement between calculations and experimental data was achieved, as the binding to the human receptor was more favoured than the corresponding binding to the rat receptor in all seven cases. All the ligands were stabilized by aromatic interactions involving the well-known binding pocket consisting of aromatic/hydrophobic residues in helices 5 and 6 [with Phe(6.51) and Phe(6.52) in direct contact with the ligand, while Trp(6.48) and Phe(5.47) interact with the former]. The observed effect of having Phe(7.38) as an additional member of the aromatic cluster in the human receptor can be due either to a direct interaction with the ligand, or to an indirect effect, by contributing to the stabilization of the aromatic cluster and therefore to strongly tighten the binding of ligand to the site.
Most residues proposed to participate in the binding of our series of compounds have been shown to be of crucial relevance for the binding of agonists (Choudhary et al., 1995; Boess et al., 1997; Braden and Nichols, 2007). Hydrophobic interactions with Leu(7.39) are frequently observed for compound mr25040, in agreement with recently proposed models of the same receptor (Medina et al., 2009). Another interesting finding is the proposed interaction between the benzyl nitrile of this compound and Tyr374(7.43). A wide search for interactions between Tyr residues and nitrile groups at the Protein Data Bank (PDB) using IsoStar (Bruno et al., 1997) results in several entries from the PDB (as PDB codes: 1N95 and 1MZC), showing that this type of interaction is present in other protein–ligand complexes.
It is worth noting that the functional behaviour of the seven compounds under study is dependent on the chemical nature of the ligand. The longer compounds (mr25020 and mr25040) are characterized as antagonists at the human 5-HT7 receptor, whereas the tricyclic compounds (mr22423, mr22423 and mr22433) display a partial agonist profile. Finally, the biphenyl compounds are somehow special, as mr25053 was characterized in the functional assays as an antagonist, while mr25052 increased cAMP accumulation but did not shift the EC50 of the agonist 5-CT. As expected, the binding mode of the three classes of ligands is not the same, probably accounting for this different behaviour: the longer compounds occupy all of the antagonist binding site, as defined with the docking of SB656104 A, thus occupying the cavity that extends from helix 7 till helix 5. On the contrary, the tricyclic compounds are located on the cavity defined between helices 3, 5 and 6, which has been defined as the ‘agonist binding site’ of amine GPCRs (Brea et al., 2002). Thus the partial agonist behaviour of these compounds is consistent with our modelling exploration. However, in order to properly model the differences between ligands with different functional effects, the conformational effects on the receptor model should be taken into account (de Graaf and Rognan, 2008), which is out of the scope of the present study. The case of the biphenyl compounds is more intriguing, since even when they occupy the same ‘agonist binding mode’, the particular pharmacological profile of this particular pair of compounds cannot be explained from the structural or the chemical points of view. We hypothesize that a mixed allosteric profile could be an alternative explanation for the binding of this class of compounds.
Most importantly, molecular modelling results identifying the importance of position Phe(7.38) in the binding of the class of compounds reported here have been validated by site-directed mutagenesis studies. A rat 5-HT7 receptor mutant (Cys372Phe) was experimentally created bearing Phe (the amino acid present in human 5-HT7 receptor) instead of Cys at position 7.38. As predicted, the affinity values for all the compounds tested at the mutant rat receptor increased by a factor near 3 or higher (Table 1), approaching to the values obtained for the wild-type human receptor.
The multidisciplinary approach followed in the current study provides a consistent picture of the problem of the interspecies selectivity. The different analysis reported here, molecular modelling, site-directed mutagenesis and affinity studies all point in the same direction, enhancing our confidence in the final conclusion, which is the involvement of Phe(7.38) of the human receptor in ligand binding of the families of 5-HT7 ligands under consideration. To our knowledge, this is the first published report detailing the importance of this amino acid, Phe(7.38), in human 5-HT7 receptor for ligand binding. Altogether, the refined 3D models of human and rat 5-HT7 receptors in complex with these antagonists and partial agonists have shed light on putative ligand-receptor molecular interactions. These results should facilitate the identification of selective human 5-HT7 receptor ligands, which would be invaluable pharmacological tools.
