

Abstract
Background and purpose:
The present study reports on the preparation and testing of a sustained delivery system for the immunomodulatory peptide P10 aimed at reducing the in vivo degradation of the peptide and the amount required to elicit a protective immune response against paracoccidioidomycosis.
Experimental approach:
BALB/c mice were infected with the yeast Paracoccidioides brasiliensis to mimic the chronic form of paracoccidioidomycosis. The animals were treated daily with sulfamethoxazole/trimethoprim alone or combined with peptide P10, either emulsified in Freund's adjuvant or entrapped in poly(lactic acid-glycolic acid) (PLGA) nanoparticles at different concentrations (1 µg, 5 µg, 10 µg, 20 µg or 40 µg·50 µL−1). Therapeutic efficacy was assessed as fungal burden in tissues and the immune response by quantitative determination of cytokines.
Key results:
Animals given combined chemotherapy and P10 nanotherapy presented a marked reduction of fungal load in the lungs, compared with the non-treated animals. After 30 days of treatment, P10 entrapped within PLGA (1 µg·50 µL−1) was more effective than ‘free’ P10 emulsified in Freund's adjuvant (20 µg·50 µL−1), as an adjuvant to chemotherapy. After treatment for 90 days, the higher doses of P10 entrapped within PLGA (5 or 10 µg·50 µL−1) were most effective. Treatment with P10 emulsified in Freund's adjuvant (20 µg·50 µL−1) or P10 entrapped within PLGA (1 µg·50 µL−1) were accompanied by high levels of interferon-gamma in lung.
Conclusions and implications:
Combination of sulfamethoxazole/trimethoprim with the P10 peptide entrapped within PLGA demonstrated increased therapeutic efficacy against paracoccidioidomycosis. P10 incorporation into PLGA nanoparticles dramatically reduced the peptide amount necessary to elicit a protective effect.
Keywords: immunomodulatory peptide, antifungal therapy, biodegradable polymers, drug delivery, nanobiotechnology
Introduction
Extensive research has focused on the design of efficient adjuvants to vaccines for immunological protection in several diseases. More recently, because of the dramatic increase in the incidence of systemic mycoses, attention has been given to the development of antifungal vaccines (Cutler et al., 2007). Peptide antigens, among other immunogens, are especially promising for triggering effective immune protective responses against these infections (Taborda et al., 1998; Travassos et al., 2008a). Incorporation of peptides into controlled release systems is an approach used to avoid degradation and promote their release at predetermined rates (Johansen et al., 2000).
Biodegradable polymers are attractive delivery systems for vaccines because of their property of slowly and gradually controlling the release of an antigen (Commandeur et al., 2006). After in vivo administration of biocompatible polymers, they are broken down into molecules that take part in normal metabolic pathways and are then eliminated. Any polymer-entrapped molecule (a drug or a peptide) is released upon polymer biodegradation, which depends on polymer constitution and occurs under certain conditions, such as pH and temperature changes (Commandeur et al., 2006; Vicent and Duncan, 2006).
Polymeric systems, such as nano- and micro-particles, are appropriate as adjuvants to prepare a single-shot vaccine. Antigens encapsulated within polymers can be released for a prolonged period at a controlled rate of polymer degradation (Vicent and Duncan, 2006). Polymers have been shown to be effective adjuvants for various antigenic proteins, including ovalbumin, cholera and tetanus toxoid, and malarial and pneumotrophic bacterial antigens (Dhiman and Khuller, 1998; Jaganathan et al., 2005). Poly(lactic acid-glycolic acid) (PLGA) is a polymer whose degradation rate and antigen release can be predicted (Jiang et al., 2005). By releasing an immunomodulatory peptide at a predetermined rate, it can significantly reduce the amount and number of doses of antigen required for protection.
Taborda et al. (1998) identified a 15-amino acid peptide that carries the T-cell epitope of the glycoprotein 43 kDa glycoprotein (Gp43), the major diagnostic antigen secreted by Paracoccidioides brasiliensis (Puccia et al., 1986). This peptide, named P10, elicits an interferon-gamma (IFN-γ)-dependent immune protection against experimental paracoccidioidomycosis (PCM; Taborda et al., 1998; Travassos et al., 2004). Immunization with P10 improved the therapeutic efficacy against PCM when combined with sulfamethoxazole/trimethoprim, among other antifungals, suggesting an important contribution of P10 in improving outcome and reducing the time of treatment against PCM (Marques et al., 2006).
