

Abstract
Background and purpose:
In humans and non-human primates, the 7TM receptor GPR17 exists in two isoforms differing only by the length of the N-terminus. Of these, only the short isoform has previously been characterized. Hence, we investigated gene expression and ligand-binding profiles of both splice variants and furthermore uncovered and characterized constitutive activity of both isoforms.
Experimental approach:
Expression levels of the hGPR17 isoforms were determined in several brain regions as well as heart and kidney using quantitative RT-PCR. A CREB reporter assay and [35S]-GTPγS binding were employed to assess the constitutive activity and the activation by UDP, UDP-glucose and -galactose and the cysteinyl leukotrienes LTC4 and LTD4. Leukotriene binding and induction of internalization were furthermore tested using homologous competition binding and antibody-feeding experiments respectively.
Key results:
The short isoform (hGPR17-S) was expressed more abundantly (eight- to 23-fold) in the brain than the long isoform (hGPR17-L), whereas the opposite was observed in heart and kidney. As previously reported, the uracil nucleotides activated hGPR17-S with micromolar potencies. However, much lower potencies were observed for hGPR17-L with a 50- to 170-fold increase in EC50. Furthermore, contrary to previous reports, neither of the isoforms was activated or bound by the cysteinyl leukotrienes. Finally, both receptors were demonstrated to be constitutively active through Gαi.
Conclusions and implications:
We present the first isoform-specific characterization of GPR17 and show that differences exist between the isoforms, in both expression pattern and pharmacological profile. In turn, our results indicate that the two human isoforms might serve tissue-specific functions.
Keywords: 7TM receptor, GPCR, splice variants, GPR17, constitutive activity, differential expression
Introduction
The group of rhodopsin-like seven transmembrane-spanning (7TM) receptors constitutes one of the largest superfamilies of proteins in humans and vertebrates in general. These receptors, also known as G protein-coupled receptors, are targets for an extraordinarily large repertoire of endogenous ligands, which shows a vast variation in structure, size and chemical properties. The diverse group of ligands illustrates the involvement of 7TM receptors in a great variety of physiological processes, which ranges from regulation of blood pressure, over modulation of leukocyte trafficking and appetite control to visualization (Schwartz and Holst, 2003). The existence of splice variants further diversifies 7TM receptor signal transduction and expression patterns. Thus, several 7TM receptor splice variants have been shown to differ by their pharmacological profiles, constitutive activity, internalization pattern or tissue distribution (Kilpatrick et al., 1999). Importantly, a study using dopamine D2 receptor knockout mice demonstrated that two splice variants of this receptor indeed play specific roles in vivo effectively demonstrating the relevance of receptor splice variants (Usiello et al., 2000).
The 7TM receptor GPR17 was cloned in the search of new chemokine receptors, using human genomic DNA as template (Raport et al., 1996). The same finding was later obtained by another group (Blasius et al., 1998). However, having used cDNA as template, a GPR17 splice variant was also found encoding a receptor with a 28 amino acid longer N-terminus (Blasius et al., 1998). Today it is known that in humans and other primates, but not in lower species, alternative splicing results in two GPR17 isoforms that differ solely with regard to the length of the N-terminus. Of these, only the short isoform has been a subject of study so far (Ciana et al., 2006; Lecca et al., 2008; Parravicini et al., 2008; Ceruti et al., 2009; Maekawa et al., 2009; Temporini et al., 2009). Phylogenetically, GPR17 is most closely related to members of the P2Y nucleotide receptor subfamily containing, among others, the P2Y-specific sequence ‘fingerprint’ in transmembrane (TM) 3 (FLFYxNLYxSILFLTCISx). Besides the P2Y receptors, GPR17 is also structurally related to the two cysteinyl receptors CysLT1 and CysLT2 and is the closest human homologue to the Epstein–Barr virus-induced receptor 2 (EBI2).
Given the structural resemblance to both P2Y and cysteinyl leukotriene receptors, the short human GPR17 isoform as well as the rat and murine GPR17 were recently suggested to be targeted by two distinct types of agonists, namely cysteinyl leukotrienes and uracil nucleotides (Ciana et al., 2006; Lecca et al., 2008). According to these studies, LTC4 and LTD4 activated the receptors with EC50 values in the nM range, while UDP, UDP-glucose and UDP-galactose activated them with EC50 values in the µM range (Ciana et al., 2006). It was furthermore shown that GPR17 expression was up-regulated in a rat model of ischaemic damage and that knock-down of the receptor prevented ischaemia, suggesting that GPR17 putatively could function as a mediator of brain damage (Ciana et al., 2006). The same phenomenon was later observed in the equivalent murine model of ischaemia (Lecca et al., 2008) and in a model of spinal cord injury (Ceruti et al., 2009). Very recently, GPR17 was shown to function as a negative regulator of CysLT1 receptor activation in stable cell lines by inhibiting LTD4 binding to CysLT1. This regulatory effect was furthermore confirmed both in bone marrow-derived macrophages by shRNA-mediated knock-down and in GPR17-deficient mice. Of note, no binding of LTC4 and LTD4 to GPR17 was seen in this study (Maekawa et al., 2009)
In the present study, we carry out a detailed expression analysis of the two human GPR17 isoforms in a range of brain regions as well as the heart and kidney. Using several different assays, we furthermore demonstrate that both of these isoforms and the murine GPR17 are constitutively active via Gαi. In addition, we uncovered a difference in the pharmacological profiles of the two human isoforms by showing that the short isoform is activated more potently by uracil nucleotides than the long isoform. Finally, by employing [35S]-GTPγS binding and competition binding, in line with the very recent report by Maekawa et al. (2009), we demonstrated that neither of the human isoforms nor the mGPR17 are activated by or bound by the leukotrienes LTD4 and LTC4.
Methods
Materials
Receptor nomenclature throughout the manuscript conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2008). The long isoform of hGPR17, hGPR17-L (GenBank accession number NM_005291) was kindly provided by H.R. Luttichau, Laboratory for Molecular Pharmacology. The promiscuous chimeric G protein GαΔ6qi4myr (abbreviated Gqi4myr) was kindly provided by Evi Kostenis (Rheinische Friedrich-Wilhelm University, Bonn, Germany). Lipofectamine™ 2000 reagent and OPTIMEM were purchased from Life Technologies. SteadyLite (Lyophilized Substrate Solution) was from Packard (Boston, MA, USA). Goat anti-mouse horseradish peroxidase-conjugated antibody was from Pierce (Rockford, IL, USA), while mouse anti-M1-FLAG antibody, Ticlopidine (P2Y12 receptor antagonist), forskolin and pertussis toxin were from Sigma Chemicals Co. (St. Louis, MO, USA). Montelukast (CysLT1 receptor antagonist) was from Sequoia Research Products (Oxford, UK). Both the SlowFade Antifade reagent and goat anti-mouse Alexa Fluor 488-conjugated and Alexa Flour 568-conjugated antibodies were from Molecular Probes (Carlsbad, CA, USA). The TMB (3,3',5,5'-tetramethylbenzidine) substrate was purchased from KemEnTech (Taastrup, Denmark).
DMEM was purchased from Invitrogen; the Complete protease inhibitor mixture from Roche (Mannheim, Germany); BCA protein assay kit from Pierce; the 96-well plates from Nunc and the transparent 96-well plates from Costar. [35S]-GTPγS, SteadyLite substrate, [3H]-LTC4 and [3H]-LTD4 were purchased from Perkin Elmer; WGA-coupled SPA-beads from Amersham Biosciences; the trans-reporter CREB-luciferase assay was obtained from Stratagene (La Jolla, CA, USA); the ImProm II Reverse Transcriptase kit from Promega; the SYBR Premix Ex Taq RT-PCR kit from Takara Bio Inc. and the MX3000p cycler from Stratagene.
Site-directed mutagenesis and cloning
All GPR17 constructs, CysLT1 and CysLT2 were inserted into a modified pcDNA3 vector, kindly provided by Kate Hansen (7TM-Pharma, Denmark), which contained an upstream sequence encoding a haemagglutinin signal peptide fused to the M1-FLAG tag. Cloning of the murine GPR17 was achieved by nested PCR using genomic DNA from mice as template and verified by sequencing.
Transfection and tissue culture
Both HEK293 and 1321N1 cells were grown in DMEM adjusted to contain 4500 mg·L–1 glucose, 10% FBS (fetal bovine serum), 180 u·mL–1 penicillin and 45 ug·mL–1 streptomycin (PenStrep) at 10% CO2 and 37°C. For CREB-luciferase and ELISA assays, transient transfections were carried out using Lipofectamine™ 2000 reagent and the serum-free medium OPTIMEM as described previously (Benned-Jensen and Rosenkilde, 2008). Cells were always transfected in parallel for the CREB luciferase and ELISA assays. For membrane preparations, transient transfections were performed using the calcium precipitation method as described previously (Rosenkilde et al., 1999).
Membrane preparation
Membranes were prepared from HEK293 and 1321N1 cells transiently transfected with pcDNA3 vector control, hGPR17-L, hGPR17-S, mGPR17 and CysLT2 wt. The cells were manually harvested with a rubber policeman in ice-cold PBS and homogenized using a Dounce on ice. The homogenate was centrifuged at 45 g for 3 min at 4°C. Subsequently, the supernatants were collected and centrifuged at 47 800 g at 4°C. The resulting membrane pellets were resuspended in 20 mM HEPES buffer containing 2 mM MgCl2 and Complete protease inhibitor mixture and kept at –80°C until subjected to [35S]-GTPγS binding experiments. The protein concentrations in each preparation were determined using the BCA protein assay kit.