Acknowledgments
The Computational Centers at Montpellier (CINES) and Galicia (CESGA) are gratefully acknowledged for allocation of computing time. This work was supported by a fellowship of the ‘Region Basse Normandie’ to T.V. M.C. and H.G.T. received financial support from ‘Programa Isidro Parga Pondal’ and J.B. received financial support from the ‘Program Isabel Barreto’, both from XUNTA de Galicia (Spain). Financial support from the Integrated action programs coordinated between French («Partenariats Hubert Curien») and Spanish Ministries of Education permitted the mobility of researchers between labs.
Glossary
Abbreviations:
- 5-CT
5-carboxytryptamine
- GPCRs
G protein-coupled receptors
- KD
dissociation constant
- Ki
inhibitory dissociation constant
- LIE
linear interaction energy
- MD
molecular dynamics
- PDB
protein data bank
- TM
transmembrane
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Functional characterization of selected compounds as partial agonists at human 5-HT7 receptors. Concentration–response curves for 5-CT in the absence (▪) and in the presence (▴) of 3 μM mr22423 (A), 10 μM mr22433 (B), 10 μM mr23284 (C) or 0.3 μM mr25052 (D). Data are expressed as the % of the maximal cAMP formation induced by 5-CT and represent the mean ± SEM (vertical bars) of two independent (n = 2) experiments performed in duplicate.
Figure S2 Docking of the 5-HT7 antagonist SB656104 A on the h5-HT7 receptor.
Figure S3 MD snapshots of seven ligands reported in this work on h5HT7 (left) and r5HT7 (right).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adham N, Zgombick JM, Bard J, Branchek TA. Functional characterization of the recombinant human 5-hydroxytryptamine7(a) receptor isoform coupled to adenylate cyclase stimulation. J Pharmacol Exp Ther. 1998;287:508–514. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl 2):S1, S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ander M, Luzhkov VB, Aqvist J. Ligand binding to the voltage-gated Kv1.5 potassium channel in the open state–docking and computer simulations of a homology model. Biophys J. 2008;94:820–831. doi: 10.1529/biophysj.107.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Attwood TK. A compendium of specific motifs for diagnosing GPCR subtypes. Trends Pharmacol Sci. 2001;22:162–165. doi: 10.1016/s0165-6147(00)01658-8. [DOI] [PubMed] [Google Scholar]
- Baldwin JM, Schertler GF, Unger VM. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, et al. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Methods in Neurosciences. San Diego: Academic Press; 1994. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors; pp. 366–428. [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Bissantz C. Conformational changes of G protein-coupled receptors during their activation by agonist binding. J Recept Signal Transduct Res. 2003;23:123–153. doi: 10.1081/rrs-120025192. [DOI] [PubMed] [Google Scholar]
- Bissantz C, Logean A, Rognan D. High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J Chem Inf Comput Sci. 2004;44:1162–1176. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- Boess FG, Monsma FJ, Jr., Meyer V, Zwingelstein C, Sleight AJ. Interaction of tryptamine and ergoline compounds with threonine 196 in the ligand binding site of the 5-hydroxytryptamine6 receptor. Mol Pharmacol. 1997;52:515–523. doi: 10.1124/mol.52.3.515. [DOI] [PubMed] [Google Scholar]
- Bonaventure P, Kelly L, Aluisio L, Shelton J, Lord B, Galici R, et al. Selective blockade of 5-hydroxytryptamine (5-HT)7 receptors enhances 5-HT transmission, antidepressant-like behavior, and rapid eye movement sleep suppression induced by citalopram in rodents. J Pharmacol Exp Ther. 2007;321:690–698. doi: 10.1124/jpet.107.119404. [DOI] [PubMed] [Google Scholar]
- Braden MR, Nichols DE. Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol Pharmacol. 2007;72:1200–1209. doi: 10.1124/mol.107.039255. [DOI] [PubMed] [Google Scholar]