Paracoccidioidomycosis is a health problem in Latin America, where around 10 million individuals may be infected by the dimorphic human pathogenic fungus P. brasiliensis and 2% of them may develop acute or chronic forms of PCM (Brummer et al., 1993). The acute form involves the lymphoreticular system and may be lethal; in chronic PCM, the lung is mainly affected with a granulomatous inflammatory response, which represents an effective defence against fungal spread (Brummer et al., 1993; de Camargo and de Franco, 2000). The usual therapy for PCM is based on polyenes, azoles and sulphonamides. Amphotericin B is indicated in severe disseminated cases and must be followed by a prolonged treatment with azoles and sulfamethoxazole/trimethoprim (Brummer et al., 1993; Lortholary et al., 1999).
The present study reports on the design, preparation and in vivo testing of a sustained delivery system containing the immunoprotective peptide P10 loaded on nanoparticles of the biodegradable polymer PLGA. This preparation was administered in combination with sulfamethoxazole/trimethoprim during the treatment of a murine model of systemic PCM.
Methods
Preparation of PLGA particles loaded with P10 peptide
The particles were prepared using PLGA polymeric blends with 50:50 poly-lactic acid (PLA) : poly-glycolic acid (PGA). The polymers were first dissolved in dichloromethane and then added to an aqueous solution containing 1% polyvinyl alcohol and the P10 peptide, aiming for the final preparation to contain 1, 5, 10, 20 or 40 µg·50 µL−1 of the peptide. The mixture was submitted to vigorous agitation in a blender (10 000–15 000 rpm) to obtain the water-in-oil emulsification. The organic solvent was removed from the solution by stirring at room temperature and evaporation under reduced pressure. The particles were centrifuged (4–10°C; 1100–4600× g.) for 10- to 20-min intervals. The preparation was washed three times in distilled water, suspended in 1.0 mL phosphate-buffered saline (PBS), stored at 4°C and used for up to 1 week. All procedures were performed in a sterile room with all the manipulations in a sterile hood.
Animals
All animal handling and experimental procedures performed in this study were approved by the Animal Care and Use Committee of the Universidade de São Paulo. Male BALB/c mice (6–8 weeks old) from the Universidade de São Paulo (USP), Brazil, were used in this study. Animals were housed in polypropylene cages under specific pathogen-free conditions and were provided with food and water ad libitum.
Fungal inoculum for in vivo experiments
P. brasiliensis, isolate Pb18, was used to infect the animals. The strain was sub-cultured in defined liquid medium McVeigh-Morton culture medium at 35°C in a rotatory shaker (220 rpm) (Restrepo and Arango, 1980). After 5 or 7 days of culture, the yeast cells were collected by centrifugation, the supernatant was discarded, and the cells were washed three times in sterile PBS, pH 7.4. We determined the cell count in a haematocytometer and adjusted the inoculum suspension to 3 × 105 viable fungi per 50 µL. The cellular viability was 90–95% in these experiments, as determined by vital staining with Evans blue (Sigma, St. Louis, MO, USA).
Intratracheal infection
To mimic the most common infection in the chronic form of PCM, BALB/c mice were inoculated intratracheally with P. brasiliensis Pb18, and after 30 days of infection, the animals were subjected to a combined therapy of sulfamethoxazole/trimethoprim and a range of doses of P10 (1–40 µg) within PLGA. The animals were anaesthetized by intraperitoneal injection of 200 µL of a solution of 80 mg·kg−1 ketamine and 10 mg·kg−1 xylazine (União Química, Brazil). After 10 min, their necks were hyperextended and incised to barely expose the trachea. Each animal received 3 × 105 viable fungi in 50 µL PBS using a 26.5-gauge needle. The incisions were sutured immediately after the delivery of the fungi.
Antifungal treatment
The infected mice were randomly divided into nine groups of six animals each and subjected to antifungal treatment with sulfamethoxazole/trimethoprim (15 mg·kg−1 and 3 mg·kg−1 respectively) in PBS, pH 7.4, with or without 20 µg P10 solubilized in 50 µL Freund's adjuvant named ‘free’ or P10 entrapped within PLGA at 1 µg, 5 µg, 10 µg, 20 µg or 40 µg·50 µL−1. Group (i) was the control group treated only with PBS, group (ii) was treated with sulfamethoxazole/trimethoprim alone, group (iii) was treated with sulfamethoxazole/trimethoprim and 50 µL of ‘empty’ PLGA nanoparticles, group (iv) was treated with 20 µg·50 µL−1 of ‘free’ P10, and groups (v) (vi) (vii) (viii) and (ix) were treated with 1 µg, 5 µg, 10 µg, 20 µg and 40 µg·50 µL−1, respectively, of P10 entrapped within PLGA. The treatment regimens started 30 days after fungal challenge and were continued for 30 days. The empty PLGA nanoparticles given to group (iii) were prepared using the same procedure, methodology and ratio amount of PLA : PGA (50:50) that was used to prepare the nanoparticles associated with P10, which gave the same final concentration in both preparations with or without P10. The particle size and size distribution were measured by laser light scattering using a particle size analyser (Zetasizer, Malvern, UK). Size distribution was analysed over the range of 1–1000 nm, and the mean diameter was calculated for each sample. The PLGA particles with and without peptide P10 at various concentrations were measured, and the average diameter found was 430 ± 5.1 nm for the loaded particle and 410 ± 4.9 nm for the empty one.