[35S]-GTPγS binding assay
[35S]-GTPγS binding experiments were carried out in white 96-well plates using the SPA-based method. A volume of membrane preparation (corresponding to 20 µg protein per well) was diluted in assay buffer (50 mM HEPES, 3 mM MgCl2, 100 mM NaCl, 1 mM EGTA, 3 µM GDP, 10 µg·mL–1 saponin and Complete protease inhibitor mix, pH 7.4). [35S]-GTPγS (1250 Ci·mmol–1, 12.5 mCi·mL–1) diluted in assay buffer was added to a final concentration of 1 nM and incubated for 1 h at 30°C. When used, LTD4 was added at 1 µM along with a vehicle control (DMSO) at this step. Subsequently, WGA-coupled SPA-beads was added (final concentration of 2.8 mg·mL–1) followed by 30 min incubation at room temperature on a plate shaker. Finally, the plates were centrifuged at 400 g for 5 min and the amount of radioactivity determined using a Top Count scintillation counter (Packard Instruments). The level of non-specific binding was determined by adding unlabelled GTPγS at a final concentration of 40 µM. All experiments were carried out at least three times and in triplicates.
Competition binding assay
Competition binding experiments were carried out in transparent 96-well plates employing [3H]-LTC4 and [3H]-LTD4 as radioligands. A volume of membrane preparation (corresponding to 25 µg protein per well) was diluted in binding buffer (final concentration: 50 mM Tris-HCl, 5 mM MgCl2, 100 µg·mL–1 Bacitracin and Complete protease inhibitor mix, pH 7.4). [3H]-LTC4 or [3H]-LTD4 (122.7 Ci·mmol–1, 0.01 mCi·mL–1) diluted in binding buffer was added to a final concentration of 0.4 nM and subsequently unlabelled LTC4 or LTD4 was added in the concentration range of 0.1 nM to 1 µM. Following 2 h incubation at room temperature, the membranes were captured on Skatron 11731 FilterMATs using a Skatron cell harvester and GF/C filters. The harvested membranes were washed in buffer (50 mM Tris-HCl, 5 mM MgCl2 and 0.1% BSA) and dried for 30 min at 60°C. The amount of radioactivity was determined using EcoScint™ XR and a Beckman scintillation counter. The specific binding accounted for approximately 8% of total binding (∼1600 cpm). All experiments were carried out at least three times and in duplicates.
cAMP response element binding protein trans-reporter luciferase assay
The level of constitutive activity was determined using the trans-reporter cAMP response element binding protein (CREB)-luciferase assay according to the manufacturer's recommendations. HEK293 cells were seeded at 35 000 cells per well in 96-well plates and were transiently transfected the following day with FLAG-tagged receptor constructs or pcDNA3 at the indicated concentrations along with the trans-activator plasmid pFA2-CREB and the reporter plasmid pFRLUC at 6 ng per well and 50 ng per well respectively. In some experiments, the cells were also co-transfected with the chimeric G-protein, GαΔ6qi4myr (abbreviated Gqi4myr) at 30 ng per well (Kostenis, 2001). The CREB activity was determined 48 h after transfection using the SteadyLite substrate. Briefly, the cells were washed twice in Dulbecco's PBS (0.9 mM CaCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl2, 137 mM NaCl, and 8.1 mM Na2HPO4) and the luminescence measured 10 min after addition of the substrate using a TopCounter (Packard). When used, forskolin (15 µM), UDP, UDP-glucose, UDP-galactose (all in range of 10 nM to 100 µM), LTC4 or LTD4 (0.1 nM to 1 µM) was added 5 h prior to substrate addition, while pertussis toxin (100 ng·mL–1) was added 24 h before substrate addition. For the antagonism test of Montelukast and Ticlopidine, the compounds were given 15 min prior to the agonists (i.e. 5 h and 15 min before substrate addition), whereas the same two compounds, when tested for inverse agonism, were given 24 h before substrate addition (i.e. right after termination of transfection). Each receptor construct was tested at least three times in quadruplicates.
Enzyme-linked immunosorbent assay
HEK293 cells were transiently transfected with the indicated FLAG-tagged receptor constructs as described above; 48 h after transfection, the cells were fixed in 4% formaldehyde for 10 min, washed three times in TBS (50 mM Tris and 150 mM NaCl, pH 7.6) and blocked for 30 min with TBS containing 2% BSA. Subsequently, the cells were incubated with mouse anti-FLAG M1 antibody at 2 ug·mL–1 for 2 h in TBS supplemented with 1% BSA and 1 mM CaCl2. After three washes in TBS containing 1% BSA and 1 mM CaCl2, the cells were incubated for 1 h with goat anti-mouse horseradish peroxidase-conjugated antibody diluted 1:1000 in the same buffer as the primary antibody. Following washing, the immune reactivity was determined by addition of TMB according to manufacturer's instructions. All steps were carried out at room temperature.
Quantitative real-time PCR
Total RNA from heart, kidney, total brain and eight different brain regions (amygdala, cerebellar hemisphere, cerebellum, frontal cortex, hippocampus, hypothalamus, putamen and thalamus) were acquired from Ambion. cDNA was synthesized from 1 ug of RNA using the ImProm II Reverse Transcriptase kit and random hexamer primers. Quantification of hGPR17-L and -S transcript levels were performed using the SYBR Premix Ex Taq RT-PCR kit and a MX3000p cycler. Expression levels of β-actin were used for copy normalization. All experiments were performed four times in triplicates. The forward primers for the two GPR17 transcripts were isoform-specific and did both span exon-exon junctions preventing amplification of contaminant genomic DNA, while the reverse primer was shared giving amplicon lengths of 189 bp for hGPR17-L and 187 bp for hGPR17-S. The β-actin primers did also span an exon-exon junction and gave an amplicon length of 80 bp. The sequence of the primers were: forward hGPR17-L: GCTGAAACTCTCAGGCTCTGAC, forward hGPR17-S: CCAGCAGCTAGAGGCTCTGA, reverse hGPR17-L/-S: GCCAGGGTATTGCCAACTAA, forward β-actin: CGTCTTCCCCTCCATCGT and reverse β-actin: CGCCCACATAGGAATCCTTC. Amplifications of the primer pairs were validated using serial dilution of the total brain cDNA and specificity by dissociation curve analysis being satisfactory in all cases.
Antibody-feeding internalization assay
HEK293 cells were seeded on poly-L-lysine-coated coverslips in six-well plates at 5 × 105 cells per well. The following day the cells were transfected with the indicated FLAG-tagged receptor constructs at 150 ng per well using Lipofectamine™ 2000 as described above. Forty-eight hours after transfection, the cells were incubated in cold DMEM medium containing mouse M1 anti-FLAG antibody at 2 µg·mL–1 and incubated 1 h at 4°C. After being washed in cold DMEM medium, the specimens were either immediately fixed in 4% paraformaldehyde or incubated in pre-warmed DMEM medium containing either vehicle (DMSO) or LTD4 at 1 µM at 37°C for 30 min, to induce internalization, and then fixed. Subsequently, the coverslips were blocked with TBS containing 2% BSA. To specifically detect labelled receptors still residing at the cell surface, the cells were incubated with goat anti-mouse Alexa Flour 488-conjugated antibody diluted 1:1000 in TBS containing 1% BSA for 30 min. After being washed, the cells were permeabilized using TBS containing 1% BSA and 0.2% saponin for 30 min. To detect internalized labelled receptors, the coverslips were incubated with goat anti-mouse Alexa-Fluor 568-conjugated antibodies diluted 1:1000 in TBS containing 1% BSA for 30 min. Subsequent to washing, the specimens were mounted in SlowFade Antifade reagent using nail polish as a sealant. All steps following the internalization step were carried out at room temperature. Mock transfected cells were included to ensure no non-specific binding of any of the antibodies.
Confocal microscopy
Confocal microscopy was performed using a LSM 510 laser scanning unit coupled to an inverted microscope with a 63 × 1.4 numerical aperture oil immersion Plan-Apochromat objective (Carl Zeiss). Alexa-Fluor 488 was excited using an argon-krypton laser (λ = 488 nm) and the emission collected with a 505 nm long-pass filter. Images were recorded in 1024 × 1024 pixels and averaged over 16 whole frame scans.
Statistical analysis
All statistical analyses were carried out using GraphPad Prism software. EC50 and IC50 values were determined using non-linear regression curve fits. Differences in expression levels (Figure 1D), expression ratios (Figure 2C), constitutive activity measured using [35S]-GTPγS binding (Figure 4) and receptor activation by LTD4 using [35S]-GTPγS binding (Figure 7C) were analysed by using Student's t-test.
Figure 1.
