

Abstract
Background and purpose:
Recent clinical guidelines advocate the use of the isosorbide dinitrate/hydralazine combination in treatment for heart failure. However, clinical and laboratory evidence suggest that some vasodilators may induce cardiac hypertrophy under uncertain conditions. This study investigated the effects and underlying mechanism of action of the vasodilator hydralazine on cardiac growth.
Experimental approach:
Wild-type mice and animals deficient in guanylyl cyclase-A (GCA) and/or angiotensin receptors (AT1 and AT2 subtypes) were treated with hydralazine (≈24 mg·kg−1·day−1 in drinking water) for 5 weeks. Cardiac mass and/or cardiomyocyte cross-sectional area, fibrosis (van Giessen-staining) and cardiac gene expression (real-time RT-PCR) were measured.
Key results:
Hydralazine lowered blood pressure in mice of all genotypes. However, this treatment increased the heart and left ventricular to body weight ratios, as well as cardiomyocyte cross-sectional area, and cardiac expression of atrial natriuretic peptide mRNA in mice lacking GCA. Hydralazine did not affect cardiac hypertrophy in wild-type mice and mice lacking either AT1 or AT2 receptors alone. However, the pro-hypertrophic effect of hydralazine was prevented in mice lacking both GCA and AT2, but not GCA and AT1 receptors. However, hydralazine did decrease cardiac collagen deposition and collagen I mRNA (signs of cardiac fibrosis) in mice that were deficient in GCA, or both GCA and AT2 receptors.
Conclusions and implications:
The vasodilator hydralazine induced AT2 receptor-mediated cardiomyocyte growth under conditions of GCA deficiency. However, attenuation of cardiac fibrosis by hydralazine could be beneficial in the management of cardiac diseases.
Keywords: angiotensin; receptor; vasodilator, hydralazine; natriuretic peptide; hypertrophy; heart
Introduction
Despite the advances in pharmacotherapy for chronic heart failure due to reduced left ventricular function, mortality still remains high and many patients are hospitalized because of worsening symptoms of heart failure. Hydralazine is an established antihypertensive drug that exerts its actions via arterial dilation. Hydralazine is not used as a primary drug for treating hypertension because it also elicits sympathetic stimulation and salt retention, which may lead to the development of congestive heart failure, and increases plasma renin activity (Gerber and Nies, 1990). However, concomitant use of hydralazine together with a venodilatory nitrate has been shown to prevent the development of nitrate tolerance and to maintain the favourable haemodynamic effect of nitrates in animals (Bauer and Fung, 1991) and in patients (Gogia et al., 1995) with congestive heart failure. Recent findings from the African American Heart Failure Trial (A-HeFT) have demonstrated a survival benefit in African-American patients with chronic congestive heart failure treated with BiDil (NitroMed; Lexington, MA, USA), a fixed-dose combination of isosorbide dinitrate and hydralazine (Taylor et al., 2004). As a consequence, recent clinical guidelines advocate the isosorbide dinitrate/hydralazine combination as ‘a reasonable option’ in the treatment of patients with advanced, but stable heart failure and who remain symptomatic despite optimal standard therapy (Hunt et al., 2005; 2009a,b; Heart Failure Society of America, 2006).
It has been reported that long-term treatment with arterial vasodilators such as hydralazine and minoxidil is associated with progression (or absence of regression) of cardiac hypertrophy in spontaneously hypertensive rats, as well as in other models of hypertension [e.g. two-kidney, one-clip (2K1C)] and in normotensive rats (Sen et al., 1974; 1977; Pegram et al., 1982; Fenje and Leenen, 1985; Tsoporis and Leenen, 1986; Leenen and Prowse, 1987). In the clinic, it has been also reported that long-term treatment with hydralazine increased left ventricular mass in patients with chronic asymptomatic aortic regurgitation (Kleaveland et al., 1986). The underlying molecular mechanisms, however, are still unclear.