Animals from groups (ii) to (ix) received daily injections of sulfamethoxazole/trimethoprim (15 mg·kg−1 and 3 mg·kg−1 respectively). Mice from groups (iv) to (ix) received four doses of peptide P10 administered once a week during 4 weeks as follows: 20 µg·50 µL−1 of ‘free’ or 1 µg, 5 µg, 10 µg, 20 µg and 40 µg·50 µL−1 entrapped within PLGA. ‘Free’ P10 was administered with complete Freund's adjuvant in the hind paw, and P10 entrapped within PLGA at various concentrations was administered i.p. without adjuvant.
Fungal burden assay
The combined treatment effects were assessed by the residual lung, liver and spleen fungal burden. The animals from all experimental groups were killed by cervical rupture 30 and 90 days after the beginning of therapy, and the lungs, liver and spleen were aseptically removed and weighed. The organs were rinsed and homogenized in sterile PBS, pH 7.4, and 100 µL of the homogenates was cultured in BHI agar supplemented with 4% horse serum, 5% P. brasiliensis 192 culture filtrate, 10 000 IU penicillin (Cultilab, Brazil) and 10 mg·L−1 streptomycin (Cultilab, Brazil) in duplicate. The Pb192 culture filtrate was prepared according to a previously published method (Singer-Vermes et al., 1992). The plates were incubated at 37°C and colony-forming units (CFUs) were counted 10 days post-plating to determine the CFU·g−1 of tissue.
Cytokine assays
To assess cytokine production, we assayed IFN-γ and interleukins-4 (IL-4), -10 (IL-10) and -12 (IL-12) in lung homogenates. Lung fragments were aseptically removed, weighed and homogenized in sterile PBS with protease inhibitor (Roche, USA). The cytokines were determined using commercial ELISA kits (BD Biosciences – Pharmingen, San Diego, CA, USA).
Statistical analysis
Statistical Package for Social Sciences (SPSS) version 15 was used to analyse our data. All results are expressed as means ± standard error. A one-way analysis of variance (anova) with Tukey's post-test was applied to test inter-group differences. Differences between paired groups were analysed by the Mann-Whitney test. P-values less than 0.05 were considered significant.
Materials
Sulfamethoxazole was purchased from Sigma (St. Louis, MO, USA) and trimethoprim from Ducto (Bac-sulfitrin, Ducto). The poly-lactic acid (PLA) and poly-glycolic acid (PGA) used to prepare the nanoparticles were purchased from Sigma (St. Louis, MO, USA). The P10 peptide was synthesized by the 9-fluoroenylmethoxy-carbonyl technique (Huang et al., 1993) and provided by Dr Maria A. Juliano from the Department of Biophysics of the Federal University of São Paulo. Drug and molecular target nomenclature conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2008).
Results
Fungal burden in treated animals
To evaluate the efficacy of the combined therapy using the P10 peptide entrapped within PLGA in association with sulfamethoxazole/trimethoprim, we carried out in vivo antifungal drug experiments in BALB/c mice infected with the fungus P. brasiliensis, isolate Pb18. The treatment regimens started after 30 days of infection and were evaluated 30 and 90 days after the beginning of treatment. The fungal load was investigated in the lungs, liver and spleen of the animals. No significant number of fungi in the liver and a very low number in the spleen were detected 30 days after intratracheal (i.t.) infection (data not shown). This result was expected as the liver or/spleen infection is only detectable in the early stages of this animal model of mycosis. In contrast, the main focus of the infection was the lungs. A high load of fungal cells was recovered in the lungs of the control animals that received only PBS, both 30 and 90 days after beginning the treatment (Figure 1). In the group treated only with sulfamethoxazole/trimethoprim, the infection was controlled during the first 30 days of treatment followed by a high number of fungal cells after 90 days, comparable to the untreated group (Figure 1).
Figure 1.