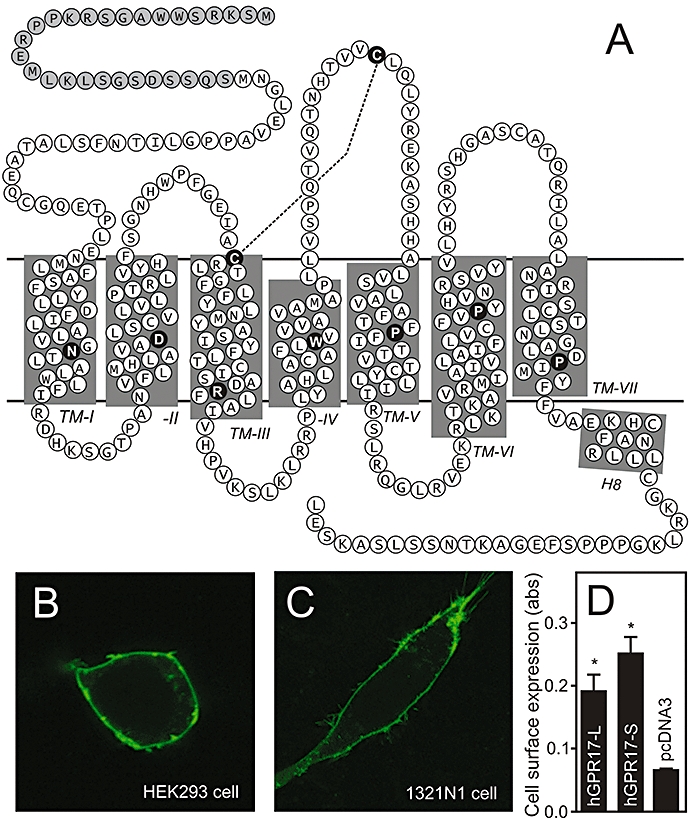
Primary structure and cell surface expression of hGPR17. (A) Serpentine model of hGPR17. The highly conserved residues among rhodopsin-like 7TM receptors are indicated as black circles with white letters in each transmembrane helix and the conserved disulphide-bridge between the extracellular loop 2 and the conserved cysteine residue in TM3 denoted as a stapled line. The 28 amino acids comprising the longer N-terminus of the hGPR17-L splice variant are indicated as grey circles. (B) Expression of C-terminally eGFP-tagged hGPR17-L in transiently transfected HEK293 cells as detected by confocal microscopy. (C) Expression of C-terminally eGFP-tagged hGPR17-L in transiently transfected 1321N1 cells as detected by confocal microscopy. (D) Cell surface expression of FLAG-tagged hGPR17 isoforms in HEK293 cells transiently transfected with hGPR17-L, -S or pcDNA3 vector control as measured by ELISA. The results represent mean ± SEM of raw data from three independent experiments performed in quadruplicates. *P<0.05 by Student's t-test. 7TM, seven transmembrane; hGPR17-L, long GPR17 isoform.
Figure 2.
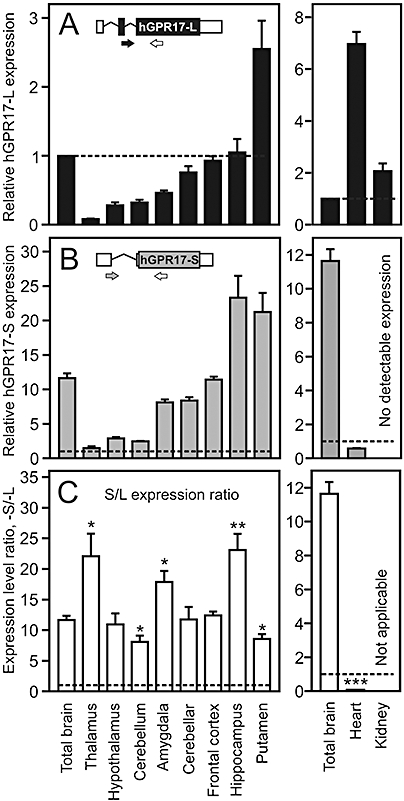
Gene expression levels of the two hGPR17 isoforms and the ratio of these in brain, heart and kidney. (A) Levels of hGPR17-L transcript in total brain and eight different brain regions (left panel) and kidney and heart (right panel). The expression levels are given relative (in fold) to that in total brain, which is indicated by the stippled line. The insert is a schematic representation of the hGPR17-L mRNA species. Translated and untranslated regions are indicated in black and white respectively. The isoform-specific forward primer spans the exon 2–3 border and is depicted in black, whereas the shared reverse primer is depicted in white. (B) Levels of hGPR17-S the same tissues as in (A). The expression levels are given relative (in fold) to that of hGPR17-L in total brain (stippled line). No expression was detected in kidney. The insert is a schematic representation of the hGPR17-S mRNA species. Translated and untranslated regions are indicated in grey and white respectively. The isoform-specific forward primer spans the exon 1–3 border and is depicted in grey, whereas the shared reverse primer is depicted in white. (C) Ratio of hGPR17-S and -L expression levels from (B) and (A) respectively. The stippled line indicates a ratio of 1, i.e. equal expression levels. Ratios significantly different from that in total brain are indicated; *P<0.05, **P<0.01 and ***P<0.0001 (Student's t-test). Given the absence of hGPR17-S expression in kidney the ratio is not calculable in this instance. hGPR17-L, long GPR17 isoform; hGPR17-S, short GPR17 isoform.
Figure 4.
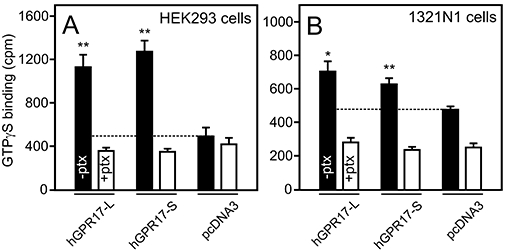
Constitutive signalling of hGPR17-L and -S as measured by [35S]-GTPγS binding. (A) [35S]-GTPγS binding to membranes isolated from HEK293 cells transiently transfected with FLAG-tagged hGPR17-L, -S or pcDNA3. The cells were cultured in the absence (solid columns) or presence (open columns) of 100 ng·mL–1 pertussis toxin (ptx). The level of binding to pcDNA3-membranes is indicated by the stippled line. The results represent mean ± SEM of raw data from four independent experiments performed in triplicates. **P<0.01 by Student's t-test. (B) [35S]-GTPγS binding to membranes isolated from 1321N1 cells transiently transfected with hGPR17-L, -S or pcDNA3; performed and presented as in (A). *P<0.05, **P<0.01 by Student's t-test. hGPR17-L, long GPR17 isoform; hGPR17-S, short GPR17 isoform.
Figure 7.
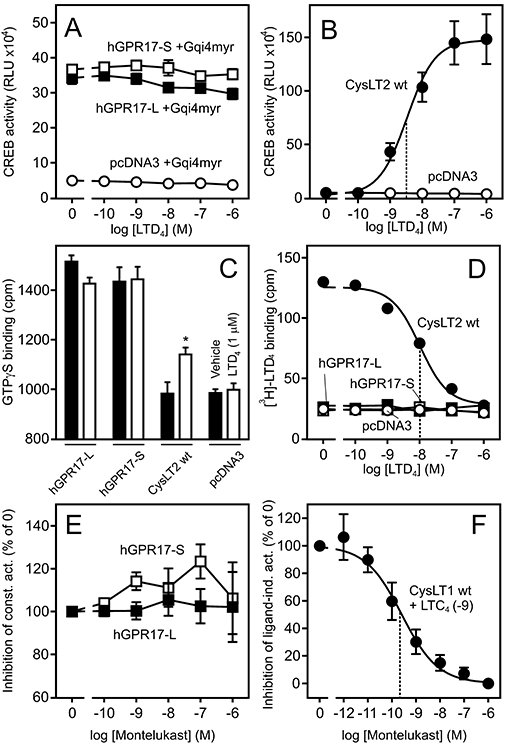
LTD4 does not activate or bind to either of the hGPR17 isoforms. (A) Response to LTD4 in HEK293 cells transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and FLAG-tagged hGPR17-L, -S or pcDNA3 at 15 ng per well. The results represent mean ± SEM of raw data from four independent experiments performed in quadruplicates. (B) Activation of FLAG-tagged CysLT2 by LTD4; performed and presented as in (A). The EC50 value is indicated by the stippled line. (C) Effects of LTD4 1 µM (open columns) or vehicle (DMSO, solid columns) on [35S]-GTPγS binding to membranes isolated from HEK293 cells transiently transfected with FLAG-tagged hGPR17-L, -S, CysLT2 or pcDNA. The data represent mean ± SEM of raw data from four independent experiments performed in triplicates. *P<0.05 by Student's t-test. (D) Homologous competition assay for [3H]-LTD4 binding to the membranes from HEK293 cells transiently transfected with hGPR17-L, -S, CysLT2 or pcDNA3; these were similar to those used in (C). The data represent mean ± SEM of raw data from four independent experiments performed in duplicates. ipThe IC50 value for CysLT2 is indicated by the stapled line. (E) Putative inverse agonism of Montelukast. HEK293 cells were transfected as in (A) and incubated with Montelukast in varying concentrations. The results represent mean ± SEM of three independent experiments performed in triplicates and are given as % relative to the activity in absence of the ligand. (F) CysLT1 control. HEK293 cells were transfected as described in (A) and incubated with LTC4 at 1 nM and Montelukast at varying concentrations. The EC50 value is indicated by the stippled line. CREB, cAMP response element binding protein; hGPR17-L, long GPR17 isoform; hGPR17-S, short GPR17 isoform.
Results
The human GPR17 exists in two different isoforms, that are both expressed at the cell surface
While searching the Ensembl Genome Browser we noticed that GPR17 exists in two splice variations in human and non-human primates. These differ only with respect of the N-terminus, where the long human isoform, here referred to as hGPR17-L, is 28 amino acids longer than the short, here referred to as hGPR17-S (Figure 1A). The outline of the mRNAs encoding the human receptor isoforms are depicted in the inserts in Figure 2A,B. Initially, to test whether hGPR17 is expressed at the cell surface, we generated a fusion construct of hGPR17-L and eGFP. The cellular localization of the construct was evaluated using confocal microscopy in HEK293 cells and, as GPR17 is mainly expressed in the brain, 1321N1 astrocytoma cells. In both cell lines, the receptor was predominantly localized at the cell surface as seen for the majority of endogenous 7TM receptors (Figure 1B,C). The surface expression was confirmed by an ELISA-based assay employing N-terminal FLAG-tagged versions of both human isoforms (Figure 1D). Thus, in HEK293 cells transiently transfected with M1-GPR17-L and -S both were expressed at the cell surface with the short isoform being present at sligthly higher levels (Figure 1D). The expression levels were comparable with that previously seen for other 7TM receptors, e.g. the M1-FLAG tagged EBI2 receptor (Rosenkilde et al., 2006).
