

Abstract
Background and purpose:
This study was undertaken to compare the analgesic activity of antagonists acting at P2X1, P2X7, and P2Y12 receptors and agonists acting at P2Y1, P2Y2, P2Y4, and P2Y6 receptors in neuropathic, acute, and inflammatory pain.
Experimental approach:
The effect of the wide spectrum P2 receptor antagonist PPADS, the selective P2X7 receptor antagonist Brilliant Blue G (BBG), the P2X1 receptor antagonist (4,4′,4″,4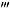 -[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt (NF449) and (8,8′-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-1,3,5-naphthalene-trisulphonic acid, hexasodium salt (NF023), the P2Y12 receptor antagonist (2,2-dimethyl-propionic acid 3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyloxymethyl)-propylester (MRS2395), the selective P2Y1 receptor agonist ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt (MRS2365), the P2Y2/P2Y4 agonist uridine-5′-triphosphate (UTP), and the P2Y4/P2Y6 agonist uridine-5′-diphosphate (UDP) were examined on mechanical allodynia in the Seltzer model of neuropathic pain, on acute thermal nociception, and on the inflammatory pain and oedema induced by complete Freund's adjuvant (CFA).
-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt (NF449) and (8,8′-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-1,3,5-naphthalene-trisulphonic acid, hexasodium salt (NF023), the P2Y12 receptor antagonist (2,2-dimethyl-propionic acid 3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyloxymethyl)-propylester (MRS2395), the selective P2Y1 receptor agonist ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt (MRS2365), the P2Y2/P2Y4 agonist uridine-5′-triphosphate (UTP), and the P2Y4/P2Y6 agonist uridine-5′-diphosphate (UDP) were examined on mechanical allodynia in the Seltzer model of neuropathic pain, on acute thermal nociception, and on the inflammatory pain and oedema induced by complete Freund's adjuvant (CFA).
Key results:
MRS2365, MRS2395 and UTP, but not the other compounds, significantly alleviated mechanical allodynia in the neuropathic pain model, with the following rank order of minimal effective dose (mED) values: MRS2365 > MRS2395 > UTP. All compounds had a dose-dependent analgesic action in acute pain except BBG, which elicited hyperalgesia at a single dose. The rank order of mED values in acute pain was the following: MRS2365 > MRS2395 > NF449 > NF023 > UDP = UTP > PPADS. MRS2365 and MRS2395 had a profound, while BBG had a mild effect on inflammatory pain, with a following rank order of mED values: MRS2395 > MRS2365 > BBG. None of the tested compounds had significant action on oedema evoked by intraplantar injection of CFA.
Conclusions and implications:
Our results show that antagonism at P2X1, P2Y12, and P2X7 receptors and agonism at P2Y1 receptors define promising therapeutic strategies in acute, neuropathic, and inflammatory pain respectively.
Keywords: neuropathic pain, mechanical allodynia, ATP, purinergic receptors, P2X7, P2X1, P2Y12, analgesia, nociception, sciatic nerve injury, oedema, MRS2365, MRS2395, inflammation
Introduction
Neuropathic pain is a chronic pain syndrome that occurs and persists in a heterogeneous group of etiologically different diseases and is caused by a primary lesion or dysfunction of the peripheral and central nerves. It affects 0.6–3% of the adult population and is qualified as a devastating condition because of its severity, chronicity, and resistance to traditional analgesics (Gilron et al., 2006). Unfortunately, the treatment of neuropathic pain has not been resolved yet: although substantial efforts have been made in recent years to develop efficacious medication and several new anti-neuropathic drugs have been introduced into the therapy, a real breakthrough has not been achieved (Kennedy, 2007; Jarvis and Boyce-Rustay, 2009). Consequently, there is still a considerable need to explore novel treatment modalities.
The extracellular mediator ATP, by activating ionotropic P2X and metabotropic P2Y receptors (nomenclature follows Alexander et al., 2008), participates significantly in the generation and modulation of various forms of pain (Chizh and Illes, 2001; Kennedy et al., 2003; Gerevich and Illes, 2004; Burnstock, 2006; Donnelly-Roberts et al., 2008). ATP itself has an algogenic action, which was recognized early (Collier et al., 1966), but the molecular mechanism underlying this action is still not completely understood. It has been postulated that ATP is released from the sensory neurons themselves (Holton, 1959) or from damaged or stressed cells (Cook and McCleskey, 2002) and activates various subtypes of P2X and P2Y receptors expressed along the nociceptive pathways, both at their peripheral and central terminals, in the spinal cord (Dunn et al., 2001; Ruan and Burnstock, 2003). Accordingly, the analgesic activity of ligands acting at various subtypes of P2X and P2Y receptors have been shown in acute and chronic pain modalities, including neuropathic pain models. Among P2X receptor subtypes, P2X3 and P2X2/3 receptors are expressed relatively selectively in the sensory system and therefore their role in pain transmission has been extensively characterized in previous studies (Wirkner et al., 2007). Thus, P2X3 receptor agonists, like ATP, elicit hyperalgesia (Barclay et al., 2002; Dorn et al., 2004; Chen et al., 2005), whereas P2X3 receptor antagonists have analgesic action in inflammatory and neuropathic pain models, whereas they do not affect acute pain sensitivity (Jarvis et al., 2002; Sharp et al., 2006). Similar effects were obtained by the genetic deletion of P2X2 and P2X3 receptors (Souslova et al., 2000; Cockayne et al., 2005) using the siRNA strategy (Dorn et al., 2004) and anti-sense oligonucleotides against P2X3 receptor (Barclay et al., 2002; Honore et al., 2002). Also, the anti-neuropathic action of P2X7 receptor antagonism is well documented (see Donnelly-Roberts and Jarvis, 2007) and is revealed using both genetic knockout (Chessell et al., 2005) and receptor selective antagonists (Honore et al., 2006; 2009; McGaraughty et al., 2007). In addition, anti-sense oligonucleotides and antagonists acting at P2X4 receptors confer protection against neuropathic pain (Tsuda et al., 2003; Inoue et al., 2004), while other studies shed light on the role of P2Y receptors in pain transmission. Thus, activation of P2Y1 receptors, which are expressed in both dorsal root ganglia (DRG) and in the spinal cord, increased tail flick latency (Gerevich et al., 2004), and intrathecal administration of uridine-5′-diphosphate (UDP) and uridine-5′-triphosphate (UTP), agonists of P2Y2, P2Y4, and P2Y6 receptors had analgesic potential in a neuropathic pain model (Okada et al., 2002). Finally, the profound anti-allodynic action of P2Y12 receptor antagonism has been demonstrated in two recent studies (Kobayashi et al., 2008; Tozaki-Saitoh et al., 2008). Nevertheless, studies using purinergic ligands are heterogeneous with respect to the in vivo animal model and the route of administration of drugs used. Therefore, it is difficult to compare the analgesic activity of purinergic ligands acting on different receptor subtypes. To fulfil this aim, in this study we compared the activity of antagonists of P2X1, P2X7, and P2Y12 and agonists of P2Y1, P2Y2, P2Y4, and P2Y6 receptors in the Seltzer model of neuropathic pain and in the hot-plate test of acute thermal nociception. In addition, the effect of the three most potent ligands were also examined in the inflammatory pain model induced by complete Freund's adjuvant (CFA).
Methods
Animals
All animal care and experimental studies were conducted in accordance with the principles and procedures outlined in the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the local Animal Care Committee of the Institute of Experimental Medicine (13/1999). Animals were kept under standard laboratory conditions with food and water ad libitum. All efforts were made to minimize animal suffering and reduce the number of animals used. All the experiments were carried out between 9:00 and 14:00 in the housing room of the animals.