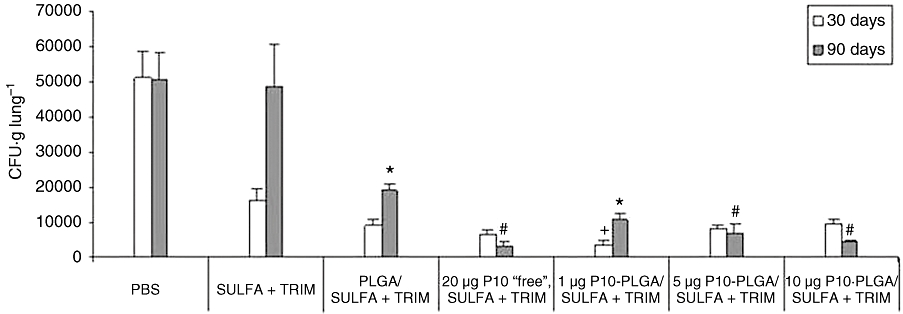
Fungal burden recovery assessed by colony-forming units [CFU (g·lung·tissue)−1] in mice infected with P. brasiliensis Pb18 and subjected to a combined therapy of sulfamethoxazole/trimethoprim (Sulfa + Trim; 15 mg·kg−1 and 3 mg·kg−1 respectively) and either 20 µg·50 µL−1 P10 peptide solubilized in Freund's adjuvant (‘free’) or P10 peptide (1, 5 or 10 µg·50 µL−1) entrapped within PLGA. Each bar represents the average CFU (g·tissue)−1 with standard deviations. After 30 days of treatment, 1 µg·50 µL−1 of P10-PLGA/Sulfa + Trim yielded the best response (lowest fungal CFU recovery) of all groups (+P < 0.05). After 90 days of treatment, no significant differences were found between the responses of the ‘free’ P10 (20 µg·50 µL−1) and the P10-PLGA (5–10 µg·50 µL−1)-treated groups (marked with #). At day 90, a significantly lower number of fungal cells were recovered from mice treated with 1 µg·50 µL−1 P10-PLGA compared with the PLGA alone treated group (*P < 0.05). PLGA, poly(lactic acid-glycolic acid).
A reduction of CFUs in comparison with untreated animals was confirmed using 20 µg of ‘free’ P10 emulsified in Freund's adjuvant (Marques et al., 2006), reversing the relapse observed in the group treated only with sulfamethoxazole/trimethoprim. The dose of 20 µg used for the ‘free’ form of P10 peptide was chosen on the basis of earlier results (Taborda et al., 1998; Taborda et al., 2004). Under this condition, lymphoproliferative and cell-mediated immune responses protecting mice from the P. brasiliensis infection were observed. In the groups subjected to the combined therapy of sulfamethoxazole/trimethoprim and P10 entrapped within PLGA, there was always a significant protection. After 30 days of treatment, amounts in the range of 1–10 µg·50 µL−1 of P10 encapsulated with PLGA were efficient as an adjuvant to chemotherapy, reducing the dose of the peptide necessary to decrease the fungal load and to avoid disease relapse by at least 20-fold. The groups that received 20 or 40 µg·50 µL−1 of P10 entrapped within PLGA, did not present a marked additive action as adjuvants to chemotherapy (data not shown), although they responded better than the mice given sulfamethoxazole/trimethoprim alone. The PLGA nanoparticles without P10 showed a protective effect after 30 days, proving to be an adjuvant to chemotherapy by themselves, but these ‘empty’ nanoparticles were unable to prevent the relapse observed by sulfamethoxazole/trimethoprim treatment after 90 days of treatment. Thus,the fungal burden recovery was greater after treatment with empty nanoparticles than after any of the loaded nanoparticles of PLGA (containing 1, 5 or 10 µg·50 µL−1 of P10 peptide; Figure 1).
Cytokine production induced by P10-PLGA
Cytokine production was examined by monitoring the levels of IFN-γ, IL-4, IL-10 and IL-12 levels in lung tissue homogenates from the animals given the combined therapy of sulfamethoxazole/trimethoprim and peptide P10.