Isoform-specific analysis of GPR17 gene expression in brain, heart and kidneys
Earlier studies of GPR17 gene expression in humans, rats and mice have shown that the receptor is primarily expressed in brain. In addition, minor expression has also been observed in heart and kidney (Blasius et al., 1998; Ciana et al., 2006; Lecca et al., 2008). However, these analyses were only qualitative (Northern blotting and end-point RT-PCR), performed on whole organs and not isoform-specific. In order to obtain a more detailed picture of the expression pattern, using quantitative RT-PCR, we determined the relative expression levels of the two GPR17 isoforms in eight different brain regions as well as whole brain, heart and kidney. The forward primers were designed to span the exon2-exon3 junction and the exon1-exon3 junction for hGPR17-L and hGPR17-S, respectively, ensuring that the amplifications were isoform-specific, while the reverse primer was shared ensuring similar amplicon lengths (189 and 187 bp respectively) (inserts, Figure 2A,B). All primer sequences are given in the Methods section. The expression levels were normalized to that of β-actin and presented relative to the normalized hGPR17-L expression in whole brain.
As shown in Figure 2A,B, the expression patterns of hGPR17-L and -S were remarkably similar in the different brain tissues. Thus, for both isoforms, the lowest expression was observed in thalamus with gradually increasing expression levels through the hypothalamus, cerebellum, amygdala, cerebellar hemisphere, frontal cortex, hippocampus and putamen. For both isoforms, the putamen region exhibited relatively higher expression than in whole brain, while this was also observed for hGPR17-S in hippocampus. Expression levels equal to those in whole brain were seen in frontal cortex and for hGPR17-L in hippocampus, whereas lower levels were present in all other brain tissues for both isoforms in varying degrees (Figure 2A,B). To facilitate a comparison of the expression levels of the two isoforms, the ratios of these are presented in Figure 2C. In the brain, the short isoform was expressed at much higher levels than the long and, given the similar expression pattern, the ratios were largely similar. Thus, in whole brain, hGPR17-S was expressed at approximately 10-fold higher levels than hGPR17-L, which was also observed in hypothalamus, cerebellar hemisphere and frontal cortex. Slightly lower ratios (compared with that in whole brain) were observed in cerebellum and putamen. However, although significant, the differences are small with ratios of approximately 8 in both cases. In contrast, considerably higher ratios of 22, 18 and 23 were observed in thalamus, amygdala and hippocampus respectively. Interestingly, the reverse picture was observed in heart and kidney. In both tissues, hGPR17-L was expressed at higher levels compared with total brain (approximately seven- and twofold respectively), while the opposite was the case for hGPR17-S. In fact, no detectable expression was present in kidneys for the latter (Figure 2A,B, right panels). Thus, in the heart, hGPR17-L was expressed at approximately 12-fold higher levels than hGPR17-S. Given the absence of hGPR17-S expression in kidneys, the similar comparison was not applicable for this organ (Figure 2C, right).
Both hGPR17 isoforms are constitutively active through Gαi
To examine whether both isoforms are functional, we initially tested for putative signalling using the CREB reporter assay. The activity of this transcription factor is primarily regulated by the intracellular cAMP level via cAMP-regulated kinases and consequently reflects the Gαi or Gαs coupling of a given receptor when used as a reporter. In addition, kinases downstream of Gαq also mediate CREB activation (Shaywitz and Greenberg, 1999). Gene-dosage experiments in transiently transfected HEK293 cells demonstrated that both human isoforms of GPR17 inhibited a forskolin-induced CREB activity with approximately 75% (Figure 3A,B). Importantly, empty pcDNA3 vector (negative control) at similar doses did not have any effect, indicating that both isoforms are constitutively active through Gαi, This was further corroborated by administration of the Gαi inhibitor pertussis toxin, which abolished the effect in both cases (Figure 3A,B). Also, as shown in Figure 3C, co-transfection with the chimeric Gα-subunit Gqi4myr, resulted in dose-dependent increases in CREB activity. This Gα fusion protein recognizes Gαi-coupled receptors but transduces a Gαq signal (Kostenis, 2001). Thus, the observed increase in CREB activity upon hGPR17-L and -S transfection substantiates the notion that both human GPR17 isoforms couple to and signal constitutively via Gαi. In the absence of forskolin, increasing doses of hGPR17-L and -S, as opposed to the positive control ORF74, did not result in any increase in CREB activity or differ from that of pcDNA3 transfected cells (Figure S1A–D) demonstrating that neither of the hGPR17 isoforms signals constitutively through either Gαs or Gαq. We also tested putative constitutive activation of the transcription factors NF-κB and NFAT; neither, however, was activated dose-dependently by any of the isoforms (Figure S1E–F). To ensure that both receptors were present at the cell surface, ELISA was performed on HEK293 cells transfected in parallel with the functional experiments (Figure 3D). Both isoforms were expressed well with hGPR17-S being present at approximately 30–40% higher levels than hGPR17-L at corresponding doses (Figure 3D). The above-mentioned experiments were all carried out using the M1-tagged receptor constructs in order to compare receptor activity with receptor expression in parallel (Figure 3). The M1-tag did not influence receptor activity, as similar levels of constitutive activity were obtained for the untagged receptor constructs (data not shown).
Figure 3.
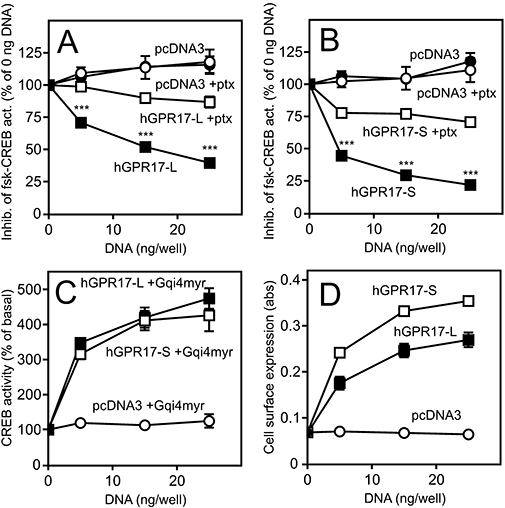
Constitutive signalling (A–C) and surface expression (D) of the two hGPR17 isoforms. (A) Inhibition of forskolin-induced CREB activity (abbreviated fsk-CREB act.) in HEK293 cells transfected with hGPR17-L or pcDNA3 vector control in the presence or absence of pertussis toxin (ptx, 100 ng·mL–1). The cells were transiently transfected with CREB-Luc reporter vector and FLAG-tagged hGPR17-L or pcDNA3 at 0, 5, 15 and 25 ng per well. Production of cAMP was stimulated with 15 µM forskolin. The results are given relative to the activity at 0 ng per well in % and are presented as mean ± SEM from at least four independent experiments performed in quadruplicates. (B) Inhibition of forskolin-induced cAMP production in HEK293 cells transfected with FLAG-tagged hGPR17-S or pcDNA3; performed and presented as in (A). Statistical analyses of data in (A) and (B): ***P<0.0001 (Student's t-test). (C) CREB activity in HEK293 cells transiently transfected with the chimeric Gα subunit Gqi4myr, CREB-Luc reporter vector and hGPR17-L, -S or pcDNA3 at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well in % and are presented as mean ± SEM from four independent experiments performed in quadruplicates. (D) Cell surface expression of FLAG-tagged hGPR17-L and -S in HEK293 cells as measured by ELISA. The cells were transiently transfected with hGPR17-L, -S or pcDNA3 in parallel with the experiments presented in (C). The results represent mean ± SEM of raw data from four independent experiments performed in quadruplicates. CREB, cAMP response element binding protein; hGPR17-L, long GPR17 isoform; hGPR17-S, short GPR17 isoform.
As activation of a transcription factor is an event relatively far downstream from the initial receptor activation and can be influenced by a variety of factors, we decided to assess the constitutive activity of the two hGPR17 isoforms using the GTPγS assay. The measurement of [35S]-GTPγS binding was performed on membranes purified from HEK293 and 1321N1 cells transfected with hGPR17-L, -S or vector control (pcDNA3). Prior to membrane purification, proper cell surface expression of the receptors was verified by ELISA experiments with the relative expression levels being similar to those shown in Figure 3D (data not shown). As presented in Figure 4A, [35S]-GTPγS binding was significantly elevated in membranes from HEK293 cells containing either of the hGPR17 isoforms compared with membranes from empty pcDNA3 vector transfected cells. In both cases, an approximately threefold increase was observed with no significant difference between the two isoforms (Figure 4A). To confirm the suggestion that hGPR17-L and -S are Gαi-coupled, [35S]-GTPγS binding was performed on membranes purified from hGPR17-L-, -S- or pcDNA3-transfected cells that had been exposed to pertussis toxin (100 ng·mL–1). Importantly, the presence of this toxin completely reduced the level of [35S]-GTPγS binding to hGPR17-L and -S membranes to the level observed in pcDNA3 control membranes (Figure 4A). As hGPR17-S mainly is expressed in the brain, [35S]-GTPγS binding was also evaluated in membranes from transiently transfected 1321N1 astrocytoma cells. As shown in Figure 4B, the presence of either isoform led to a significant increase in [35S]-GTPγS binding compared with pcDNA3-transfected membranes. The magnitude of this increase, however, was less marked than in membranes from HEK293 cells, which probably reflects the lower transfection efficiency of 1321N1 cells compared with HEK293 cells (as judged by confocal microscopy; data not shown). As observed in HEK293 cells, the increase in [35S]-GTPγS binding was abolished upon treatment with pertussis toxin (Figure 4B), corroborating the observation that both isoforms are Gαi-coupled. In addition, the basal binding to pcDNA3-transfected membranes was reduced upon administration of pertussis toxin, suggesting that 1321N1 cells express endogenous constitutively active Gαi-coupled receptors.