Myocardial hypertrophy is prevalent in a substantial portion of individuals with essential hypertension, and it is generally accepted as an independent risk factor for congestive heart failure and sudden cardiac death. Recently, several lines of evidence have suggested that activity of an isoform of guanylyl cyclase (GC), GCA, which is also the receptor for atrial natriuretic peptide (ANP), could be impaired in a subpopulation of patients with essential hypertension and heart failure (Tsutamoto et al., 1992; 1993; Nakayama et al., 2000; Rubattu et al., 2006; Usami et al., 2008). Moreover, mice lacking GCA also exhibit hypertension, cardiac hypertrophy and fibrosis and sudden death (Lopez et al., 1995; Oliver et al., 1997). Although heart failure has not been reported under basal conditions, the incidence of heart failure was more pronounced in mice genetically deficient in GCA [GCA knockout (KO) mice] than in wild-type (WT) mice, after myocardial infarction induced by ligation of the left coronary artery (Nakanishi et al., 2005). This finding is consistent with increased susceptibility to heart failure in GCA deficiency. Recently, we have also demonstrated that genetic or pharmacological blockade of angiotensin AT1 receptors (Li et al., 2002) or AT2 receptors (Li et al., 2009) attenuated cardiac hypertrophy in GCA KO mice, indicating that both AT1 and AT2 receptors mediate hypertrophy under conditions of GCA deficiency.
Cardiac hypertrophy may be an adaptive response of the heart to haemodynamic overload, as in hypertension. To evaluate the contribution of increased blood pressure to cardiac hypertrophy and fibrosis, GCA KO mice were treated with hydralazine. Unexpectedly, we found that, despite its antihypertensive actions, hydralazine potentiated cardiac hypertrophy in GCA KO mice. We further investigated the underlying mechanism of this effect by genetically deleting AT1 or AT2 receptors.
Methods
Animals and treatments
The animal care and all experimental protocols were reviewed and approved by the Animal Research Committee in Kyoto University Graduate School of Medicine.
Experiment 1
Male homozygous GCA KO and WT mice used in this experiment (n = 7–14 each group) were generated by methods described previously (Lopez et al., 1995). The genetic background of the mice was C57BL/6. Treatments were started when mice were 12 weeks of age. Hydralazine (Sigma, Osaka, Japan) was administered in drinking water (≈24 mg·kg−1·day−1) over a 5 week period; the solution was replaced on alternate days. Control mice received drinking water alone. Systolic blood pressure (SBP) was measured prior to commencement and at weekly intervals during the 5 week experiment after which animals were killed.
Experiment 2
The genetic background of the mice lacking the AT1a gene (AT1 KO) mice was C57BL/6. Male homozygous AT1 KO mice and mice with both GCA and AT1 KO (‘double’ KO; GCA/AT1 DKO) used in this experiment (n = 6−7 each group) were generated by methods described previously (Li et al., 2002; 2004;). The other experimental procedures were the same as those described in Experiment 1.
Experiment 3
The genetic background of the AT2-deficient (AT2 KO) mice was FVB/N. Male homozygous AT2 KO and GCA and AT2 double KO (GCA/AT2 DKO) mice used in this experiment (n = 7–8 each group) were generated by methods described previously (Li et al., 2009). The other experimental procedures were the same as those described in Experiment 1.
Heart rate and SBP measurement
Heart rate (HR) and SBP were measured in conscious mice using a computerized tail-cuff method (Softron, Co., Ltd., Tokyo, Japan) at 10:00–14:00. The validity of this system has been established in our laboratory (Li et al., 2002; 2004; 2009; Nakanishi et al., 2005).
Determination of heart and left ventricular weights
Hearts were dissected and the weights of the total heart (HW) and left ventricle (LVW) were measured. The ratios of HW or LVW to the body weight (HW/BW and LVW/BW) were calculated and used as an index of cardiac hypertrophy.
Measurement of cardiomyocyte cross-sectional areas
A section from each left ventricle was fixed in 10% neutral formalin over several days and dehydrated with graded concentrations of alcohol for embedding in paraffin. Paraffin slices from each heart were stained with haematoxylin-eosin. Morphometry of left ventricular myocytes was performed according to the method described in previous reports (Sanada et al., 2003). The cross-sectional area was measured using an image analysing system (KS 400 Imaging System; Carl Zeiss Vision, Eching, Germany) in cardiomyocytes that were cut transversely and had a visible nucleus and an unbroken cellular membrane. The outer borders of the cardiomyocytes (original magnification: ×400) were traced and the cardiomyocyte areas were calculated. One hundred cells per left ventricle were counted, and the averaged value was used for analysis.
Determination of cardiac fibrosis
To determine the extent of collagen fibre accumulation, paraffin slices from each heart were subjected to van Giessen-staining. Forty fields in three individual sections were selected randomly and the ratio of the areas of van Giessen-stained interstitial fibrosis to the total left ventricular area was calculated using image analysis software and a Zeiss KS400 system (Carl Zeiss Vision, Eching, Germany) (Li et al., 2002; 2004;).