Experimental neuropathy and measurement of mechanical allodynia
Male Wistar rats weighing 250–350 g were submitted to partial ligation of the sciatic nerve, following the method of Seltzer et al. (1990). Briefly, rats were deeply anaesthetized with ketamine and xylazine (50 mg·kg−1 i.p. each), and the sciatic nerve of one of the hind paws was exposed at the mid-thigh level. Then one-half to one-third of the nerve was tightly ligated with a 7-0 siliconized silk suture (Ethicon 7.0, Johnson & Johnson, Somerville, NJ, USA), the wound was closed with sutures and the animals were allowed to recover. Before and 7–14 days after surgery, mechanical sensitivity was measured on both paws (sham and operated) using a dynamic plantar von Frey aesthesiometer (Ugo Basile Instruments Model 37 400, Stoelting, Wood Dale, IL, USA).
The animals were placed into the observation chamber and allowed a habituation period of 10 min. They were then submitted to five to eight individual consecutive measurements, and the average was taken as the value for mechanical sensitivity for the paw of each animal [paw withdrawal threshold (PWT)] expressed in grams. Seven days after surgery, most animals showed an increased mechanical sensitivity of the operated paws, as compared with the pre-surgery levels. Only those animals were included into the study, which showed a minimum of 25% change in mechanical sensitivity.
Animals with manifest mechanical allodynia received intraperitoneal injections of several doses of pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS; 1–100 mg·kg−1), UDP (12.5–100 mg·kg−1), UTP (12.5–100 mg·kg−1), Brilliant Blue G (BBG; 1–100 mg·kg−1), ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt (MRS2365; 0.03–0.3 mg·kg−1), (2,2-dimethyl-propionic acid 3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyloxymethyl)-propylester (MRS2395; 0.03–1 mg·kg−1), (8,8′-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-1,3,5-naphthalene-trisulphonic acid, hexasodium salt (NF023; 0.003–3 mg/kg), and (4,4′,4″,4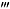 -[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt (NF449; 0.001–1 mg·kg−1) or their vehicle, and 30 min after treatment, mechanical allodynia was measured. The doses of drugs were chosen based on previous studies (Soto et al., 1999; Okada et al., 2002; Ravi et al., 2002; Rettinger et al., 2005; Kobayashi et al., 2008; Diaz-Hernandez et al., 2009).
-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt (NF449; 0.001–1 mg·kg−1) or their vehicle, and 30 min after treatment, mechanical allodynia was measured. The doses of drugs were chosen based on previous studies (Soto et al., 1999; Okada et al., 2002; Ravi et al., 2002; Rettinger et al., 2005; Kobayashi et al., 2008; Diaz-Hernandez et al., 2009).
Acute thermal nociception (hot-plate test)
The effects of purinergic ligands on acute thermal nociception were also investigated using an increasing-temperature hot-plate system. This has an advantage over the conventional (constant temperature) hot-plate system because no sensitization or desensitization occurs after repeated experiments, which enables repeated testing on the same animal (Almasi et al., 2003).
In the experiments, male Wistar rats (weighing 170–300 g, six per group) were used. On the day of testing, animals were placed in the hot-plate apparatus (IITC Life Science, Woodland Hills, CA, USA), and after a period of habituation (about 10 min), the plate was heated from the starting temperature of 30°C with a constant rate of 6 degrees min−1 until the animals showed nocifensive behaviour (frequent lifting and/or licking of the hind paw).
Then heating was instantly stopped; the animal was removed from the cage and the plate was rapidly cooled down. The temperature at which the animal showed any sign of nocifensive behaviour was taken as PWT, expressed in °C (PWT). Thirty minutes later, the measurement was repeated and the average of two values was taken as the baseline thermal nociceptive threshold.
After the second measurement, the animals received treatments with P2 receptor agonists and antagonists as described above, and 30 min after drug administrations, post-drug nociceptive threshold was measured.
Measurement of inflammatory oedema and mechanical sensitivity in CFA-induced arthritis model
In this set of experiments, experimental inflammation was induced with CFA, and the extent of oedema was evaluated by measuring paw volumes using a plethysmometer (Ugo Basile 7140, Italy, Stoelting, Wood Dale, IL, USA). Also, the mechanical sensitivity of the hind paws was measured using dynamic plantar aesthesiometry on each animal using the same procedure as above and expressed in g.
Briefly, after consecutively submitting the animals (male Wistar rats, 200–250 g, six to eight per group) to both tests for baseline measurements, freshly prepared CFA (100 µL, 50% in saline) was injected intradermally into the plantar surface of the right hind paw.
Forty-eight hours after treatment with CFA, the extent of oedema and mechanical sensitivity were measured on both hind paws. One to two hours later, the animals received intraperitoneal injections of P2 receptor ligands (MRS2365, MRS2395, BBG or vehicle), and post-drug measurements were carried out 30 and 60 min later (respectively for paw volume and mechanical sensitivity). The results of inflammatory oedema measurements for both paws were expressed in millilitres or as a percentage of baseline paw volumes.
Statistics
All data were expressed as means ± the standard error of the mean of n observations. For statistical comparison of data obtained on mechanical sensitivity (neuropathic and CFA-induced allodynia), anova followed by Tukey's post hoc test was used, and post-operative PWT values of vehicle treated rats were compared with values treated with drugs. The identical statistical method was used for oedema measurements. For statistical analysis of the hot-plate test results, corresponding baseline and post-drug data were compared using the paired Student's t-test.
The minimal effective dose (mED) was determined as the lowest dose necessary to obtain significant change in the post-operative value/heat threshold as compared with the vehicle-treated/baseline controls. The maximal (Emax) effect is defined as the extent of maximal effect obtained in the dose-range examined and is expressed in g or °C for mechanical and thermal sensitivity respectively.
Materials
CFA, BBG, UDP, UTP and MRS2395 were from Sigma (Sigma-Aldrich, Budapest, Hungary). MRS2365, PPADS, NF449, NF023 were from Tocris Bioscience, Bristol, UK, and morphine hydrochloride was from Teva (Hungary). Ketamine was obtained from CP-Ketamine, CP-Pharma (Germany) and xylazine was obtained from CEVA Phylaxia (Hungary). All drugs were dissolved in sterile saline except MRS2395, which was dissolved in 25% dimethylsulfoxide + 75% polyethyleneglycol 300 as a stock solution, and were freshly prepared on the day of use.
Results
Effects of P2 receptor agonists and antagonists on the mechanical allodynia in neuropathic pain model
When mechanical sensitivity was evaluated after 1, 7, 9, 13 and 15 days after the surgery, the PWT value started to decline on the 1st day, reached its minimum at the 7th day, and remained significantly low at the 9th and 13th days and then started to recover (Figure 1, n = 10). Therefore in subsequent experiments mechanical sensitivity measurements were performed in the 7–14-day period after surgery.
Figure 1.