- Brea J, Rodrigo J, Carrieri A, Sanz F, Cadavid MI, Enguix MJ, et al. New serotonin 5-HT(2A), 5-HT(2B), and 5-HT(2C) receptor antagonists: synthesis, pharmacology, 3D-QSAR, and molecular modeling of (aminoalkyl)benzo and heterocycloalkanones. J Med Chem. 2002;45:54–71. doi: 10.1021/jm011014y. [DOI] [PubMed] [Google Scholar]
- Brenchat A, Romero L, Garcia M, Pujol M, Burgueno J, Torrens A, et al. 5-HT7 receptor activation inhibits mechanical hypersensitivity secondary to capsaicin sensitization in mice. Pain. 2009;141:239–247. doi: 10.1016/j.pain.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Bruno IJ, Cole JC, Lommerse JP, Rowland RS, Taylor R, Verdonk ML. IsoStar: a library of information about nonbonded interactions. J Comput Aided Mol Des. 1997;11:525–537. doi: 10.1023/a:1007934413448. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, et al. >San Francisco: University of California; 2004. AMBER 8.0. [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary MS, Sachs N, Uluer A, Glennon RA, Westkaemper RB, Roth BL. Differential ergoline and ergopeptine binding to 5-hydroxytryptamine2A receptors: ergolines require an aromatic residue at position 340 for high affinity binding. Mol Pharmacol. 1995;47:450–457. [PubMed] [Google Scholar]
- Cullen BR. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- Czeh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M, et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA. 2001;98:12796–12801. doi: 10.1073/pnas.211427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:6620. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- Deng Q, Clemas JA, Chrebet G, Fischer P, Hale JJ, Li Z, et al. Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol Pharmacol. 2007;71:724–735. doi: 10.1124/mol.106.029223. [DOI] [PubMed] [Google Scholar]
- Dezi C, Brea J, Alvarado M, Ravina E, Masaguer CF, Loza MI, et al. Multistructure 3D-QSAR studies on a series of conformationally constrained butyrophenones docked into a new homology model of the 5-HT2A receptor. J Med Chem. 2007;50:3242–3255. doi: 10.1021/jm070277a. [DOI] [PubMed] [Google Scholar]
- Guscott M, Bristow LJ, Hadingham K, Rosahl TW, Beer MS, Stanton JA, et al. Genetic knockout and pharmacological blockade studies of the 5-HT7 receptor suggest therapeutic potential in depression. Neuropharmacology. 2005;48:492–502. doi: 10.1016/j.neuropharm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H, Correia C, Rodríguez D, Carvalho MA, Brea J, Cadavid MI, et al. Identification of Novel Scaffolds from an Original Chemical Library as Potential Antipsychotics. QSAR Comb Sci. 2009 doi: 10.1002/qsar200860198. [Google Scholar]
- Gutiérrez-de-Terán H, Nervall M, Ersmark K, Dunn BM, Hallberg A, Åqvist J. Inhibitor binding to the plasmepsin IV aspartic protease from plasmodium falciparum. Biochemistry. 2006;45:10529–10541. doi: 10.1021/bi0609669. [DOI] [PubMed] [Google Scholar]
- Hedlund PB, Huitron-Resendiz S, Henriksen SJ, Sutcliffe JG. 5-HT7 receptor inhibition and inactivation induce antidepressantlike behavior and sleep pattern. Biol Psychiatry. 2005;58:831–837. doi: 10.1016/j.biopsych.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Hedlund PB, Sutcliffe JG. Functional, molecular and pharmacological advances in 5-HT7 receptor research. Trends Pharmacol Sci. 2004;25:481–486. doi: 10.1016/j.tips.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res. 2003;31:294–297. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, et al. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin) Pharmacol Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav. 2002;71:533–554. doi: 10.1016/s0091-3057(01)00746-8. [DOI] [PubMed] [Google Scholar]
- Huang XP, Nagy PI, Williams FE, Peseckis SM, Messer WS., Jr Roles of threonine 192 and asparagine 382 in agonist and antagonist interactions with M1 muscarinic receptors. Br J Pharmacol. 1999;126:735–745. doi: 10.1038/sj.bjp.0702301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- King G, Warshel A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J Chem Phys. 1989;91:3647–3661. [Google Scholar]
- Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, et al. A highly conserved aspartic acid (Asp-155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT(2A) serotonin receptor but does not participate in activation via a ‘salt-bridge disruption’ mechanism. J Pharmacol Exp Ther. 2000;293:735–746. [PubMed] [Google Scholar]
- Laurila JM, Xhaard H, Ruuskanen JO, Rantanen MJ, Karlsson HK, Johnson MS, et al. The second extracellular loop of alpha2A-adrenoceptors contributes to the binding of yohimbine analogues. Br J Pharmacol. 2007;151:1293–1304. doi: 10.1038/sj.bjp.0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepailleur A, Bureau R, Lemaîtra S, Dauphin F, Lancelot JC, Contesse V, et al. Molecular design based on 3D pharmacophores. Applications to 5-HT7 receptors. J Chem Inf Comput Sci. 2004;44:1148–1152. doi: 10.1021/ci030036l. Correction added after online publication 25 February 2010: the above reference was added to the list. [DOI] [PubMed] [Google Scholar]
- Lepailleur A, Bureau R, Paillet-Loilier M, Fabis F, Saettel N, Lemaitre S, et al. Molecular modeling studies focused on 5-HT7 versus 5-HT1A selectivity. Discovery of novel phenylpyrrole derivatives with high affinity for 5-HT7 receptors. J Chem Inf Model. 2005;45:1075–1081. doi: 10.1021/ci050045p. [DOI] [PubMed] [Google Scholar]
- Li JH, Choe H, Wang AF, Maiti K, Wang C, Salam A, et al. Extracellular loop 3 (EL3) and EL3-proximal transmembrane helix 7 of the mammalian type I and type II gonadotropin-releasing hormone (GnRH) receptors determine differential ligand selectivity to GnRH-I and GnRH-II. Mol Pharmacol. 2005;67:1099–1110. doi: 10.1124/mol.104.004887. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez ML, Porras E, Benhamu B, Ramos JA, Morcillo MJ, Lavandera JL. First pharmacophoric hypothesis for 5-HT7 antagonism. Bioorg Med Chem Lett. 2000;10:1097–1100. doi: 10.1016/s0960-894x(00)00166-9. [DOI] [PubMed] [Google Scholar]
- Lopez-Rodriguez ML, Porras E, Morcillo MJ, Benhamu B, Soto LJ, Lavandera JL, et al. Optimization of the pharmacophore model for 5-HT7R antagonism. Design and synthesis of new naphtholactam and naphthosultam derivatives. J Med Chem. 2003;46:5638–5650. doi: 10.1021/jm030841r. [DOI] [PubMed] [Google Scholar]
- Manivet P, Schneider B, Smith JC, Choi DS, Maroteaux L, Kellermann O, et al. The serotonin binding site of human and murine 5-HT2B receptors: molecular modeling and site-directed mutagenesis. J Biol Chem. 2002;277:17170–17178. doi: 10.1074/jbc.M200195200. [DOI] [PubMed] [Google Scholar]
- Marelius J, Hansson T, Åqvist J. Calculation of ligand binding free energies from molecular dynamics simulations. Int J Quantum Chem. 1998;69:77–88. [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Martin-Cora FJ, Pazos A. Autoradiographic distribution of 5-HT7 receptors in the human brain using [3H]mesulergine: comparison to other mammalian species. Br J Pharmacol. 2004;141:92–104. doi: 10.1038/sj.bjp.0705576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JM, Martinez L. Packing optimization for automated generation of complex system's initial configurations for molecular dynamics and docking. J Comput Chem. 2003;24:819–825. doi: 10.1002/jcc.10216. [DOI] [PubMed] [Google Scholar]
- Medina RA, Sallander J, Benhamu B, Porras E, Campillo M, Pardo L, et al. Synthesis of new serotonin 5-HT7 receptor ligands. Determinants of 5-HT7/5-HT1A receptor selectivity. J Med Chem. 2009;52:2384–2392. doi: 10.1021/jm8014553. [DOI] [PubMed] [Google Scholar]
- Mialet J, Dahmoune Y, Lezoualc'h F, Berque-Bestel I, Eftekhari P, Hoebeke J, et al. Exploration of the ligand binding site of the human 5-HT(4) receptor by site-directed mutagenesis and molecular modeling. Br J Pharmacol. 2000;130:527–538. doi: 10.1038/sj.bjp.0703356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Clay WC, Ng SW, Roman S, Thompson K, Condreay JP, et al. Identification of regions of the P2X(7) receptor that contribute to human and rat species differences in antagonist effects. Br J Pharmacol. 2008;155:738–751. doi: 10.1038/bjp.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Abola E, Participants GA, Brooks, III CL, Dixon JS, Moult J, et al. Community-wide blind assessment of methods for GPCR structure modeling and docking. Nat Rev Drug Discov. 2009 doi: 10.1038/nrd2877. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandam LS, Jhaveri D, Bartlett P. 5-HT7, neurogenesis and antidepressants: a promising therapeutic axis for treating depression. Clin Exp Pharmacol Physiol. 2007;34:546–551. doi: 10.1111/j.1440-1681.2007.04608.x. [DOI] [PubMed] [Google Scholar]