The data presented in Table 1 show the production of type 1 cytokines IL-12 and IFN-γ. The treatment with 20 µg of P10 emulsified in Freund's adjuvant or 1 µg of P10 entrapped within PLGA induced higher levels of IFN-γ than in the respective controls after 30 and 90 days of the treatment. IFN-γ is an important Th1 cytokine, which elicits protection against P. brasiliensis infection. The levels of IL-12 were not similarly affected. Table 1 also shows changes in the cytokines more characteristic of Th2 responses (IL-4 and IL-10). After 30 and 90 days of starting treatment, the group that received sulfamethoxazole/trimethoprim and 20 µg·50 µL−1 of ‘free’ P10 emulsified in Freund's adjuvant showed a significant decrease of IL-10, thus confirming earlier results. P10 entrapped within PLGA at 1 µg also reduced IL-10 after 30 days and less after 90 days. Decrease of IL-10 with increasing P10 amounts in PLGA was less evident, and no decrease was observed in the levels of IL-4.
Table 1.
Determination of cytokine production (pg·mL−1) on days 30 and 90 after starting treatment with sulfamethoxazole/trimethoprim and P10
|
Treatment Groups |
Th1 |
Th2 |
||||||
|---|---|---|---|---|---|---|---|---|
|
IL-12 |
IFN-γ |
IL-4 |
IL-10 |
|||||
| 30 | 90 | 30 | 90 | 30 | 90 | 30 | 90 | |
| PBS | 11.2 ± 2.5 | 6.2 ± 2.4 | 3.2 ± 1.1 | 1.8 ± 0.6 | 13.9 ± 0.5 | 11.4 ± 6.0 | 2.5 ± 0.7 | 3.7 ± 0.4# |
| Sulfa + Trim | 7.3 ± 0.7 | 6.4 ± 0.1 | 2.3 ± 0.3 | 2.5 ± 0.1 | 11.4 ± 2.6 | 11.2 ± 0.3 | 4.1 ± 0.2 | 3.7 ± 0.2# |
| PLGA/Sulfa + Trim | 6.7 ± 0.7 | 6.8 ± 2.0 | 1.3 ± 0.4 | 1.8 ± 0.2 | 10.7 ± 0.4 | 8.6 ± 0.2 | 2.8 ± 0.9 | 3.2 ± 0.4 |
| Sulfa + Trim/Freund/P10: 20 µg | 10.9 ± 1.0 | 6.1 ± 1.9 | 7.5 ± 1.0 | 10.6 ± 2.4* | 12.7 ± 1.2 | 9.7 ± 0.9 | 0.6 ± 0.2 | 0.6 ± 0.1 |
| Sulfa + Trim/PLGA/P10: 1 µg | 7.0 ± 1.9 | 5.5 ± 2.7 | 5.1 ± 0.8 | 7.7 ± 1.1* | 11.6 ± 0.5 | 15.1 ± 1.2 | 0.8 ± 0.3 | 2.1 ± 0.3 |
| Sulfa + Trim/PLGA/P10: 5 µg | 8.4 ± 2.5 | 8.7 ± 3.5 | 1.0 ± 0.1 | 2.9 ± 1.2 | 17.9 ± 6.7 | 12.1 ± 2.2 | 2.0 ± 0.8 | 2.0 ± 0.5 |
| Sulfa + Trim/PLGA/P10: 10 µg | 7.6 ± 3.4 | 13.4 ± 7.6 | 2.9 ± 0.5 | 3.0 ± 0.3 | 14.8 ± 0.5 | 17.8 ± 0.3 | 2.5 ± 0.4 | 1.9 ± 0.3# |
Not significant between groups,
different for 10 µg P10-PLGA compared with PBS and sulfamethoxazole/trimethoprim groups (P < 0.05).
Values are means ± standard deviations of measurements from each group.
Bold: show high levels of IFN-γ compared with the group treated with sulfamethoxazole/trimethoprim. These two groups also showed lower CFUs (see Figure 1).
Italic: decrease in the production of IL-10 in the groups treated with free P10 and encapsulated P10 (1 µg) compared with the group treated with sulfamethoxazole/trimethoprim.
CFUs, colony-forming units; IFN-gamma, interferon-γ; IL, interleukin; PBS, phosphate-buffered saline; PLGA, poly(lactic acid-glycolic acid); Sulfa + Trim, sulfamethoxazole/trimethoprim.
Discussion
In the present work, we examined the antifungal effects and the protection elicited by a therapy using sulfamethoxazole/trimethoprim combined with the immunostimulatory peptide P10 encapsulated into PLGA-dimercaptosuccinic acid (DMSA) for treatment of experimental PCM. The therapeutic efficacy and cytokine production were examined in a murine model of chronic PCM. Sulfamethoxazole/trimethoprim is a low-cost sulphonamide treatment that has been used in the initial therapy of non-disseminated cases of PCM (Lortholary et al., 1999; Travassos et al., 2008b) and it is recommended for long-term treatments, but several cases of fungal resistance and relapsing disease have been reported (Travassos et al., 2008a).