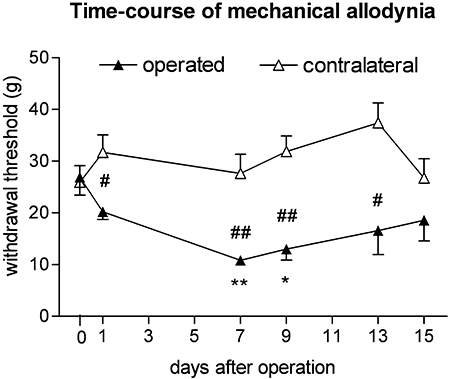
Evaluation of the time-course of mechanical allodynia 1–15 days after partial ligation of the sciatic nerve (Seltzer operation). Mechanical allodynia was most markedly present seven days after operation. Nerve ligated but not sham operated paws showed a significant drop of withdrawal threshold compared to pre-operated values. *P < 0.05 and **P < 0.01; significant difference between pre-operated and post-operative thresholds: #P < 0.05 and ##P < 0.01; significant difference between sham and ligated paws.
Before the partial ligation of sciatic nerve, the PWT in all experiments was 42.8 ± 0.43 g (n = 119). This value decreased to 25.2 ± 0.46 g until the seventh day after surgery, which represents a significant, approximately 42% decrease (n = 119, P < 0.0001). On the other hand, the mechanical sensitivity of the contralateral paw did not change significantly during this time interval (43.1 ± 0.52 g, n = 119, P > 0.05). These values are in good in agreement with those obtained by other studies using the same experimental model (see Bolcskei et al., 2005).
None of the drugs, at all doses tested, had any effect on the withdrawal threshold of the unoperated contralateral paw. PPADS, an antagonist of P2X1, P2X2, P2X3, P2X5, and P2X7 receptors and of P2Y1 and P2Y4 receptors, was given i.p. over a dose range of 6.25–50 mg·kg−1. Although it slightly attenuated the allodynia developed on operated paws, its effect did not reach the level of significance with respect to the post-operative values of vehicle-treated controls (Figure 2A).
Figure 2.
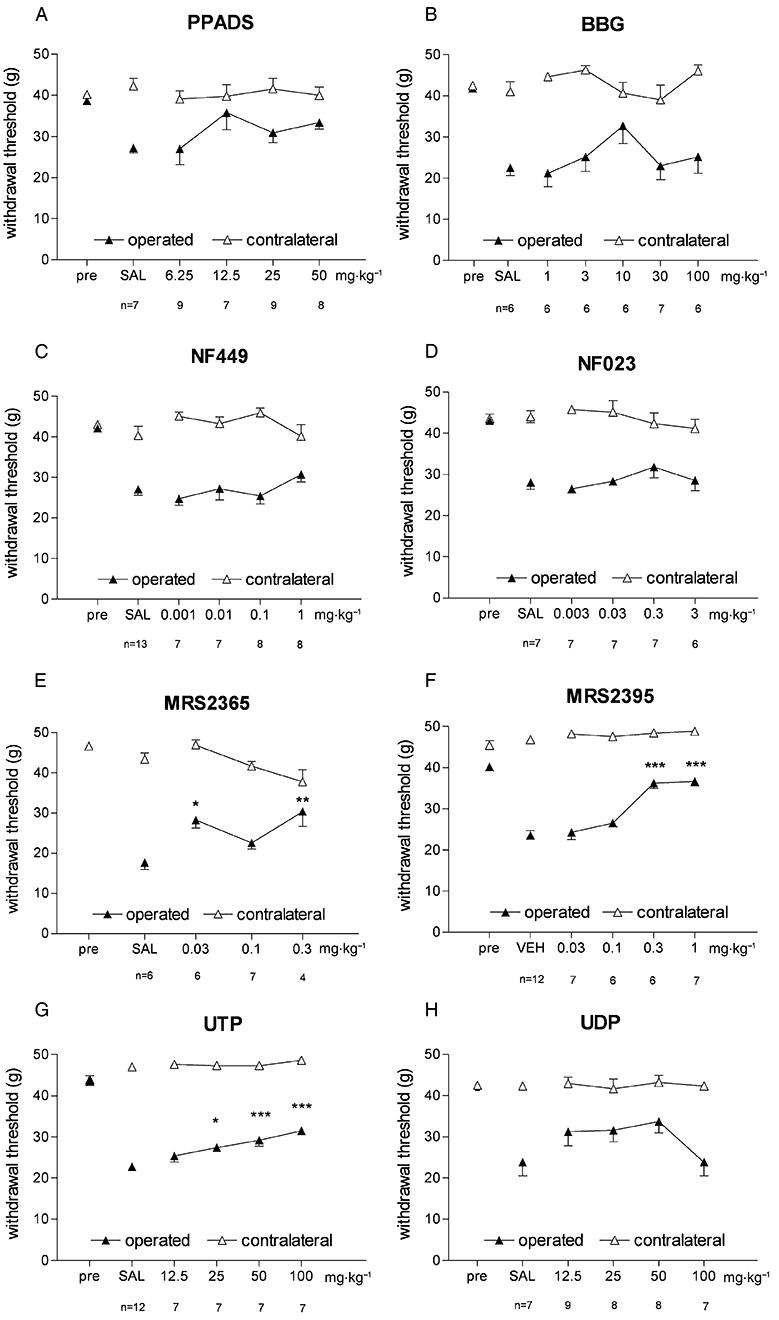
Effects of P2X and P2Y ligands on mechanical allodynia in the experimental neuropathy (Seltzer) model. Values shown are the withdrawal threshold (g) of ligated and sham paws assessed with dynamic plantar aesthesiometry. *P < 0.05 and ***P < 0.001 denote significant changes (Tukey's post hoc test) to vehicle (saline, SAL, or 25% dimethylsulfoxide + 75% polyethyleneglycol 300, VEH) treatment of ligated hindpaws respectively. Note that ‘pre’ values overlap in some cases.
Intraperitoneal administration of the selective P2X7 receptor antagonist BBG (Jiang et al., 2000; 1–100 mg·kg−1) before testing had a biphasic effect on post-surgery allodynia. In lower doses (1–10 mg·kg−1), BBG showed a non-significant tendency of reversal of mechanical allodynia, but in higher doses (>10 mg·kg−1), this tendency was lost (Figure 2B).
NF449 has been reported as a potent and selective antagonist of P2X1 receptors (Rettinger et al., 2005). In our experiments, NF449 (0.001–0.1 mg·kg−1 i.p.) did not affect mechanical allodynia after partial ligation of sciatic nerve (Figure 2C). Likewise, NF023 (0.003–0.3 mg·kg−1 i.p.), another antagonist of P2X1 receptors that also has some activity on P2X3 receptors (Soto et al., 1999), was not effective in alleviating mechanical allodynia (Figure 2D).
Among the compounds acting at P2Y receptors, MRS2365 (0.03–0.3 mg·kg−1 i.p.) is a potent, selective agonist acting at P2Y1 receptors (Ravi et al., 2002), and this compound reversed mechanical allodynia after surgery. Its mED was 0.03 mg·kg−1, the lowest among all the tested compounds (Figure 2E).
It was recently reported that continuous intrathecal infusion of MRS2395, a selective P2Y12 receptor antagonist, reverses mechanical allodynia in the partial sciatic nerve ligation model. We therefore examined the effects of this antagonist after systemic administration. MRS2395 (0.03–1 mg·kg−1) exhibited dose-dependent analgesic effects, with a mED value of 0.3 mg·kg−1 (Figure 2F). Its maximal effect was obtained at 1 mg·kg−1 where the PWT value almost reached the preoperative value (40.29 ± 1.0 g, n = 10 vs. 36.7 ± 1.1 g, n = 7, for operated paws, before surgery and after MRS2395, respectively, P < 0.05).