Analysis of gene expression
Total RNA was isolated from left ventricles with the TRIzol reagent (Life Technologies Inc., Rockville, MD, USA). Expression of mRNAs encoding ANP and collagen I was evaluated using real-time RT-PCR in an ABI PRISM™ 7700 Sequence Detector (Applied Biosystems, Foster city, CA, USA). The primers and probes of the genes examined were as follows: ANP: sense, 5′-GCCATATTGGAGCAAATCCT-3′; antisense, 5′-GCAGGTTCTTGAAATCCATCA-3′; oligonucleotide probe, 5′-TGTACAGTGCGGTGTCCAACACAGAT-3′; collagen I: sense, 5′-GTCCCAACCCCCAAAGAC-3′; antisense, 5′-CATCTTCTGAGTTTGGTGATACGT-3′; oligonucleotide probe, 5′-CACGGCTGTGTGCGATGACG-3′. To verify that equal amounts of mRNA were subjected to real-time RT-PCR, GAPDH mRNA was also amplified using the same method with specific primers and probe (Applied Biosystems, Hammonton, NJ, USA).
Statistical analysis
All data are expressed as means ± SEM of values obtained in individual animals. Data were analysed by single-factor anova. If a significant effect was found, the Student-Newman-Keuls test was performed to isolate the difference between the groups. Values of P < 0.05 were considered statistically significant.
Results
HR, SBP, cardiac mass, collagen accumulation and expression of hypertrophic and fibrogenic genes in WT and GCA KO mice
The antihypertensive agent hydralazine was used to assess the role of blood pressure in the development of cardiac hypertrophy in GCA-deficient mice. HR was not different between the four groups of WT and GCA KO mice at baseline (data not shown). Consistent with enhancement of cardiac sympathetic activity (Tsoporis and Leenen, 1988), hydralazine (≈24 mg·kg−1·day−1 in drinking water) significantly increased HR in WT and GCA KO mice over the course of the experiment, but there was no difference between the treated groups (data not shown). In contrast, SBP was increased in GCA KO mice compared with WT mice (Figure 1A). In accord with previous findings, hydralazine elicited a potent and stable antihypertensive effect in WT mice (Figure 1A). Although hydralazine also lowered SBP in GCA KO mice after 1 week of treatment, its efficacy diminished over the course of the 5 week experimental period.
Figure 1.

Systolic blood pressure (SBP, A) and the ratios of heart weight to body weight (HW/BW, B) and left ventricular weight to body weight (LVW/BW, C) in wild-type (WT) and guanylyl cyclase-A-deficient [GCA knockout (KO) mice] mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. SBP was measured in conscious mice prior to, and at weekly intervals after, the commencement of treatment using a computerized tail-cuff method. Hearts and left ventricles were weighed at the conclusion of the experiments (week 5), and HW/BW and LVW/BW were calculated. Values are means ± SEM (n = 7−14). *P < 0.05; $different from WT control (HYD −) or #different from GCA KO (HYD −) at corresponding time-points (P < 0.05). HYD −: control, without HYD; HYD +, with HYD.
Hydralazine did not alter BW in mice (WT control: 33.9 ± 1.4 g; WT + hydralazine: 32.4 ± 0.8 g; GCA KO control: 33.9 ± 2.1g; GCA KO + hydralazine: 34.4 ± 1.0 g). The HW/BW (Figure 1B) and the LVW/BW (Figure 1C) ratios were higher in GCA KO than in WT mice. The cross-sectional area of cardiac myocytes was also increased in GCA KO mice (Figure 2). Somewhat surprisingly, treatment with hydralazine over 5 weeks markedly increased the ratios of HW/BW and LVW/BW, and the cross-sectional area in GCA KO mice, but not in WT mice (Figures 1B,C and 2A).
Figure 2.