- Nordling E, Homan E. Generalization of a targeted library design protocol: application to 5-HT7 receptor ligands. J Chem Inf Comput Sci. 2004;44:2207–2215. doi: 10.1021/ci049822w. [DOI] [PubMed] [Google Scholar]
- Paillet-Loilier M, Fabis F, Lepailleur A, Bureau R, Butt-Gueulle S, Dauphin F, et al. Phenylpyrroles, a new chemolibrary virtual screening class of 5-HT7 receptor ligands. Bioorg Med Chem Lett. 2005;15:3753–3757. doi: 10.1016/j.bmcl.2005.05.059. [DOI] [PubMed] [Google Scholar]
- Paillet-Loilier M, Fabis F, Lepailleur A, Bureau R, Butt-Gueulle S, Dauphin F, et al. Novel aminoethylbiphenyls as 5-HT7 receptor ligands. Bioorg Med Chem Lett. 2007;17:3018–3022. doi: 10.1016/j.bmcl.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rezmann-Vitti LA, Louis SN, Nero TL, Jackman GP, Machida CA, Louis WJ. Site-directed mutagenesis of the rat beta1-adrenoceptor. Involvement of Tyr356 (7.43) in (+/−)cyanopindolol but not (+/−)[125Iodo]cyanopindolol binding. Eur J Med Chem. 2004;39:625–631. doi: 10.1016/j.ejmech.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Selent J, Lopez L, Sanz F, Pastor M. Multi-receptor binding profile of clozapine and olanzapine: a structural study based on the new beta2 adrenergic receptor template. ChemMedChem. 2008;3:1194–1198. doi: 10.1002/cmdc.200800074. [DOI] [PubMed] [Google Scholar]
- Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a model of agonist-induced activation of 5-hydroxytryptamine 2A serotonin receptors that involves the disruption of a strong ionic interaction between helices 3 and 6. J Biol Chem. 2002;277:11441–11449. doi: 10.1074/jbc.M111675200. [DOI] [PubMed] [Google Scholar]
- Sylte I, Bronowska A, Dahl SG. Ligand induced conformational states of the 5-HT(1A) receptor. Eur J Pharmacol. 2001;416:33–41. doi: 10.1016/s0014-2999(01)00860-3. [DOI] [PubMed] [Google Scholar]
- Thomas DR, Hagan JJ. 5-HT7 receptors. Curr Drug Targets CNS Neurol Disord. 2004;3:81–90. doi: 10.2174/1568007043482633. [DOI] [PubMed] [Google Scholar]
- Thomas DR, Melotto S, Massagrande M, Gribble AD, Jeffrey P, Stevens AJ, et al. SB-656104-A, a novel selective 5-HT7 receptor antagonist, modulates REM sleep in rats. Br J Pharmacol. 2003;139:705–714. doi: 10.1038/sj.bjp.0705290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, et al. A1 adenosine receptors. Two amino acids are responsible for species differences in ligand recognition. J Biol Chem. 1994;269:27900–27906. [PubMed] [Google Scholar]
- Vermeulen ES, Schmidt AW, Sprouse JS, Wikstrom HV, Grol CJ. Characterization of the 5-HT(7) receptor. Determination of the pharmacophore for 5-HT(7) receptor agonism and CoMFA-based modeling of the agonist binding site. J Med Chem. 2003;46:5365–5374. doi: 10.1021/jm030826m. [DOI] [PubMed] [Google Scholar]
- Vermeulen ES, van Smeden M, Schmidt AW, Sprouse JS, Wikstrom HV, Grol CJ. Novel 5-HT7 receptor inverse agonists. Synthesis and molecular modeling of arylpiperazine- and 1,2,3,4-tetrahydroisoquinoline-based arylsulfonamides. J Med Chem. 2004;47:5451–5466. doi: 10.1021/jm049743b. [DOI] [PubMed] [Google Scholar]
- Wilcox RE, Ragan JE, Pearlman RS, Brusniak MY, Eglen RM, Bonhaus DW, et al. High-affinity interactions of ligands at recombinant guinea pig 5HT7 receptors. J Comput Aided Mol Des. 2001;15:883–909. doi: 10.1023/a:1014319812972. [DOI] [PubMed] [Google Scholar]
- Wolf S, Bockmann M, Howeler U, Schlitter J, Gerwert K. Simulations of a G protein-coupled receptor homology model predict dynamic features and a ligand binding site. FEBS Lett. 2008;582:3335–3342. doi: 10.1016/j.febslet.2008.08.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.