The pyrimidine nucleotide UTP acts as a P2Y2 and P2Y4 receptor agonist (Abbracchio et al., 2006). In our experiments UTP (12.5–100 mg·kg−1 i.p.) reduced allodynia, but even at the highest tested dose (100 mg·kg−1), only a partial attenuation was observed (P < 0.05, Figure 2G). UDP (a P2Y4 and P2Y6 agonist) did not significantly affect mechanical allodynia in the dose-range tested (12.5–100 mg·kg−1) (Figure 2H).
Based on these data mED and Emax values of different test compounds were determined (Table 1). The rank order of the mED values of the effective ligands was the following: MRS2365 > MRS2395 > UTP.
Table 1.
Minimal effective doses (mED), maximal effects (Emax) attenuating mechanical allodynia and doses of maximal effect (dose of Emax) of purinergic ligands in the Seltzer model of neuropathic pain
| mED (mg·kg−1) | Emax (g) | Dose of Emax (mg·kg−1) | Number of animals per group | |
|---|---|---|---|---|
| MRS2365 | 0.03 | 30.4 ± 3.7 | 0.3 | 7 |
| MRS2395 | 0.3 | 36.7 ± 1.1 | 1 | 7 |
| UTP | 25 | 31.5 ± 0.7 | 100 | 7 |
The mED was determined as the lowest dose necessary to obtain significant change in the postoperative value as compared to the vehicle treated controls. The Emax is defined as the extent of maximal effect obtained in the dose-range examined.
Effects of P2 receptor agonists and antagonists on the nociceptive threshold in the hot-plate test
In the hot-plate test, several compounds were found to have anti-nociceptive efficacy. The baseline nociceptive threshold in our experiments in untreated animals was 46.27 ± 0.10°C (n = 102), which could be well reproduced during the subsequent test period (46.64 ± 0.09°C, n = 102). Intraperitoneal injection of the opiate analgesic morphine (10 mg·kg−1) elicited a remarkable elevation of the thermal pain threshold (45.53 ± 0.35°C vs. 50.35 ± 0.52°C, respectively, for saline and morphine-treated groups, n = 6, P < 0.001).
For the sake of comparison, the effect of different P2 receptor agonists and antagonists were examined in identical doses that were used in the previous experiments. PPADS significantly elevated the heat threshold at the dose of 25 and 50 mg·kg−1 (n = 6, P < 0.05; Figure 3A).
Figure 3.
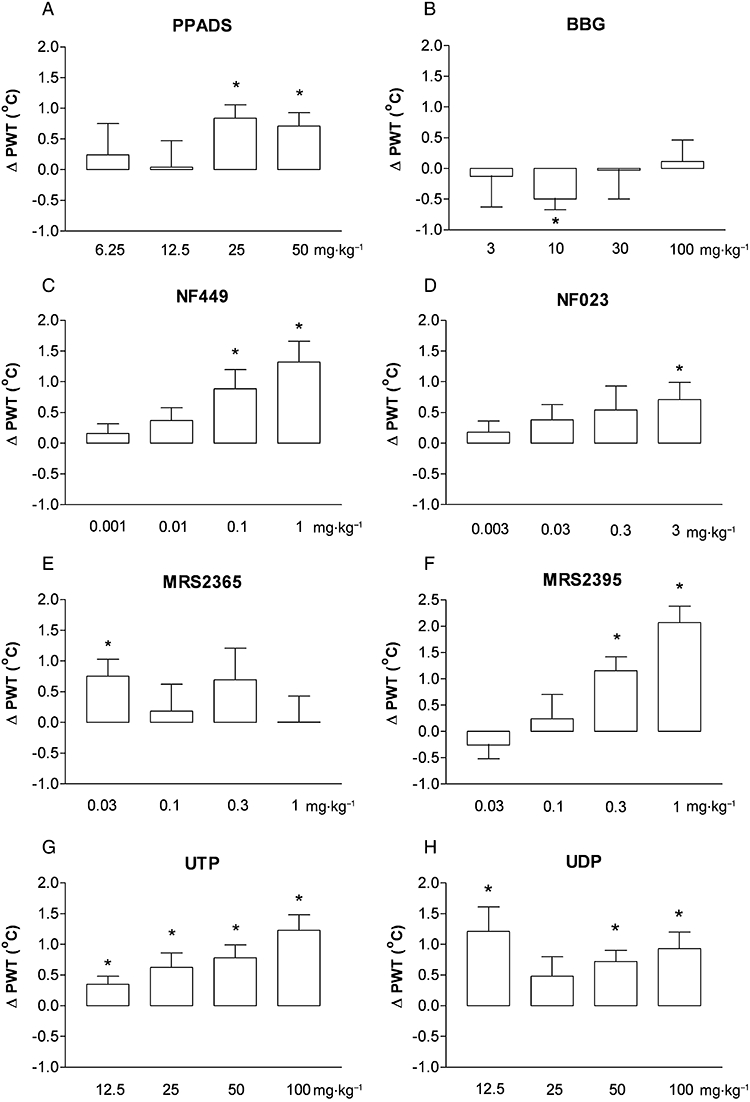
Effects of P2X and P2Y ligands on acute thermal nociception. The changes in nociceptive threshold (post-drug minus baseline values) were represented on the graphs (ΔPWT, °C). *P < 0.05 (paired t-test); significant difference between baseline and post-drug nociceptive threshold.
Interestingly, the selective P2X7 receptor antagonist BBG did not have any analgesic effects in this model and even caused a minor thermal hyperalgesia at 10 mg·kg−1 (−0.5 ± 0.17°C, n = 6, P < 0.05; Figure 3B).
NF449 and NF023 increased the nocifensive threshold (Figure 3C and 3D). However, significant effect was found with NF023 only at the highest dose (3 mg/kg, n = 6, P < 0.05, Figure 3D), while NF449 was more potent, causing hypoalgesia at 0.1 and 1 mg/kg (n = 6, P < 0.05; Figure 3C).
Although the selective P2Y1 receptor agonist MRS2365 had efficacy at the lowest dose used (0.03 mg·kg−1, n = 6, P < 0.05; Figure 3E), higher doses had no significant effect on thermal pain threshold. In contrast, the P2Y12 antagonist MRS2395 had a robust analgesic effect in the 0.03–1 mg·kg−1 dose range (n = 6, P < 0.05; Figure 3F).
Both UDP and UTP elevated the nocifensive threshold in the hot-plate test. UTP displayed analgesia in all four doses tested (Figure 3G), while UDP elevated PWT in the lowest dose and was also effective in the higher dose range (n = 6, P < 0.05; Figure 3H).
Based on these data, the rank order of the mED values of the ligands was the following (Table 2): MRS2365 > MRS2395 > NF449 > NF023 > UDP = UTP > PPADS.
Table 2.
Minimal effective doses (mED), maximal effects (Emax) attenuating thermal pain and doses of maximal effect (dose of Emax) of purinergic ligands in the hot plate test
| mED (mg·kg−1) | Emax (°C) | Dose of Emax (mg·kg−1) | Number of animals per group | |
|---|---|---|---|---|
| PPADS | 25 | 0.84 ± 0.22 | 25 | 6 |
| NF449 | 0.1 | 1.32 ± 0.34 | 1 | 6 |
| NF023 | 3 | 0.71 ± 0.28 | 3 | 6 |
| MRS2365 | 0.03 | 0.75 ± 0.28 | 0.03 | 6 |
| MRS2395 | 0.3 | 2.07 ± 0.31 | 1 | 6 |
| UTP | 12.5 | 1.23 ± 0.25 | 100 | 6 |
| UDP | 12.5 | 1.21 ± 0.4 | 12.5 | 6 |
The mED was determined as the dose necessary to obtain significant change in the thermal threshold compared with the baseline controls, while the Emax is defined as the extent of maximal effect obtained in the dose-range examined.