The development of immunotherapies using peptides as adjuvant molecules is a promising procedure that could be used to improve chemotherapy of fungal diseases. The use of P10 peptide has been effective in reducing the fungal burden in animals infected with P. brasiliensis, thus eliciting an efficient cellular immune response to combat the fungus (Taborda et al., 1998). The association of P10 with sulfamethoxazole/trimethoprim was effective in treating PCM and in avoiding the time-dependent relapse of the mycosis in the murine model (Marques et al., 2006). The disadvantage of the use of the free peptide in vivo could be its lack of metabolic stability and the requirement for adjuvants not allowed for human use. A multiple antigenic peptide (MAP) construct carrying four truncated P10 branches has been synthesized and shown to be protective without additional adjuvants. However, the chemical synthesis of MAPs is a complex procedure, and it is subject to errors during the process, making their purification and characterization difficult (Taborda et al., 2004). Thus, efforts to prepare simpler and more efficient delivery systems for immunostimulating peptides are still needed.
In the present work, PLGA-DMSA acid nanoparticles containing P10 in amounts ranging from 1 µg·50 µL−1 to 40 µg·50 µL−1 were used combined with sulfamethoxazole/trimethoprim. The use of PLGA-based formulations for the encapsulation of these molecules aimed at reducing the frequency of injections and also protecting the peptide against in vivo degradation while helping drug administration (Amaral et al., 2009). Nanoparticles composed of PLGA are appropriate devices for delivering the peptide due to the low rate of co-polymer degradation and a constant release of the peptide, which would be better at stimulating the immune cells. The proportion of PLA and PGA (50:50) used in this type of drug delivery system presented a sustained release of the active component over an average time of 72 h, as observed in other studies (Mittal et al., 2007; Amaral et al., 2009). Nanoparticulated PLGA as a drug delivery system has been used for some time based on its high stability, ready uptake into the cell by endocytosis and targeting of specific organs, with many different mixtures of PLA and PGA (Dhiman et al., 2000). There is no toxicity associated with this drug delivery system by itself and its use has been approved by drug regulatory agencies, such as the Food and Drug Administration (Khang et al., 2003; Gabler et al., 2007). In vitro tests using different cell lines showed no toxicity for nanoparticulated PLGA (Khandare and Minko, 2006; Gomes et al., 2008). PLGA by itself does have the advantage of eliciting cytotoxic T-cell responses and also of inducing a mild inflammatory response, which could be involved in its adjuvant characteristics (Jiang et al., 2005).
In our study, incorporating peptide P10 into PLGA reduced the amount of this peptide necessary to decrease the fungal load in the infected animals and avoid disease relapse. A small amount (1 µg·50 µL−1) of P10 in nanoparticles had the same immunotherapeutic effect as 20 µg·50 µL−1 of ‘free’ P10 emulsified in Freund's adjuvant over the first 30 days of treatment. However, the best protective effect of the PLGA-encapsulated peptide after 90 days of treatment was shown by the groups that received 5 or 10 µg·50 µL−1 of P10 in PLGA, making it possible to reduce the amount of the peptide by at least fourfold and preserving the protective capacity to avoid relapse of the infection. Based on a previous report (Johansen et al., 2000), the entrapment of P10 within PLGA nanoparticles probably effectively protected it from in vivo enzymatic destruction, which could explain the effectiveness of the lower doses of P10-PLGA.
Interaction of PLGA with antigen-presenting cells could underlie the improvement of immunomodulatory effects (Jaganathan et al., 2005). Further, the use of PLGA for controlled release of the peptide eliminates the need for an adjuvant, which becomes an advantage as only a limited number of adjuvants are accepted for human administration (Jiang et al., 2005). The protection elicited by 20 µg·50 µL−1 of ‘free’ P10 in Freund's adjuvant as well as P10-PLGA at 1 µg·50 µL−1 most probably depended on the induction of the high levels of IFN-γ, as noted in previous studies (Marques et al., 2006). Although the 10 µg P10-PLGA treatment showed low IFN-γ and high IL-10 cytokine values after 90 days of therapy, it still managed to have a good protective effect. This may be explained by the maintenance of high levels of IL-12 and low production of IL-10 at 90 days, almost twofold lower than that produced in PBS and Sulfa + Trim-treated groups (Table 1), which could compensate for the low IFN-γ level noted for this group. Most patients with the severe form of PCM present a polarized Th2 response, with production of IL-4 and IL-10 (Oliveira et al., 2002). In contrast, individuals exhibiting a polarized Th1 response, with production of IFN-γ, tend to resolve P. brasiliensis infections (Oliveira et al., 2002; Calich et al., 2008).