To determine whether the murine GPR17 displays a similar signalling phenotype to the human homologue, we carried out the exact same experiments as described above for this receptor. As shown in Figure S2, mGPR17 was indeed also constitutively active through Gαi and in this regard completely similar to its human counterpart.
The uracil nucleotides UDP-glucose, UDP-galactose and UDP activate hGPR17-S and -L with different potencies
GPR17 was recently deorphanized as the target for two chemically very distinct types of agonists, namely uracil nucleotides and cysteinyl leukotrienes (Ciana et al., 2006; Lecca et al., 2008; Parravicini et al., 2008). The former group of ligands (consisting of UDP-glucose, UDP-galactose and UDP) has been found to have potencies in the µM range and the latter group (LTC4 and LTD4) in the nM range (Ciana et al., 2006; Lecca et al., 2008; Parravicini et al., 2008). However, these studies included the rat and murine GPR17 and the hGPR17-S, but not hGPR17-L. To determine whether the ligands also activated the long isoform, we conducted dose–response experiments in transiently transfected HEK293 cells, using CREB activity as readout. For the uracil nucleotides, we included the hP2Y14 and the hP2Y6 receptors as positive controls for the UDP-sugars and UDP respectively. As seen in Figure 5, UDP-glucose, UDP-galactose and UDP activated hGPR17-S dose-dependently with EC50 values of 1.2, 0.3 and 1.6 µM, respectively, which closely match those previously reported for this isoform (Ciana et al., 2006). However, these ligands were much less potent at hGPR17-L. Thus, for the UDP-sugars, a robust activation was observed only at 100 µM. Assuming equal efficacies for both isoforms, UDP-glucose (Figure 5A) and UDP-galactose (Figure 5C) were approximately 50- and 170-fold more potent for hGPR17-S as compared with hGPR17-L respectively. Contrary to the UDP-sugars, UDP did not activate hGPR17-L at concentrations up to 100 µM (Figure 5E). In all cases, the positive controls were activated by the ligands with EC50 values matching those previously reported (Communi et al., 1996; Chambers et al., 2000) (Figure 5B, D and F). In contrast, the efficacies were slightly lower (1.4-fold lower for the UDP-sugars and 2.4-fold lower for UDP) on GPR17-S (Figure 5A, C and E) compared with the corresponding efficacies on the positive controls (Figure 5B, D and F). In order to determine whether the constitutive nature of GPR17 was the reason behind this phenotype (due to putative saturation of intracellular signalling pathways), we repeated all dose–response experiments in cells expressing lower levels of receptor (by lowering the receptor DNA during transfection from 15 ng per well, to 5 and 1 ng per well). No increases in the efficacies were observed under these settings, indicating that the slightly lower efficacies are not an artifact of the increased basal receptor activity.
Figure 5.
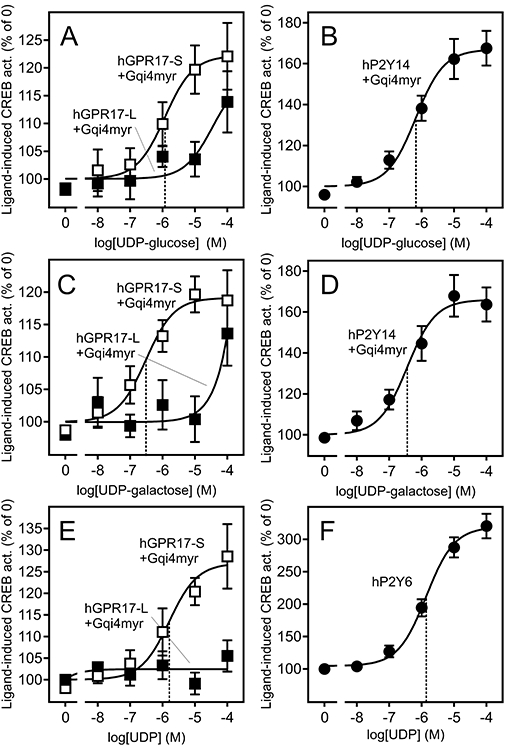
Uracil nucleotide ligands activate hGPR17-L and -S with different potencies. (A) Activation of FLAG-tagged hGPR17-L and -S by UDP-glucose in transiently transfected HEK293 cells. The cells were transfected with Gqi4myr, CREB-Luc reporter plasmid and receptor or pcDNA3 at 15 ng per well. The results are corrected for background (pcDNA3-transfected cells) and given as % relative to the activity in absence of the ligand. The data represent mean ± SEM of four independent experiments performed in quadruplicates. The EC50 of the hGPR17-S response is indicated by the stippled line. (B) Activation of hP2Y14 by UDP-glucose in transiently transfected HEK293 cells; performed and presented as in (A). (C) Activation of FLAG-tagged hGPR17-L and -S by UDP-galactose in transiently transfected HEK293 cells; performed and presented as in (A). (D) Activation of hP2Y14 by UDP-galactose in transiently transfected HEK293 cells; performed and presented as in (A). (E) Activation of FLAG-tagged hGPR17-L and -S by UDP in transiently transfected HEK293 cells; performed and presented as in (A). (F) Activation of hP2Y6 by UDP in transiently transfected HEK293 cells; performed and presented as in (A) with the exception that Gqi4myr was not co-transfected in this case. CREB, cAMP response element binding protein; hGPR17-L, long GPR17 isoform; hGPR17-S, short GPR17 isoform.
The above-mentioned experiments were all carried out using the M1-tagged receptor constructs. In order to assure that the N-terminal M1-tag was not affecting ligand-binding, we tested the same ligands on untagged GPR17-S and GPR17-L, and found that the M1-tag had no effect (data not shown). In conclusion, these results reveal a functional difference between the two isoforms. In turn, this aligns well with the differential expression pattern presented in Figure 2 and supports the notion that each isoform probably has a tissue-specific role.
As the activation of GPR17 by UDP ligands has previously been shown to be antagonized by several P2Y compounds (including a P2Y12 antagonist) (Pugliese et al., 2009), we decided to test whether the P2Y12 antagonist Ticlopidine had any inverse agonistic or antagonistic effects on GPR17. However, Ticlopidine did not suppress the constitutive activity of any of the hGPR17 isoforms (Figure 6A) or antagonize the activation by UDP or UDP-glucose (Figure 6B) as opposed to that seen in the MeSADP-mediated activation of hP2Y12 in the parallel control experiment (Figure 6C).
Figure 6.
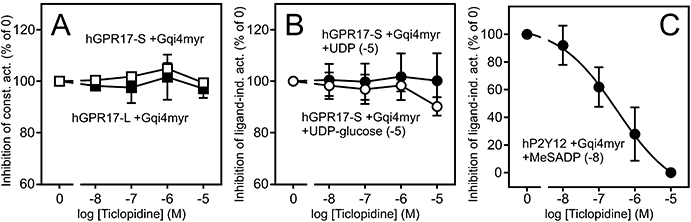
Ticlopidine does not display inverse agonism or antagonism at hGPR17 isoforms. (A) Putative inverse agonism of Ticlopidine. HEK293 cells were transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and receptor or pcDNA3 at 15 ng per well.and incubated with the ligand in varying concentrations. The results are corrected for background (pcDNA3-transfected cells) and given as % relative to the activity in absence of the ligand. The data represent mean ± SEM of two independent experiments performed in quadruplicates. (B) Putative antagonism of Ticlopidine. HEK293 cells were transfected as described in (A) and incubated with agonist (UDP or UDP-glucose) at 10 µM and Ticlopidine in varying concentrations. The results are presented as in (A). (C) hP2Y12 control. HEK293 cells were transfected as described in (A) and were incubated with the agonist MeSADP at 10 nM and Ticlopidine in varying concentrations. The results are presented as in (A). CREB, cAMP response element binding protein; hGPR17, GPR17 isoform.
The cysteinyl leukotrienes LTD4 and LTC4 do not activate or bind to hGPR17-L or -S
As mentioned earlier, the short isoform of GPR17 has also been reported to be activated by the cysteinyl leukotrienes LTC4 and LTD4 with EC50 values in the nM range (Ciana et al., 2006; Lecca et al., 2008). Analogous to the uracil nucleotides, we wanted to assess whether the cysteinyl leukotrienes also activate hGPR17-L, initially focusing on LTD4. However, contrary to previous findings, LTD4 did not activate any of the human isoforms in transiently transfected HEK293 cells, using CREB activity as read out (Figure 7A). Both receptors were present at the cell surface, as evident by the apparent constitutive activity when compared with pcDNA3 transfected cells (Figure 7A). In contrast to this observation, in the parallel control experiment LTD4 dose-dependently activated CysLT2 with an EC50 value of 3.1 nM (Figure 7B). Similarly, in the GTPγS assay, LTD4 at 1 µM did not activate either of the isoforms, whereas it resulted in a significant increase in [35S]-GTPγS binding in membranes from CysLT2-transfected HEK293 cells (Figure 7C). The magnitude of this relatively small increase is probably due to the endogenous stoichiometry of Gα-subunits, as the GTPγS assay is most suited for Gαi-coupled receptors (such as GPR17) compared with Gαs- or Gαq-coupled receptors (such as CysLT2) given the lower expression levels of Gαs and Gαq compared with Gαi (Milligan, 2003).