Inflammatory oedema and mechanical sensitivity in CFA-induced arthritis model
The mean value of the PWT in untreated animals was 41.99 ± 0.55 g (n = 97) in our experiments, whereas the basal volume of the hind paw was 1.72 ± 0.02 mL (n = 106). Forty-eight hours after intraplantar injection of CFA (100 µL, 50% in saline) the mechanical sensitivity of the affected hind paw increased (26.38 ± 2.15 g, n = 119, P < 0.001), and oedema developed (2.23 ± 0.06 mL, n = 106, P < 0.001, 128.6 ± 5.69% increase in volume). No changes were observed in the untreated hind paw (withdrawal threshold: 42.970 ± 0.55 g, n = 119, P > 0.05; paw volume: 1.79 ± 0.02 mL, n = 106, P > 0.05). Based on published data and their efficacy in the neuropathic and acute pain models, MRS2365, MRS2395, and BBG were chosen to be tested in the CFA model. Drugs were injected i.p. 30 and 60 min before oedema and allodynia measurements respectively.
None of the drugs, at all doses tested, had any effect on the withdrawal threshold of the untreated contralateral paw. BBG exhibited a slight antiallodynic effect in the 3–100 mg·kg−1 dose range, which became significant at the highest 100 mg·kg−1 dose as compared with post-operative, saline-treated controls (Figure 4A). On the other hand, BBG did not affect oedema at any dose tested (Figure 4B). MRS2365 (0.1–2 mg·kg−1) elevated PWT and was effective at 1 and 2 mg·kg−1 compared with saline treatment (Figure 4C). Similarly to BBG, MRS2365 failed to decrease paw oedema (Figure 4D). MRS2395 alleviated mechanical sensitivity of the inflamed paws, an effect that was significantly different from vehicle treatment at 0.3, 1 and 3 mg/kg (Figure 4E). MRS2395 slightly reduced CFA-induced paw oedema at 0.3 mg·kg−1 dose, but this effect was not significant (Figure 4F).
Figure 4.
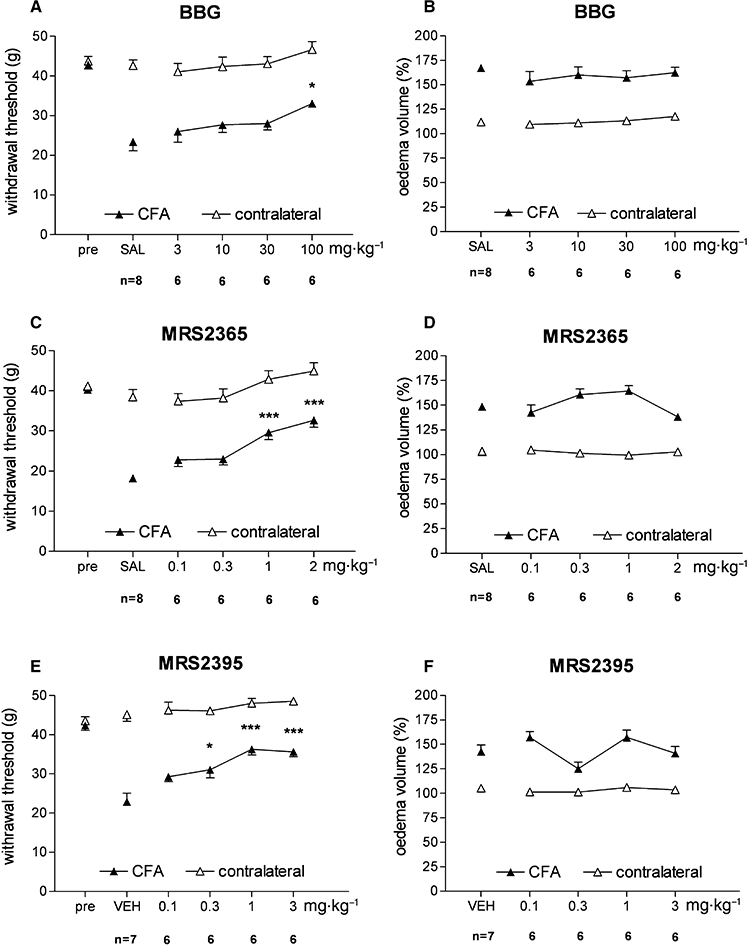
Effects of BBG (A, B), MRS2365 (C, D) and MRS2395 (E, F) on CFA-induced hyperalgesia (left column) and paw oedema (right column). *P < 0.05 and ***P < 0.001 significant difference between CFA-treated inflamed paws and vehicle-treated (saline, SAL, or 25% dimethylsulfoxide + 75% polyethyleneglycol 300, VEH) paws respectively.
Among the tested compounds the following rank order of mED values could be set based on their effect of mechanical sensitivity (Table 3): MRS2395 > MRS2365 > BBG.
Table 3.
Minimal effective doses (mED), maximal effects (Emax) attenuating mechanical allodynia and doses of maximal effect (dose of Emax) of purinergic ligands in the CFA-induced inflammatory pain model
| mED (mg·kg−1) | Emax (g) | Dose of Emax (mg·kg−1) | Number of animals per group | |
|---|---|---|---|---|
| BBG | 100 | 33.1 ± 0.68 | 100 | 6 |
| MRS2365 | 1 | 32.7 ± 1.8 | 2 | 6 |
| MRS2395 | 0.3 | 35.6 ± 1.3 | 1 | 6 |
The mED was determined as the lowest dose necessary to obtain significant change in the post-operative value as compared with the vehicle treated controls. The Emax is defined as the extent of maximal effect obtained in the dose-range examined.
Discussion and conclusions
In our study the effect of systemic application of agonists and antagonists acting at different subtypes of P2X and P2Y receptors has been evaluated on mechanical allodynia in the Seltzer model of neuropathic pain, on acute thermal nocifensive threshold, measured in the hot-plate test and on CFA-induced inflammatory pain and oedema.
As for neuropathic pain, several different animal models are used currently in pain research including the partial ligation of sciatic nerve described by Seltzer et al. (1990). According to a comparative study (Dowdall et al., 2005), this model provides reliable and reproducible results with the measurement of mechanical allodynia with the von Frey method in the post-operative period. In addition, we have used two other widely used analgesia tests for the evaluation of acute thermal pain and inflammatory pain. Because conventional latency measurements of thermal nociception are not sensitive to all kind of analgesics, the modified, increasing-temperature hot-plate test was chosen to measure analgesia in acute pain, which detects noxious heat threshold and has been proven a reliable heat allodynia model (Almasi et al., 2003). In our experiments several test compounds proved to be efficacious to influence these pain modalities. Keeping in mind that the individual potency and the pharmacokinetic properties of the ligands obviously influence the efficacy seen in our tests, some cautious conclusions could be drawn comparing the effect of applied ligands.