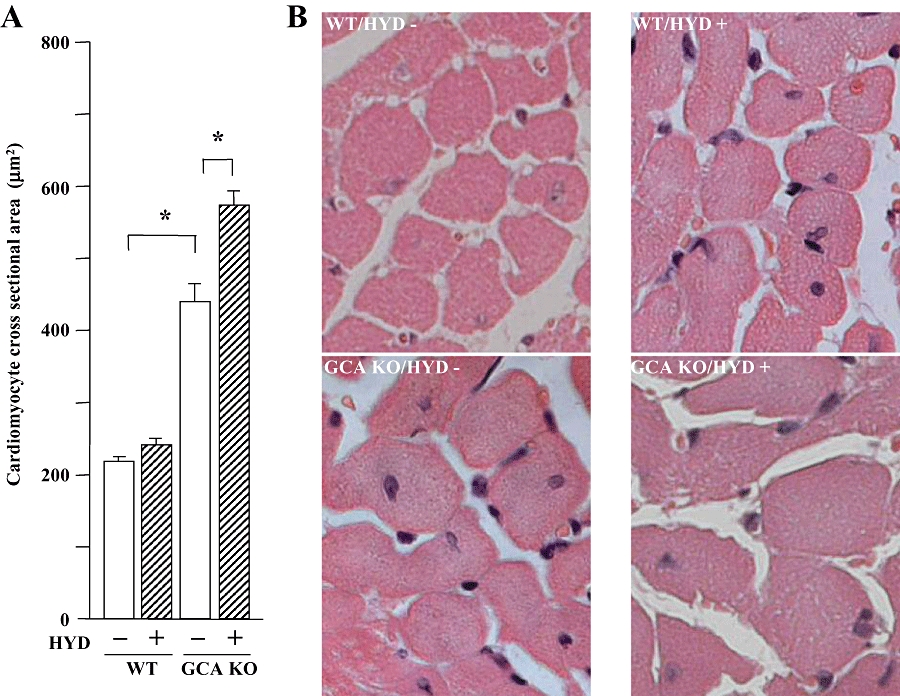
Cardiomyocyte cross-sectional areas in wild-type (WT) and guanylyl cyclase-A (GCA) knockout (KO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. Morphometry of left ventricular myocytes was performed to measure the myocyte cross-sectional area. Values are means ± SEM (n = 7−14). *P < 0.05. (A) Results of cardiomyocyte cross-sectional areas with or without HYD treatment. (B) Representative histological findings of cardiomyocytes in different experimental groups. Original magnification: ×400. HYD −: control, without HYD; HYD +, with HYD.
There was a pronounced increase in cardiac interstitial van Giessen-staining area, as a measure of fibrosis, in GCA KO mice compared with WT (Figure 3). In contrast to the effect on HW/BW and LVW/BW, hydralazine selectively decreased the extent of van Giessen-staining in cardiac sections from GCA KO mice.
Figure 3.
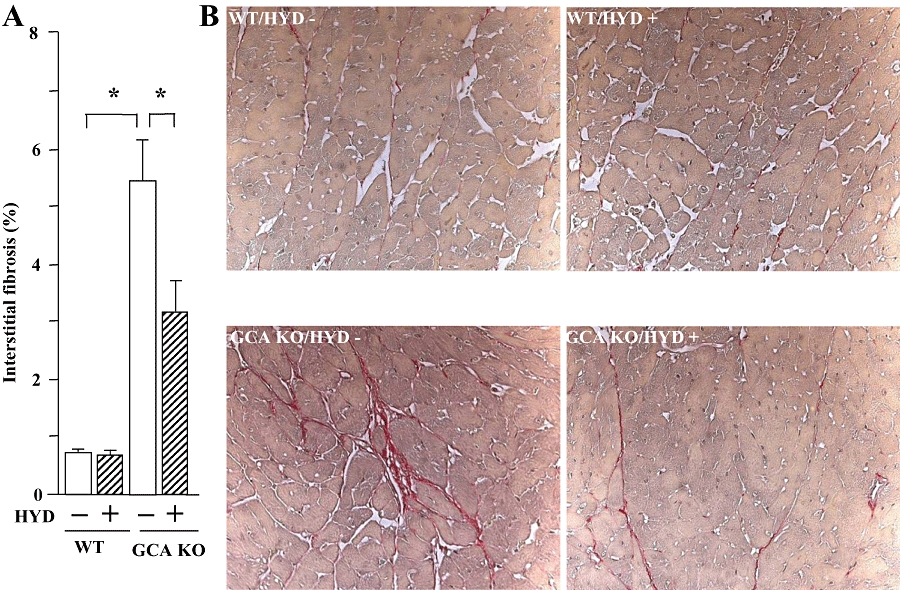
Cardiac fibrosis in wild-type (WT) and guanylyl cyclase-A (GCA) knockout (KO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. After van Giessen-staining the area of collagen deposition in the interstitial region and the total left ventricular area were quantified using an image analysing system. (A) Interstitial fibrosis (%, the ratio of the area of interstitial collagen accumulation to the total left ventricular area); (B) Representative examples of interstitial fibrosis (red) (original magnification: ×200). Values are means ± SEM (n = 7–14). *P < 0.05. HYD −: control, without HYD; HYD +, with HYD.