Given these results, we decided to carry out homologous competition binding to assess whether LTD4 binds to hGPR17-L and -S. This was performed on the same membranes used for the GTPγS assay, ensuring that all of the receptors were present in the membranes (assessed by ELISA) and were functional (assessed by [35S]-GTPγS binding). As shown in Figure 7D, [3H]-LTD4 bound to HEK293 membranes harbouring CysLT2 and was dose-dependently displaced by unlabelled LTD4 with an IC50 of 10.3 nM. In contrast to this, no binding was observed for membranes containing hGPR17-L or -S, demonstrating that LTD4 does not bind to any of these isoforms. To ensure that the lack of binding was not a matter of cell type, we repeated the binding experiment in membranes from 1321N1 cells, and as in HEK293 cells, no binding was observed to membranes containing either of the isoforms (data not shown). Analogous to the functional studies, the same experiments were carried out for mGPR17 and gave the same results as described above for the human counterparts (Figure S2).
Given that the CysLT1 selective antagonist Montelukast has previously has been suggested to function as an antagonist on GPR17 (Pugliese et al., 2009), we decided to test for inverse agonism on both GPR17-isoforms of this compound. However, as shown in Figure 7E, Montelukast did not suppress the constitutive activity of either of the isoforms. As a control, the antagonizing effect of this compound on the LTC4-mediated CysLT1 activation was evaluated in parallel and gave an IC50 value in agreement with previous reports (Figure 7F).
Finally, we determined whether LTD4 stimulates internalization of GPR17, as this is a hallmark of agonist-induced activation of 7TM receptors. To do this, we employed an antibody feeding internalization assay using CysLT2 as a positive control (Figure 8). Initially, cell surface expressed receptors were labelled with primary antibody and subsequently incubated with LTD4 to induce internalization. Following this, labelled receptors still residing at the surface and those internalized were separately detected using two different fluorophore-conjugated secondary antibodies before and after permeabilization respectively. In turn, a comparison of these two receptor populations gives a qualitative measurement of the apparent internalization rate. As shown in Figure 8B (lower panels), virtually all labelled CysLT2 receptors were internalized after 30 min incubation in the presence of 1 µM LTD4 as virtually no labelled receptors were detected before permeabilization. That this was a specific event was evident by the notion that only a small amount of (constitutive) internalization happened when vehicle (DMSO) was present (Figure 8B, upper panels). Contrary to CysLT2, LTD4 did not induce internalization of hGPR17-L as only constitutive internalization was observed when either this or vehicle was present with no apparent differences in the internalization pattern between these conditions (Figure 8A). In conclusion, these results align well with the observation that LTD4 neither activates nor binds to GPR17.
Figure 8.
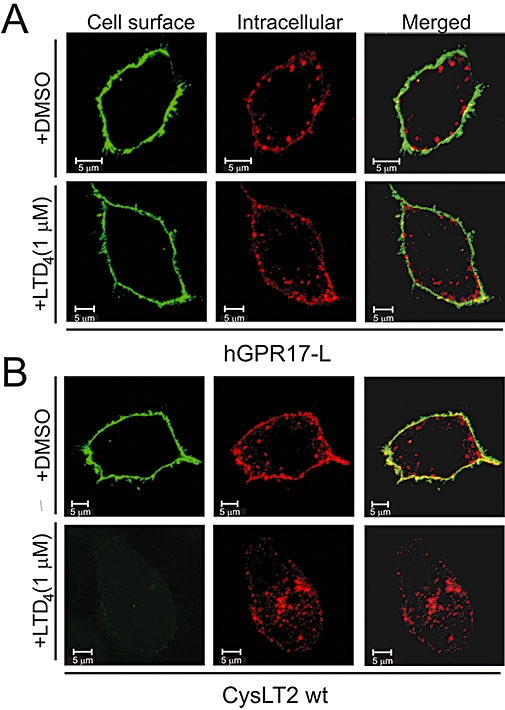
LTD4 does not induce internalization of hGPR17-L. (A) Internalization of FLAG-tagged hGPR17-L in transiently transfected HEK293 cells in the presence of vehicle (DMSO, upper panels) or LTD4 at 1 µM (lower panels). Receptors at the surface were labelled with M1 anti-FLAG antibody prior to internalization. Subsequent to this, labelled receptors still residing at the cell surface were detected before permeabilization (green, left) and internalized receptors after (red, middle) with two different secondary antibodies and analysed by confocal microscopy. Merged images are presented to the right. (B) Internalization of FLAG-tagged CysLT2 in the presence of vehicle or LTD4; performed and presented as in (A). Scale bars denote 5 µM. hGPR17-L, long GPR17 isoform.
Furthermore, we carried out similar dose–response and competition binding experiments with LTC4. As shown in Figure S3, LTC4 did not activate or bind to hGPR17-L, -S or mGPR17, whereas this was readily observed for CysLT2. Thus, in our eyes, and contrary to previous reports (Ciana et al., 2006; Lecca et al., 2008) and in line with the recent work of Maekawa et al. (Maekawa et al. 2009), the cysteinyl leukotrienes LTD4 and LTC4 are not agonists of GPR17.
Discussion
In the present study, we characterized the 7TM receptor GPR17. First, we present a detailed expression analysis of the two human isoforms, hGPR17-L and -S, in several brain regions as well as heart and kidney. Second, we showed that hGPR17-L, -S and the murine GPR17 are constitutively active through Gαi. Third, we demonstrated that uracil nucleotide ligands activate the two human isoforms with very different potencies. Finally, we demonstrated that this receptor, previously reported to be activated by cysteinyl leukotrienes (Ciana et al., 2006; Lecca et al., 2008), is not activated, internalized or bound by these molecules.
Constitutive activity of GPR17
The constitutive activity of the two human isoforms and the murine homologue was investigated both directly at the level of G protein activation using [35S]-GTPγS binding (Figure 4) and further downstream by measuring CREB transcriptional factor activity (Figure 3). We found that all three constructs signalled constitutively as evident by (i) the substantial increase in [35S]-GTPγS binding to membranes containing either of the receptors compared with membranes from pcDNA3-transfected cells (Figure 4); (ii) the dose-dependent inhibition of forskolin-induced CREB activity (Figure 3A,B); and (iii) the increase in CREB activity upon co-expression of the chimeric Gα protein subunit Gqi4myr (Figure 3C). This constitutive signalling was mediated via Gαi as both [35S]-GTPγS binding and CREB activity inhibition were highly sensitive to pertussis toxin.
Constitutive activity per se is not an unusual phenomenon among endogenous 7TM receptors. Notable examples are the ghrelin and the melanocortin 4 receptor, which have well characterized constitutive signalling patterns(Holst et al., 2003; Srinivasan et al., 2004). In addition, a range of orphan receptors exhibit constitutive signalling; these include GPR3, -6, -12, -20, -39 and EBI2 (Holst et al., 2004; Rosenkilde et al., 2006; Tanaka et al., 2007; Hase et al., 2008). Historically, the concept of constitutive activity has been questioned by primarily two factors: heterologous over-expression and the possible presence of agonists in the medium, both of which can result in apparent ligand-independent signalling. In the case of GPR17, over-expression does not seem to play a significant part as neither the Gαq-coupled CysLT2 nor the Gαi-coupled CXCR4 elicit dose-dependent constitutive CREB activity when expressed at similar levels to hGPR17-L, -S or mGPR17 (Figure S1). This is further corroborated by the equal levels of [35S]-GTPγS binding to membranes isolated from either CysLT2- or pcDNA3-transfected cells (Figure 7C). The presence of an agonist in the medium also seems unlikely as the constitutive activity of GPR17 was observed with two assay-types (both membrane and whole cell-based) in two different cell-lines, using two different media (Figures 3 and 4).
With regard to the constitutive activity of GPR17, it is interesting to note that many neurologically expressed 7TM receptors exhibit constitutive activity. This includes, among others, the receptors targeted by the neurotransmitters melatonin, dopamine, acetylcholine, opioid peptides and glutamate (Seifert and Wenzel-Seifert, 2002). In turn, this indicates that the constitutive activity of these receptors serves to support basal neuronal activity. Also, many of the orphan receptors expressed in the brain are constitutively active. This includes among others the Gαs-coupled GPR3, -6 and -12, for which it was recently shown that their constitutive activity promotes neurite outgrowth by up-regulation of cAMP levels (Tanaka et al., 2007). It is well known that the endogenous cAMP level regulates several processes in neurones including neurite outgrowth and axonal regeneration. Thus, in this regard, GPR17 (and other constitutively active Gαi-coupled receptors) could potentially balance the action of constitutively active Gαs-coupled receptors in such processes via Gαi. As the level of constitutive activation is proportional to the number of receptors on the cell surface, the intracellular cAMP level is in part regulated by the relative expression levels of the Gαs- and Gαi-coupled receptors. Notably, in two rodent models of ischaemic damage (Ciana et al., 2006; Lecca et al., 2008) and a model of spinal cord injury (Ceruti et al., 2009), GPR17 expression is up-regulated. Given the constitutive activity observed in the present study, this up-regulation would result in decreased cAMP levels, which could, by inhibiting outgrowth and regeneration, contribute to the neuronal damage.