Thus, a principal novel finding of our study is the potent analgesic action of the selective P2Y1 receptor agonist MRS2365 in neuropathic and acute pain. In addition, although it exhibited a weaker potency, MRS2365 also alleviated inflammatory pain. Although the anti-nociceptive effect of agonists of P2Y1 receptors such as ADP and ADP-γ-S have been already demonstrated in acute and neuropathic pain (Okada et al., 2002; Gerevich et al., 2004), none of these compounds display selectivity towards the P2Y1 receptor. For instance, ADP binds preferentially to P2Y12 and P2Y13 receptors as well (Abbracchio et al., 2006). MRS2365 acts in the nanomolar range and selectively activates recombinant P2Y1 receptors but not P2Y12 and P2Y13 receptors in vitro (Chhatriwala et al., 2004), which renders likely that its potent in vivo actions are mediated by P2Y1 receptors, although we cannot entirely rule out an additional action on other receptors. Moreover, it displays an enhanced stability and resistance to ectonucleotidases, which is in line with our observations that its actions are clearly detectable in vivo (Ravi et al., 2002). P2Y1 receptors are co-expressed with P2X3 receptors on IB4 containing C-type sensory neurons (Ruan and Burnstock, 2003), and their expression is profoundly increased after peripheral axotomy (Xiao et al., 2002). Activation of P2Y receptors on DRG neurons leads to the inhibition of high- but not low-voltage-activated Ca2+ currents, and this effect has been proposed to underlie a putatively P2Y1 receptor-mediated analgesic action (Gerevich et al., 2004). In addition, activation of P2Y1 receptors on DRG cells inhibits P2X3 receptor-mediated currents, accelerating their desensitization (Gerevich et al., 2005; 2007;), which could also contribute to analgesia. Interestingly, it has been recently reported that P2Y1 receptors down-regulate the expression of P2X3 receptors in DRG, a process that is controlled by the activation of P2X7 receptors on satellite cells (Chen et al., 2008). Finally, inhibition of glutamate and noradrenaline release in the spinal cord as described in our previous study (Heinrich et al., 2008) might also be responsible for P2Y1 receptor-mediated analgesia.
Our studies also confirmed and extended previous results on the anti-neuropathic effect of P2Y12 receptor antagonists (Kobayashi et al., 2008; Tozaki-Saitoh et al., 2008). Moreover, we report here that MRS2395, the selective P2Y12 receptor antagonist (Zizzo et al., 2007), exhibits analgesic action in acute and inflammatory pain as well as upon single, acute systemic application. Although there is no published report on the pharmacokinetic properties of MRS2395, because of its potent effect, it is reasonable to assume that it reached its site of action. The potential application of P2Y12 receptor antagonists as analgesic agents are especially intriguing, as they are already used clinically as anti-thrombotic agents. In contrast to P2Y1 receptors, P2Y12 receptors are expressed on the spinal microglia, and they are up-regulated after peripheral sciatic nerve injury (Kobayashi et al., 2008) or spinal nerve transection (Tozaki-Saitoh et al., 2008). The crucial role of P2Y12 receptors in the recruitment (Ohsawa et al., 2007) and activation of microglia after nerve injury has been well established (Davalos et al., 2005; Haynes et al., 2006). In addition, the activation of P2Y12 receptors on spinal microglia may participate in neuropathic hypersensitivity by the production of pro-inflammatory cytokines and mediators such as interleukin (IL)-1β, IL-6, tumour necrosis factor α and NO or by the regulation of inflammatory cascades such as cyclooxygenase (COX)-2. Nevertheless, the exact mechanism responsible for P2Y12 receptor-mediated anti-nociception awaits further investigation.
Interestingly, BBG, the selective P2X7 receptor antagonist, exhibited only a mild effect on the PWT in the neuropathic model. Although a non-significant tendency of reversal of mechanical allodynia was observed, at higher doses this tendency was lost. On the other hand, BBG elicited a slight inhibitory effect on inflammatory pain but did not affect oedema and acute pain apart from one dose where it elicited hyperalgesia. There is convincing evidence for the analgesic effect of other P2X7 receptor antagonists in neuropathic pain (Dell'Antonio et al., 2002; Honore et al., 2006; Nelson et al., 2006; McGaraughty et al., 2007; Perez-Medrano et al., 2009), although in a recent study a P2X7 receptor antagonist of a different chemical class did not exhibit anti-neuropathic effects (Broom et al., 2008). Previous studies suggested that BBG penetrated the blood brain barrier, and we used it in a dose range, which has been proven to be effective in animal models of Huntington's disease and spinal cord injury in vivo (Diaz-Hernandez et al., 2009; Peng et al., 2009). Therefore, it seems unlikely that its relatively weak activity is due to lack of penetration into the CNS. P2X7 receptors are expressed in microglia, astrocytes, and nerve terminals of the spinal cord (Deuchars et al., 2001; Sperlagh et al., 2006) and satellite cells of the DRG (Chen et al., 2008). The anti-neuropathic action of P2X7 receptor antagonists can be explained by their ability to inhibit the spontaneous activity of all classes of spinal neurons (McGaraughty et al., 2007), modulating central sensitization and by the role of P2X7 receptors in the regulation of the production of the cytokine IL-1β from proinflammatory cells, which is able to alter pain sensitivity (Donnelly-Roberts and Jarvis, 2007). Our data also support previous observations, where P2X7 receptor antagonists (Dell'Antonio et al., 2002; Honore et al., 2006; Broom et al., 2008) and genetic deletion (Chessell et al., 2005) potently inhibited inflammatory pain, although in our experiments, BBG displayed relatively mild effects. On the other hand, as found by Honore et al. (2006), BBG was not effective in preventing oedema, indicating that its analgesic action is not the consequence of its anti-inflammatory potential. A further interesting observation is the hyperalgesia, elicited by BBG, when acute thermal nociception was measured. A potential explanation of the biphasic effects of BBG is that P2X7 receptor activation in satellite cells releases ATP, which, in turn, acts on P2Y1 receptors and maintain inhibitory control over the expression of P2X3 receptors on DRG neurons, as a physiological mechanism (Chen et al., 2008). Therefore, the removal of this protective mechanism by P2X7 receptor antagonism elicits hyperalgesia in acute nociceptive reactions, as we have seen in our experiments, while chronic pathological neuropathic pain is relieved by the blockade of spinal P2X7 receptors.
In contrast to the plethora of results on P2X7 receptor-mediated nociception, there are no published data on the action of P2X1 receptor antagonists in pain models in vivo. Here we showed that the selective P2X1 receptor antagonist NF449 elicited dose-dependent analgesic action in acute pain, while it remained ineffective in the model of neuropathic pain. Similar results were obtained with NF023, another P2X1 receptor antagonist, and in agreement with their rank order of potency in vitro (Soto et al., 1999; Rettinger et al., 2005), NF449 was the more potent. These data are in good agreement with previous findings, showing that P2X1/5 receptors are primarily expressed on capsaicin insensitive Aδ fibers of DRG (Petruska et al., 2000) and their activation releases glutamate from central afferent terminals (Nakatsuka et al., 2003; Tsuzuki et al., 2003).
We have added to earlier observations with the wide spectrum P2 receptor antagonist PPADS obtained with the chronic constriction injury model in mice (Martucci et al., 2007) and revealed that PPADS has an acute analgesic action in higher doses. By contrast, in the neuropathic pain model, apart from a slight tendency of reversal, PPADS in our hands remained ineffective using a single acute injection 7 days after surgery. The most likely reason for the discrepancy is that Martucci et al. (2007) applied multiple, daily administration of PPADS from the third post-operative day. Taking into account that PPADS acts at a variety of P2 receptor subtypes involved in sensory processing (P2X1, P2X2, P2X3, P2X5, P2X7, P2Y1, P2Y4), it is not possible to identify a single receptor subtype involved in its analgesic action.