In conclusion, the use of PLGA as a carrier for peptide P10 in combination with sulfamethoxazole/trimethoprim represents a promising alternative to treat mycosis, as P10 elicits a Th1-like immune response able to control fungal infection. Sulfamethoxazole/trimethoprim is an inexpensive medicine, and its therapeutic efficacy can be increased by the cellular immunity-stimulating P10 peptide. Encapsulation of peptide P10 within PLGA reduced the amount of this peptide needed for a therapeutic response, up to 20-fold and avoided the use of an adjuvant. Additionally, in terms of application to therapy in humans, use of the PLGA nanoparticles should lead to improved antifungal protection and, at the same time, to a reduction in costs.
Acknowledgments
The authors wish to thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for their financial support. Also, ACA was supported by a PhD fellowship from CNPq.
Glossary
Abbreviations:
- CFUs
colony-forming units
- DMSA
dimercaptosuccinic acid
- Gp43
43 kDa glycoprotein
- IFN-γ
interferon-gamma
- IL-4
interleukin-4
- IL-10
interleukin-10
- IL-12
interleukin-12
- MAP
multiple antigenic peptide
- P10
peptide P10
- PBS
phosphate-buffered saline
- PCM
paracoccidioidomycosis
- PGA
poly-glycolic acid
- PLA
poly-lactic acid
- PLGA
poly(lactic acid-glycolic acid)
Conflicts of interest
None to declare.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral AC, Bocca AL, Ribeiro AM, Nunes J, Peixoto DLG, Simioni AR, et al. Amphotericin B in poly(lactic-co-glycolic acid) (PLGA) and dimercaptosuccinic acid (DMSA) nanoparticles against paracoccidioidomycosis. J Antimicrob Chemother. 2009;63:526–533. doi: 10.1093/jac/dkn539. [DOI] [PubMed] [Google Scholar]
- Brummer E, Castaneda E, Restrepo A. Paracoccidioidomycosis: an update. Clin Microbiol Rev. 1993;6:89–117. doi: 10.1128/cmr.6.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calich VLG, Costa TA, Felonato M, Arruda C, Bernardino S, Loures FV, et al. Innate immunity to Paracoccidioides brasiliensis infection. Mycopathologia. 2008;165:223–236. doi: 10.1007/s11046-007-9048-1. [DOI] [PubMed] [Google Scholar]
- Commandeur S, van Beusekom HM, van der Giessen WJ. Polymers, drug release and drug-eluting stents. J Interv Cardiol. 2006;19:500–506. doi: 10.1111/j.1540-8183.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- Cutler JE, Deepe GS, Klein BS. Advances in combating fungal diseases: vaccines on the threshold. Nat Rev Microbiol. 2007;5:13–28. doi: 10.1038/nrmicro1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Camargo ZP, de Franco MF. Current knowledge on pathogens and immunodiagnosis of paracoccidioidomycosis. Rev Iberoam Micol. 2000;17:41–48. [PubMed] [Google Scholar]
- Dhiman N, Khuller GK. Protective efficacy of mycobacterial 71-kDa cell wall associated protein using poly (DL-lactide-co-glycolide) microparticles as carrier vehicles. FEMS Immunol Med Microbiol. 1998;21:19–28. doi: 10.1111/j.1574-695X.1998.tb01145.x. [DOI] [PubMed] [Google Scholar]
- Dhiman N, Dutta M, Khuller GK. Poly (DL-lactide-co-glycolide) based delivery systems for vaccines and drugs. Indian J Exp Biol. 2000;38:746–752. [PubMed] [Google Scholar]
- Gabler F, Frauenschub S, Ringe J, Brochhausen C, Götz P, Kirkpatrick CJ, et al. Emulsion-based synthesis of PLGA-microspheres for the in vitro expansion of porcine chondrocytes. Biomol Eng. 2007;24:515–520. doi: 10.1016/j.bioeng.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Gomes AJ, Barbougli PA, Espreafico EM, Tfouni E. trans-[Ru(NO)(NH3)4(py)](BF4)3.H2O encapsulated in PLGA microparticles for delivery of nitric oxide to B16-F10 cells: cytotoxicity and phototoxicity. J Inorg Biochem. 2008;102:757–766. doi: 10.1016/j.jinorgbio.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, et al. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Jaganathan KS, Rao YUB, Singh P, Prabakaran D, Gupta S, Jain A, et al. Development of a single dose tetanus toxoid formulation based on polymeric microspheres: a comparative study of poly(D,l-lactic-co-glycolic acid) versus chitosan microspheres. Int J Pharm. 2005;294:23–32. doi: 10.1016/j.ijpharm.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57:475–482. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Johansen P, Gander B, Merkle HP, Sesardic D. Ambiguities in the preclinical quality assessment of microparticulate vaccines. Trends Biotecnol. 2000;18:203–211. doi: 10.1016/s0167-7799(00)01437-2. [DOI] [PubMed] [Google Scholar]