Consistent with the phenotypic change in cardiac mass, cardiac expression of the mRNA for ANP, an important molecular marker of cardiac hypertrophy, was greater in GCA KO mice than in WT controls (Figure 4A). The increase in cardiac ANP expression suggests that the GCA deficiency-induced cardiac hypertrophy may not be compensatory, but may be a component of a decompensation process. Hydralazine treatment further enhanced cardiac expression of mRNA for ANP in GCA KO mice, but induced no change in WT animals (P > 0.05). In contrast, the up-regulation of cardiac collagen I mRNA (the most important collagen isoform in heart) in GCA KO mice was suppressed by hydralazine treatment (Figure 4B). Hydralazine did not affect collagen I mRNA expression in WT mice.
Figure 4.
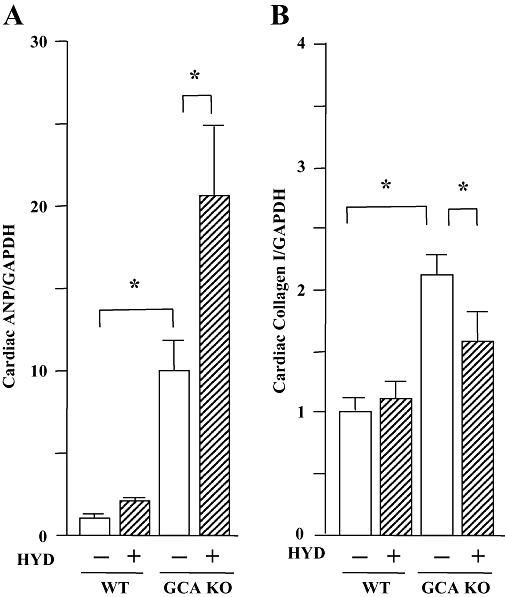
Left ventricular expression of mRNA for atrial natriuretic peptide (ANP, A) or collagen I (B) in wild-type (WT) and guanylyl cyclase-A (GCA) knockout (KO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. Total RNA was extracted from the left ventricular tissues using TRIzol. The relative levels of specific mRNAs were assessed by real-time RT-PCR. Results were normalized to GAPDH. Levels in WT group were arbitrarily assigned a value of 1. Values are means ± SEM (n = 7). *P < 0.05. HYD −: control, without HYD; HYD +, with HYD.
HR, SBP, cardiac mass, collagen accumulation and expression of cardiac hypertrophic and fibrogenic genes in AT1 receptor KO and in GCA and AT1 receptor DKO mice
The potential role of AT1 receptors in the exacerbation of cardiac hypertrophy in GCA KO mice by hydralazine was assessed. As found in WT and GCA KO mice, there was no difference in basal HR between the four groups of AT1 receptor KO and GCA/AT1 DKO mice (data not shown). Hydralazine significantly increased HR in AT1 KO and GCA/AT1 DKO mice over the experimental time course, but there was no difference between the treated groups (data not shown). SBP was considerably lower in untreated AT1 KO and GCA/AT1 DKO mice (Figure 5A) than in corresponding WT and GCA KO mice (Figure 1A) respectively. However, basal SBP in GCA/AT1 DKO mice was still higher than that in AT1 KO mice. Hydralazine treatment further decreased SBP in both AT1 KO and AT1/GCA DKO mice.
Figure 5.