Likewise, i.p. injection of UTP and UDP agonists of P2Y2, P2Y4 and P2Y6 receptors exhibited a significant, although not particularly potent, anti-nociceptive action in the acute and neuropathic pain model, supporting similar findings obtained by intrathecal administration (Okada et al., 2002), although UDP was not effective in the neuropathic model. The potential systemic breakdown of uridine nucleotides by ectonucleotidases might serve as an explanation for their relatively weak effect in our study. As a potential underlying mechanism of these effects, P2Y2 receptors are co-expressed with TRPV1 channels in small diameter DRG neurons (Moriyama et al., 2003) and ATP and UTP inhibits the slow depolarization of substantia gelatinosa neurons evoked by repetitive stimulation of C fibers of DRG neurons in the spinal cord (Yoshida et al., 2002).
In conclusion, our results show that significant anti-nociceptive action could be obtained by several agonists and antagonists acting at different subtypes of ionotropic and metabotropic P2 receptors, and in addition to the blockade of P2X7 and P2Y12 receptors, antagonism at P2X1 receptors, as well as agonism at P2Y1 receptors, also defines promising therapeutic targets in acute, neuropathic, and inflammatory pain respectively. Therefore, compounds acting at several purinergic receptors might prove to be a more successful drug design strategy than those selectively targeting individual receptors.
Acknowledgments
The authors are grateful for Zsuzsanna Helyes and Krisztián Elekes for the expert advice on in vivo pain models. This study was supported by grants of the Hungarian Research and Development Fund (Grant No. K61758 74527), Hungarian Medical Research Council (03-102/2009) and the Richter Gedeon Plc. (KK/1135/2007) to B.S.
Glossary
Abbreviations:
- BBG
Brilliant Blue G
- CFA
complete Freund's adjuvant
- DRG
dorsal root ganglia
- MRS2365
([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid mono ester trisodium salt
- MRS2395
(2,2-dimethyl-propionic acid 3-(2-chloro-6-methylaminopurin-9-yl)-2-(2,2-dimethyl-propionyloxymethyl)-propylester
- NF449
(4,4′,4″,4
 -[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt
-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulfonic acid, octasodium salt- NF023
(8,8′-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-1,3,5-naphthalene-trisulphonic acid, hexasodium salt
- PPADS
pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid
- PWT
paw withdrawal threshold
Conflict of interest
None.
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. edn. (2008 revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasi R, Petho G, Bolcskei K, Szolcsanyi J. Effect of resiniferatoxin on the noxious heat threshold temperature in the rat: a novel heat allodynia model sensitive to analgesics. Br J Pharmacol. 2003;139:49–58. doi: 10.1038/sj.bjp.0705234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J, Patel S, Dorn G, Wotherspoon G, Moffatt S, Eunson L, et al. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci. 2002;22:8139–8147. doi: 10.1523/JNEUROSCI.22-18-08139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcskei K, Helyes Z, Szabo A, Sandor K, Elekes K, Nemeth J, et al. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain. 2005;117:368–376. doi: 10.1016/j.pain.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Broom DC, Matson DJ, Bradshaw E, Buck ME, Meade R, Coombs S, et al. Characterization of N-(adamantan-1-ylmethyl)-5-[(3R-amino-pyrrolidin-1-yl)methyl]-2-chloro-ben zamide, a P2X7 antagonist in animal models of pain and inflammation. J Pharmacol Exp Ther. 2008;327:620–633. doi: 10.1124/jpet.108.141853. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic P2 receptors as targets for novel analgesics. Pharmacol Ther. 2006;110:433–454. doi: 10.1016/j.pharmthera.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li GW, Wang C, Gu Y, Huang LY. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. Pain. 2005;119:38–48. doi: 10.1016/j.pain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang LY. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc Natl Acad Sci USA. 2008;105:16773–16778. doi: 10.1073/pnas.0801793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Jacobson KA, Harden TK. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004;311:1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizh BA, Illes P. P2X receptors and nociception. Pharmacol Rev. 2001;53:553–568. [PubMed] [Google Scholar]
- Cockayne DA, Dunn PM, Zhong Y, Rong WF, Hamilton SG, Knight GE, et al. P2X2 knockout mice and P2X2/ P2X3 double knockout mice reveal a role for the P2X2 receptor subunit in mediating multiple sensory effects of ATP. J Physiol-London. 2005;567:621–639. doi: 10.1113/jphysiol.2005.088435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier HO, James GW, Schneider C. Antagonism by aspirin and fenamates of bronchoconstriction and nociception induced by adenosine-5′-triphosphate. Nature. 1966;212:411–412. doi: 10.1038/212411a0. [DOI] [PubMed] [Google Scholar]
- Cook SP, McCleskey EW. Cell damage excites nociceptors through release of cytosolic ATP. Pain. 2002;95:41–47. doi: 10.1016/s0304-3959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Dell'Antonio G, Quattrini A, Cin ED, Fulgenzi A, Ferrero ME. Relief of inflammatory pain in rats by local use of the selective P2X7 ATP receptor inhibitor, oxidized ATP. Arthritis Rheum. 2002;46:3378–3385. doi: 10.1002/art.10678. [DOI] [PubMed] [Google Scholar]
- Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TF, et al. Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci. 2001;21:7143–7152. doi: 10.1523/JNEUROSCI.21-18-07143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Diez-Zaera M, Sanchez-Nogueiro J, Gomez-Villafuertes R, Canals JM, Alberch J, et al. Altered P2X7-receptor level and function in mouse models of Huntington's disease and therapeutic efficacy of antagonist administration. FASEB J. 2009;23:1893–1906. doi: 10.1096/fj.08-122275. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts D, McGaraughty S, Shieh CC, Honore P, Jarvis MF. Painful purinergic receptors. J Pharmacol Exp Ther. 2008;324:409–415. doi: 10.1124/jpet.106.105890. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Jarvis MF. Discovery of P2X7 receptor-selective antagonists offers new insights into P2X7 receptor function and indicates a role in chronic pain states. Br J Pharmacol. 2007;151:571–579. doi: 10.1038/sj.bjp.0707265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G, Patel S, Wotherspoon G, Hemmings-Mieszczak M, Barclay J, Natt FJ, et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004;32:e49. doi: 10.1093/nar/gnh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdall T, Robinson I, Meert TF. Comparison of five different rat models of peripheral nerve injury. Pharmacol Biochem Behav. 2005;80:93–108. doi: 10.1016/j.pbb.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Dunn PM, Zhong Y, Burnstock G. P2X receptors in peripheral neurons. Prog Neurobiol. 2001;65:107–134. doi: 10.1016/s0301-0082(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Gerevich Z, Borvendeg SJ, Schroder W, Franke H, Wirkner K, Norenberg W, et al. Inhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia. J Neurosci. 2004;24:797–807. doi: 10.1523/JNEUROSCI.4019-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerevich Z, Illes P. P2Y receptors and pain transmission. Purinergic Signalling. 2004;1:3–10. doi: 10.1007/s11302-004-4740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerevich Z, Muller C, Illes P. Metabotropic P2Y1 receptors inhibit P2X3 receptor-channels in rat dorsal root ganglion neurons. Eur J Pharmacol. 2005;521:34–38. doi: 10.1016/j.ejphar.