Different expression patterns of GPR17 isoforms
Our finding that hGPR17-S is the principal isoform in brain (Figure 2) is consistent with the reported expression pattern of rat and mice GPR17 (that are homologous to the short human isoform) (Blasius et al., 1998; Ciana et al., 2006; Lecca et al., 2008). Surprisingly, we found that the long isoform hGPR17-L is expressed at higher levels in heart and kidneys compared with brain. Importantly, compared with the short isoform, hGPR17-L is expressed at much higher levels in the heart, and in the kidneys (with no detectable expression of hGPR17-S in the latter) indicating that hGPR17-S plays a principal role in the brain (like r- and mGPR17), while hGPR17-L probably adopts this role in the heart and kidneys (Figure 2). This differential expression profile suggests tissue-specific roles in humans and other primates. Inherently, the existence of 7TM receptor splice variants is puzzling and given the numerous 7TM receptor subfamilies, they seem somewhat superfluous. For example, although the α-adrenoceptor subfamily harbours six members (α1A, -1B, -1D and α2A, -2B, -2C) the α1A subtype exists (as annotated in the Ensemble Genome Browser) in 10 different splice variants. However, although our understanding of splice variants is still limited, their relevance is underlined by accumulating data showing differences in both function as well as spatial and temporal expression (Kilpatrick et al., 1999). For example, different tissue distributions have, apart from the present study of GPR17, been demonstrated for the mGluR7a and -b among others. Thus, using isoform-specific antibodies, Shigemoto et al. showed that the expression of mGluR7a was markedly different from that of mGluR7b in the rat hippocampus (Shigemoto et al., 1997). The same phenomenon has been observed for splice variants of CRF2, for which it has been shown that the a-variant is much more broadly expressed than the β- and γ-variants (Kostich et al., 1998). Direct demonstrations of the function of 7TM receptor splice variants in vivo are few. However, the two isoforms of the dopamine D2 receptor have been shown to play different roles. Specifically, it was demonstrated, by knocking out the long isoform of the D2 receptor in mice, that this isoform mainly works at postsynaptic locations, whereas the short isoform functions at presynaptic sites (Usiello et al., 2000). During revision of this manuscript, a report regarding the two same hGPR17 isoforms was published. As opposed to the results presented in this study, the gene expression of the long isoform was primarily restricted to different brain regions and was not detected in heart or kidney. A possible explanation for this discrepancy could be that the forward primer used in the study by Pugliese et al. (2009), by spanning the exon 1–2 border, only detects one out of the three different mRNA species that encodes hGPR17-L. On the contrary, the forward primer used in this study was designed to detect all three species, thus giving a more comprehensive picture of the expression pattern of the long isoform.
Different functional properties of GPR17 isoforms
Besides differences in tissue distribution, functional differences have also been identified for a range of 7TM splice variants. Among others this includes variations in G protein coupling and selectivity, ligand-binding profiles and internalization pattern (Kilpatrick et al., 1999). Although the majority of splice variations result in differences between the length of the intracellular loops or the C-terminus, a substantial number results in changes in the length of the N-terminus (as seen for GPR17) and/or sequence (Kilpatrick et al., 1999). This includes, among others, the cannabinoid receptor 1 (CB1) (Rinaldi-carmona et al., 1996), the CRF2 (Kostich et al., 1998), cholecystokinin CCKB (Miyake, 1995) and the PACAP receptor (Pantaloni et al., 1996). Splice variants that differ by their N-terminus tend to be affected primarily at the level of ligand binding. Thus, isoforms with shorter N-termini are usually activated less potently by their cognate agonists than their long counterparts. This is especially true for 7TM receptors that are activated by large peptides or proteins, which bind to extracellular epitopes often including the N-terminus. The CCKB receptor is a good example of this phenomenon, as truncation of its N-terminus reduces its affinity for both gastrin and CCK-8 (Miyake, 1995). This is further corroborated by a recent study of the chemokine receptor CCR1 demonstrating that the binding of the CCL-3 and -5 chemokines is highly dependent on an intact N-terminus (Jensen et al., 2008).
In the case of GPR17, the reverse pattern is observed. Thus, the longer isoform is only poorly (and in the case of UDP not at all) activated by uracil nucleotides, whereas the short isoform is activated with similar potencies, but slightly lower efficacies compared with the nucleotide receptors P2Y6 and P2Y14 (Figure 5). The molecular mechanism responsible for this difference remains to be identified. However, a similar observation has been noted for N-terminal splice variants of the PACAP receptor, where the short isoform is activated more potently by the neuropeptides PACAP-27 and -38 (Pantaloni et al., 1996). The endogenous ligand that activates hGPR17-L is still to be identified, and given the low efficacies observed for uracil nucleotides on GPR17-S it is questionable whether these ligands are, in fact, the most important endogenous ligands. The longer N-terminus might render hGPR17-L capable of binding other ligand types, e.g. proteins or large peptides. Another possibility is that hGPR17-L does not have a ligand at all. Putatively, the receptor activity could be regulated at the expression level as the constitutive activity is proportional to this, as suggested for, for example, the orphan 7TM receptor EBI2 (Rosenkilde et al., 2006). This could likewise be the case for Cpr2 as it was recently shown that the expression of this constitutively active receptor is induced after cell-cell fusion during the mating event in Cryptoccoccus neoformans (Hsueh et al., 2009). The notion that many constitutively active receptors are still orphan despite deorphanization projects in both industry and academia suggests that this is a possibility.
In conclusion, the discovery and elucidation of the constitutive activity of the two human GPR17 isoforms along with their differential expression and ligand-binding profiles add new information to this receptor. In turn, it opens up the possibility of selective drug targeting and also illustrates well that 7TM receptor splice variants should be considered important, as their pharmacological profiles and sites of action may differ.
Acknowledgments
We thank Inger Smith Simonsen for excellent technical assistance and Dr Ulrik Gether for use of the confocal microscope. This study was supported by the Danish Medical Research Council, the European Community's Sixth Framework Program (INNOCHEM: LSHB-CT-2005-518167), the NovoNordisk Foundation, the Lundbeck Foundation, AP-Moller foundation and the Aase and Einer Danielsen Foundation.
Glossary
Abbreviations:
- 7TM
seven transmembrane
- CB1
cannabinoid receptor
- CREB
cAMP response element binding protein
- CRF2
corticotrophin-releasing factor receptor 2
- EBI2
Epstein–Barr virus-induced receptor 2
- hGPR17-L
long GPR17 isoform
- hGPR17-S
short GPR17 isoform
Conflicts of interest
The authors declare no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 (A) The Gαq-coupled CysLT2 is not constitutively active. HEK293 cells were transiently transfected with CREB-Luc reporter plasmid and FLAG-tagged CysLT2 or pcDNA3 at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well in percent and are presented as mean ± SEM from three independent experiments performed in quadruples. (B) The Gαi-coupled CXCR4 is not constitutively active. HEK293 cells were transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and FLAG-tagged CXCR4 or pcDNA3 at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well in percent and are presented as mean ± SEM from three independent experiments performed in quadruples. (C) ORF74 and EBI2 display constitutive activity. For EBI2, HEK293 cells were transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and FLAG-tagged EBI2 or pcDNA3 at 0, 5, 15 and 25 ng per well. For the ORF74, the experiment was performed the same way but without presence of Gqi4myr. The results are given relative to the activity at 0 ng per well in percent and are presented as mean ± SEM from three independent experiments performed in quadruples. (D) Cell surface expression of CysLT2 and CXCR4. HEK293 cells were transiently transfected in the same way and in parallel to A and B. Cell surface expression of the receptors were measured by ELISA and presented relative to 0 ng per well in percent as mean ± SEM (E) hGPR17-L is not constitutively active via NFAT. HEK293 cells were transiently transfected with NFAT-Luc reporter plasmid and FLAG-tagged ORF74, hGPR17-L or pcDNA3 at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well as mean ± SEM of raw data from three independent experiments performed in quadruples. (F) hGPR17-L is not constitutively active via NFκB. HEK293 cells were transiently transfected with NFκB -Luc reporter plasmid and FLAG-tagged ORF74, hGPR17-L or pcDNA3 at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well as mean ± SEM of raw data from three independent experiments performed in quadruples.
Figure S2 mGPR17 is constitutively active through Gαi. (A) Inhibition of forskolin-induced CREB activity in HEK293 cells transfected with mGPR17 or pcDNA3 vector control in the presence or absence of pertussis toxin (ptx, 100 ng/mL). The cells were transiently transfected with CREB-Luc reporter vector and FLAG-tagged mGPR17 or pcDNA3 vector control at 0, 5, 15 and 25 ng per well. Production of cAMP was stimulated with 15 µM forskolin. The results are given relative to the activity at 0 ng per well in percent and are presented as mean ± SEM from three independent experiments performed in quadruples. The stapled line indicates the level of inhibition by hGPR17-L. (B) CREB activity in HEK293 cells transiently transfected with the chimeric Gα subunit Gqi4myr, CREB-Luc reporter vector and FLAG-tagged mGPR17 or pcDNA3 vector control at 0, 5, 15 and 25 ng per well. The results are given relative to the activity at 0 ng per well in percent and are presented as mean ± SEM from three independent experiments performed in quadruples. The stapled line indicates the activity of hGPR17-L. (C) Cell surface expression of FLAG-tagged mGPR17 in HEK293 cells as measured by ELISA. The cells were transiently transfected with mGPR17 or pcDNA3 in parallel with the experiments presented in B. The results represent mean ± SEM of raw data from three independent experiments performed in quadruples. The stapled line indicates the level of cell surface expression of hGPR17-L. (D) [35S]GTPγS binding to membranes isolated from HEK293 cells transiently transfected with FLAG-tagged mGPR17 or pcDNA3. The cells were cultured in the absence (black columns) or presence (white columns) of 100 ng/mL pertussis toxin (ptx). The level of binding to vector control membranes is indicated by the stapled line. The results represent mean ± SEM of raw data from three independent experiments performed in triplicates. *P < 0.05 by Student’s t-test. (E) [35S]GTPγS binding to membranes isolated from 1321N1 cells transiently transfected with mGPR17 or pcDNA3; performed and presented as in D. *P < 0.05 by Student’s t-test. (F) Homologous competition assay for [3H]LTD4 binding to the membranes from HEK293 cells transiently transfected with mGPR17 or pcDNA3; these were similar to those used in D. The data represent mean ± SEM of raw data from four independent experiments performed in duplicates.