- Khandare J, Minko T. Polymer-drug conjugates: progress in polymeric prodrugs. Prog Polym Sci. 2006;31:359–397. [Google Scholar]
- Khang G, Rhee JM, Jeong JK, Lee JS, Kim MS, Cho SH. Local drug delivery system using biodegradable polymers. Macromol Res. 2003;11:207–223. [Google Scholar]
- Lortholary O, Denning DW, Dupont B. Endemic mycoses: a treatment update. J Antimicrob Chemother. 1999;43:321–331. doi: 10.1093/jac/43.3.321. [DOI] [PubMed] [Google Scholar]
- Marques AF, da Silva MB, Juliano MAP, Travassos LR, Taborda CP. Peptide immunization as an adjuvant to chemotherapy in mice challenged intratracheally with virulent yeast cells of Paracoccidioides brasiliensis. Antimicrob Agents Chemother. 2006;50:2814–2819. doi: 10.1128/AAC.00220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal G, Sahana DK, Bhardwaj V, Ravi Kumar MN. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behaviour in vitro and in vivo. J Control Release. 2007;119:77–85. doi: 10.1016/j.jconrel.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Oliveira SJ, Mamoni RL, Musatti CC, Papaiordanou PM, Blotta MH. Cytokines and lymphocyte proliferation in juvenile and adult forms of paracoccidioidomycosis: comparison with infected and non-infected controls. Microbes Infect. 2002;4:139–144. doi: 10.1016/s1286-4579(01)01521-0. [DOI] [PubMed] [Google Scholar]
- Puccia R, Schenkman S, Gorin PA, Travassos LR. Exocellular components of Paracoccidioides brasiliensis: identification of a specific antigen. Infect Immun. 1986;53:199–206. doi: 10.1128/iai.53.1.199-206.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo A, Arango MD. In vitro susceptibility testing of Paracoccidioides brasiliensis to sulfonamides. Antimicrob Agents Chemother. 1980;18:190–194. doi: 10.1128/aac.18.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer-Vermes LM, Ciavaglia MC, Kashino SS, Burger E, Calich VL. The source of the growth-promoting factor(s) affects the plating efficiency of Paracoccidioides brasiliensis. J Med Vet Mycol. 1992;30:261–264. doi: 10.1080/02681219280000331. [DOI] [PubMed] [Google Scholar]
- Taborda CP, Juliano MA, Puccia R, Franco M, Travassos LR. Mapping of the T-Cell epitope in the major 3-Kilodalton glycoprotein of Paracoccidioides brasiliensis which induces a Th-1 response protective against fungal infection in BALB/c mice. Infect Immun. 1998;66:786–793. doi: 10.1128/iai.66.2.786-793.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taborda CP, Nakaie CR, Cilli EM, Rodrigues EG, Silva LS, Franco MF, et al. Synthesis and immunological activity of a branched peptide carrying the T-cell epitope of gp43, the major exocellular antigen of Paracoccidioides brasiliensis. Scan J Immunol. 2004;59:58–65. doi: 10.1111/j.0300-9475.2004.01359.x. [DOI] [PubMed] [Google Scholar]
- Travassos LR, Taborda CP, Iwai LK, Cunha-Neto E, Puccia R. The gp43 from Paracoccidioides brasiliensis: a major diagnostic antigen and vaccine candidate. In: Domer JE, Kobayashi GS, editors. The Mycota XII, Human Fungal Pathogens. Berlin-Heildeberg: Springer-Verlag; 2004. pp. 279–296. [Google Scholar]
- Travassos LR, Taborda CP, Colombo AL. Treatment options for paracoccidioidomycosis and new strategies investigated. Expert Rev Anti Infect Ther. 2008a;6:251–262. doi: 10.1586/14787210.6.2.251. [DOI] [PubMed] [Google Scholar]
- Travassos LR, Rodrigues EG, Iwai LK, et al. Attempts at a peptide vaccine against paracoccidioidomycosis, adjuvant to chemotherapy. Mycopathologia. 2008b;165:341–352. doi: 10.1007/s11046-007-9056-1. [DOI] [PubMed] [Google Scholar]
- Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24:39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]