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gerevich Z, Zadori Z, Muller C, Wirkner K, Schroder W, Rubini P, et al. Metabotropic P2Y receptors inhibit P2X3 receptor-channels via G protein-dependent facilitation of their desensitization. Br J Pharmacol. 2007;151:226–236. doi: 10.1038/sj.bjp.0707217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilron I, Watson CP, Cahill CM, Moulin DE. Neuropathic pain: a practical guide for the clinician. CMAJ. 2006;175:265–275. doi: 10.1503/cmaj.060146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Heinrich A, Kittel A, Csolle C, Sylvester Vizi E, Sperlagh B. Modulation of neurotransmitter release by P2X and P2Y receptors in the rat spinal cord. Neuropharmacology. 2008;54:375–386. doi: 10.1016/j.neuropharm.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Holton P. The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J Physiol. 1959;145:494–504. doi: 10.1113/jphysiol.1959.sp006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Kage K, Mikusa J, Watt AT, Johnston JF, Wyatt JR, et al. Analgesic profile of intrathecal P2X3 antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002;99:11–19. doi: 10.1016/s0304-3959(02)00032-5. [DOI] [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319:1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic M, Zhong C, Wade C, Chandran P, et al. The antihyperalgesic activity of a selective P2X7 receptor antagonist, A-839977, is lost in IL-1alphabeta knockout mice. Behav Brain Res. 2009;204:77–81. doi: 10.1016/j.bbr.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Inoue K, Tsuda M, Koizumi S. ATP- and adenosine-mediated signaling in the central nervous system: chronic pain and microglia: involvement of the ATP receptor P2X4. J Pharmacol Sci. 2004;94:112–114. doi: 10.1254/jphs.94.112. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Boyce-Rustay JM. Neuropathic pain: models and mechanisms. Curr Pharm Des. 2009;15:1711–1716. doi: 10.2174/138161209788186272. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Burgard EC, McGaraughty S, Honore P, Lynch K, Brennan TJ, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- Kennedy JD. Neuropathic pain:molecular complexity underlies continuing unmet medical need. J Med Chem. 2007;50:2547–2556. doi: 10.1021/jm061023c. [DOI] [PubMed] [Google Scholar]
- Kennedy C, Assis TS, Currie AJ, Rowan EG. Crossing the pain barrier: P2 receptors as targets for novel analgesics. J Physiol. 2003;553(3):683–694. doi: 10.1113/jphysiol.2003.049114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martucci C, Trovato AE, Costa B, Borsani E, Franchi S, Magnaghi V, et al. The purinergic antagonist PPADS reduces pain related behaviours and interleukin-1beta, interleukin-6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain. 2007;137:81–95. doi: 10.1016/j.pain.2007.08.017. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Namovic MT, Donnelly-Roberts DL, Harris RR, Zhang XF, et al. P2X7-related modulation of pathological nociception in rats. Neuroscience. 2007;146:1817–1828. doi: 10.1016/j.neuroscience.2007.03.035. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, et al. Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci. 2003;23:6058–6062. doi: 10.1523/JNEUROSCI.23-14-06058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuka T, Tsuzuki K, Ling JX, Sonobe H, Gu JG. Distinct roles of P2X receptors in modulating glutamate release at different primary sensory synapses in rat spinal cord. J Neurophysiol. 2003;89:3243–3252. doi: 10.1152/jn.01172.2002. [DOI] [PubMed] [Google Scholar]
- Nelson DW, Gregg RJ, Kort ME, Perez-Medrano A, Voight EA, Wang Y, et al. Structure-activity relationship studies on a series of novel, substituted 1-benzyl-5-phenyltetrazole P2X7 antagonists. J Med Chem. 2006;49:3659–3666. doi: 10.1021/jm051202e. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- Okada M, Nakagawa T, Minami M, Satoh M. Analgesic effects of intrathecal administration of P2Y nucleotide receptor agonists UTP and UDP in normal and neuropathic pain model rats. J Pharmacol Exp Ther. 2002;303:66–73. doi: 10.1124/jpet.102.036079. [DOI] [PubMed] [Google Scholar]
- Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, et al. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci USA. 2009;106:12489–12493. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Medrano A, Donnelly-Roberts DL, Honore P, Hsieh GC, Namovic MT, Peddi S, et al. Discovery and biological evaluation of novel cyanoguanidine P2X7 antagonists with analgesic activity in a rat model of neuropathic pain. J Med Chem. 2009;52:3366–3376. doi: 10.1021/jm8015848. [DOI] [PubMed] [Google Scholar]
- Petruska JC, Cooper BY, Gu JG, Rau KK, Johnson RD. Distribution of P2X1, P2X2, and P2X3 receptor subunits in rat primary afferents: relation to population markers and specific cell types. J Chem Neuroanat. 2000;20:141–162. doi: 10.1016/s0891-0618(00)00080-6. [DOI] [PubMed] [Google Scholar]
- Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, et al. Adenine nucleotide analogues locked in a Northern methanocarba conformation: enhanced stability and potency as P2Y1 receptor agonists. J Med Chem. 2002;45:2090–2100. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettinger J, Braun K, Hochmann H, Kassack MU, Ullmann H, Nickel P, et al. Profiling at recombinant homomeric and heteromeric rat P2X receptors identifies the suramin analogue NF449 as a highly potent P2X1 receptor antagonist. Neuropharmacology. 2005;48:461–468. doi: 10.1016/j.neuropharm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Ruan HZ, Burnstock G. Localisation of P2Y1 and P2Y4 receptors in dorsal root, nodose and trigeminal ganglia of the rat. Histochem Cell Biol. 2003;120:415–426. doi: 10.1007/s00418-003-0579-3. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Sharp CJ, Reeve AJ, Collins SD, Martindale JC, Summerfield SG, Sargent BS, et al. Investigation into the role of P2X3/P2X2/3 receptors in neuropathic pain following chronic constriction injury in the rat: an electrophysiological study. Br J Pharmacol. 2006;148:845–852. doi: 10.1038/sj.bjp.0706790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Lambrecht G, Nickel P, Stuhmer W, Busch AE. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R, et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Vizi ES, Wirkner K, Illes P. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–346. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Tozaki-Saitoh H, Tsuda M, Miyata H, Ueda K, Kohsaka S, Inoue K. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J Neurosci. 2008;28:4949–4956. doi: 10.1523/JNEUROSCI.0323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuzuki K, Ase A, Seguela P, Nakatsuka T, Wang CY, She JX, et al. TNP-ATP-resistant P2X ionic current on the central terminals and somata of rat primary sensory neurons. J Neurophysiol. 2003;89:3235–3242. doi: 10.1152/jn.01171.2002. [DOI] [PubMed] [Google Scholar]
- Wirkner K, Sperlagh B, Illes P. P2X3 receptor involvement in pain states. Mol Neurobiol. 2007;36:165–183. doi: 10.1007/s12035-007-0033-y. [DOI] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA. 2002;99:8360–8365. doi: 10.1073/pnas.122231899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Nakagawa T, Kaneko S, Akaike A, Satoh M. Adenosine 5′-triphosphate inhibits slow depolarization induced by repetitive dorsal root stimulation via P2Y purinoceptors in substantia gelatinosa neurons of the adult rat spinal cord slices with the dorsal root attached. Neurosci Lett. 2002;320:121–124. doi: 10.1016/s0304-3940(02)00018-6. [DOI] [PubMed] [Google Scholar]
- Zizzo MG, Mule F, Serio R. Evidence that ATP or a related purine is an excitatory neurotransmitter in the longitudinal muscle of mouse distal colon. Br J Pharmacol. 2007;151:73–81. doi: 10.1038/sj.bjp.0707188. [DOI] [PMC free article] [PubMed] [Google Scholar]