Figure S3 LTC4 does not activate or bind to either of the hGPR17 isoforms or mGPR17. (A) Response to LTC4 in HEK293 cells transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and FLAG-tagged hGPR17-L, -S or pcDNA3 at 15 ng per well. The results represent mean ± SEM of raw data from three independent experiments performed in quadruples. (B) Activation of FLAG-tagged CysLT2 by LTC4; performed and presented as in A. The EC50 value is indicated by the stapled line. (C) Homologous competition assay for [3H]LTC4 binding to the membranes from HEK293 cells transiently transfected with hGPR17-L, -S, CysLT2 or pcDNA3. The data represent mean ± SEM of raw data from three independent experiments performed in duplicates. The IC50 value for CysLT2 is indicated by the stapled line. (D) Response to LTC4 in HEK293 cells transiently transfected with Gqi4myr, CREB-Luc reporter plasmid and FLAG-tagged mGPR17 or pcDNA3 at 15 ng per well. The results represent mean ± SEM of raw data from three independent experiments performed in quadruples. (E) Homologous competition assay for [3H]LTC4 binding to the membranes from HEK293 cells transiently transfected with mGPR17, CysLT2 or pcDNA3. The data represent mean ± SEM of raw data from three independent experiments performed in duplicates. The IC50 value for CysLT2 is indicated by the stapled line.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benned-Jensen T, Rosenkilde MM. Structural motifs of importance for the constitutive activity of the orphan 7TM receptor EBI2: analysis of receptor activation in the absence of an agonist. Mol Pharmacol. 2008;74:1008–1021. doi: 10.1124/mol.108.049676. [DOI] [PubMed] [Google Scholar]
- Blasius R, Weber RG, Lichter P, Ogilvie A. A novel orphan G protein-coupled receptor primarily expressed in the brain is localized on human chromosomal band 2q21. J Neurochem. 1998;70:1357–1365. doi: 10.1046/j.1471-4159.1998.70041357.x. [DOI] [PubMed] [Google Scholar]
- Ceruti S, Villa G, Genovese T, Mazzon E, Longhi R, Rosa P, et al. The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain. 2009;132:2206–2218. doi: 10.1093/brain/awp147. [DOI] [PubMed] [Google Scholar]
- Chambers JK, Macdonald LE, Sarau HM, Ames RS, Freeman K, Foley JJ, et al. A G protein-coupled receptor for UDP-glucose. J Biol Chem. 2000;275:10767–10771. doi: 10.1074/jbc.275.15.10767. [DOI] [PubMed] [Google Scholar]
- Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006;25:4615–4627. doi: 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communi D, Parmentier M, Boeynaems JM. Cloning, functional expression and tissue distribution of the human P2Y6 receptor. Biochem Biophys Res Commun. 1996;222:303–308. doi: 10.1006/bbrc.1996.0739. [DOI] [PubMed] [Google Scholar]
- Hase M, Yokomizo T, Shimizu T, Nakamura M. Characterization of an orphan G protein-coupled receptor, GPR20, that constitutively activates Gi proteins. J Biol Chem. 2008;283:12747–12755. doi: 10.1074/jbc.M709487200. [DOI] [PubMed] [Google Scholar]
- Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor – identification of a potent inverse agonist. Mol Endocrinol. 2003;17:2201–2210. doi: 10.1210/me.2003-0069. [DOI] [PubMed] [Google Scholar]
- Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TW. Common structural basis for constitutive activity of the ghrelin receptor family. J Biol Chem. 2004;279:53806–53817. doi: 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- Hsueh YP, Xue C, Heitman J. A constitutively active GPCR governs morphogenic transitions in Cryptococcus neoformans. EMBO J. 2009;28:1220–1233. doi: 10.1038/emboj.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PC, Thiele S, Ulven T, Schwartz TW, Rosenkilde MM. Positive versus negative modulation of different endogenous chemokines for CC-chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J Biol Chem. 2008;283:23121–23128. doi: 10.1074/jbc.M803458200. [DOI] [PubMed] [Google Scholar]
- Kilpatrick GJ, Dautzenberg FM, Martin GR, Eglen RM. 7TM receptors: the splicing on the cake. Trends Pharmacol Sci. 1999;20:294–301. doi: 10.1016/s0165-6147(99)01355-3. [DOI] [PubMed] [Google Scholar]
- Kostenis E. Is Galpha16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors? Trends Pharmacol Sci. 2001;22:560–564. doi: 10.1016/s0165-6147(00)01810-1. [DOI] [PubMed] [Google Scholar]
- Kostich WA, Chen A, Sperle K, Largent BL. Molecular identification and analysis of a novel human corticotropin-releasing factor (CRF) receptor: the CRF2{gamma} receptor. Mol Endocrinol. 1998;12:1077–1085. doi: 10.1210/mend.12.8.0145. [DOI] [PubMed] [Google Scholar]
- Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, et al. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. Plos ONE. 2008;3:e3579. doi: 10.1371/journal.pone.0003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa A, Balestrieri B, Austen KF, Kanaoka Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad Sci USA. 2009;106:11685–11690. doi: 10.1073/pnas.0905364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. Principles: extending the utility of [35S]GTP[gamma]S binding assays. Trends Pharmacol Sci. 2003;24:87–90. doi: 10.1016/s0165-6147(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Miyake A. A truncated isoform of human CCK-B/gastrin receptor generated by alternative usage of a novel exon. Biochem Biophys Res Commun. 1995;208:230–237. doi: 10.1006/bbrc.1995.1328. [DOI] [PubMed] [Google Scholar]
- Pantaloni C, Brabet P, Bilanges B, Dumuis A, Houssami S, Spengler D, et al. Alternative splicing in the N-terminal extracellular domain of the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor modulates receptor selectivity and relative potencies of PACAP-27 and PACAP-38 in phospholipase C activation. J Biol Chem. 1996;271:22146–22151. doi: 10.1074/jbc.271.36.22146. [DOI] [PubMed] [Google Scholar]
- Parravicini C, Ranghino G, Abbracchio MP, Fantucci P. GPR17: molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinformatics. 2008;9:263. doi: 10.1186/1471-2105-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese AM, Trincavelli ML, Lecca D, Coppi E, Fumagalli M, Ferrario S, et al. Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTP{gamma}S binding and electrophysiological studies in 1321N1 cells. Am J Physiol Cell Physiol. 2009;297:C1028–C1040. doi: 10.1152/ajpcell.00658.2008. [DOI] [PubMed] [Google Scholar]
- Raport CJ, Schweickart VL, Chantry D, Eddy RL, Jr, Shows TB, Godiska R, et al. New members of the chemokine receptor gene family. J Leukoc Biol. 1996;59:18–23. doi: 10.1002/jlb.59.1.18. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Calandra B, Shire D, Bouaboula M, Oustric D, Barth F, et al. Characterization of two cloned human CB1 cannabinoid receptor isoforms. J Pharmacol Exp Ther. 1996;278:871–878. [PubMed] [Google Scholar]
- Rosenkilde MM, Benned-Jensen T, Andersen H, Holst PJ, Kledal TN, Luttichau HR, et al. Molecular pharmacological phenotyping of EBI2. An orphan seven-transmembrane receptor with constitutive activity. J Biol Chem. 2006;281:13199–13208. doi: 10.1074/jbc.M602245200. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Kledal TN, Brauner-Osborne H, Schwartz TW. Agonists and inverse agonists for the herpesvirus 8-encoded constitutively active seven-transmembrane oncogene product, ORF-74. J Biol Chem. 1999;274:956–961. doi: 10.1074/jbc.274.2.956. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Holst B. Molecular structure and function of 7TM G-protein-coupled receptors. In: Foreman JC, Johansen T, editors. Textbook of Receptor Pharmacology. London: CRC Press; 2003. pp. 81–110. [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, et al. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Lubrano-Berthelier C, Govaerts C, Picard F, Santiago P, Conklin BR, et al. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–1164. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Ishii K, Kasai K, Yoon SO, Saeki Y. Neural expression of G protein-coupled receptors GPR3, GPR6, and GPR12 up-regulates cyclic AMP levels and promotes neurite outgrowth. J Biol Chem. 2007;282:10506–10515. doi: 10.1074/jbc.M700911200. [DOI] [PubMed] [Google Scholar]
- Temporini C, Ceruti S, Calleri E, Ferrario S, Moaddel R, Abbracchio MP, et al. Development of an immobilized GPR17 receptor stationary phase for binding determination using frontal affinity chromatography coupled to mass spectrometry. Anal Biochem. 2009;384:123–129. doi: 10.1016/j.ab.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.