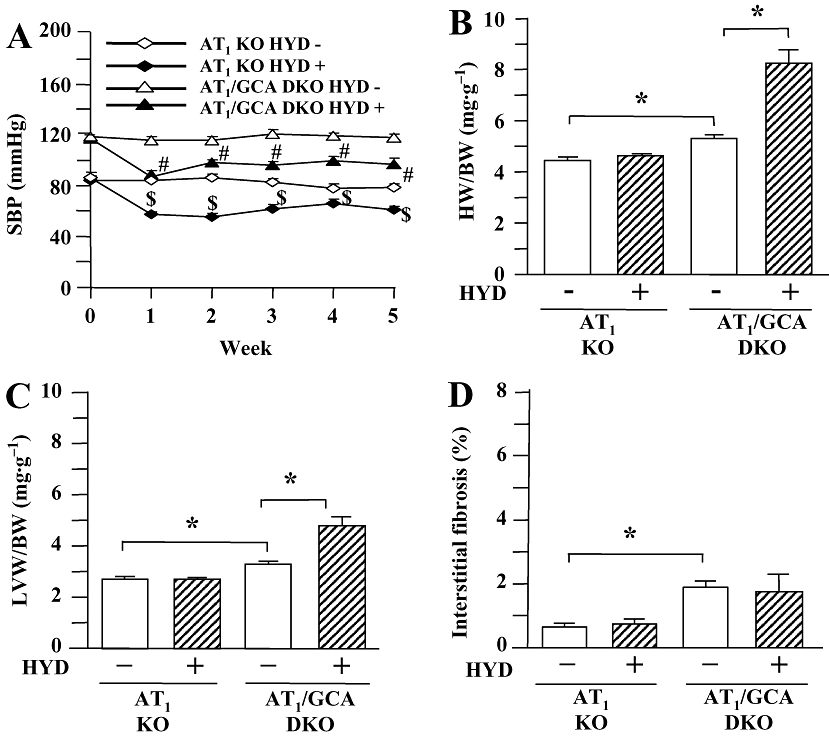
Systolic blood pressure (SBP, A), the ratios of heart weight to body weight (HW/BW, B) and left ventricular weight to body weight (LVW/BW, C) and cardiac interstitial fibrosis (D) in angiotensin II type 1 (AT1) receptor knockout (KO) and guanylyl cyclase-A (GCA)/AT1 double KO (DKO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. SBP was measured in conscious mice prior to, and at weekly intervals after, the commencement of treatment using a computerized tail-cuff method. Hearts and left ventricles were weighed at the conclusion of the experiments (week 5), and HW/BW and LVW/BW were calculated. After van Giessen-staining the area of collagen deposition on the interstitial region and the total left ventricular area were quantified using an image analysing system. Values are means ± SEM (n = 6−7). *P < 0.05; $different from AT1 KO (HYD −), or #different from GCA/AT1 DKO (HYD −) at corresponding time-points (P < 0.05). HYD −: control, without HYD; HYD +, with HYD.
Hydralazine treatment was without effect on BW in both genotypes (AT1 receptor KO control: 36.0 ± 1.3 g; AT1 KO + hydralazine: 35.3 ± 1.0 g; GCA/AT1 DKO control: 36.1 ± 1.3 g; GCA/AT1 DKO + hydralazine: 36.6 ± 1.5 g). Further, HW/BW (Figure 5B) and LVW/BW ratios (Figure 5C), as well as cardiac ANP mRNA expression (Figure 6A), were lower in untreated AT1 KO and GCA/AT1 DKO mice than untreated WT and GCA KO mice (Figures 1B,C and 4A). Hydralazine increased the HW/BW and LVW/BW ratios (Figure 5B,C) and cardiac ANP mRNA expression (Figure 6A) in GCA/AT1 DKO mice, whereas this treatment was without effect on these parameters in AT1 receptor KO mice.
Figure 6.
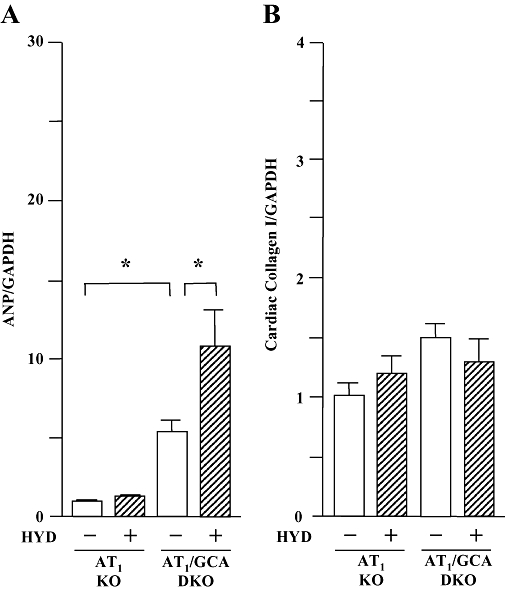
Left ventricular expression of mRNA for atrial natriuretic peptide (ANP, A) and collagen I (B) in angiotensin II type 1 (AT1) receptor knockout (KO) and guanylyl cyclase-A (GCA)/AT1 double KO (DKO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. Total RNA was extracted from the left ventricular tissues using TRIzol. The relative levels of specific mRNAs were assessed by real-time RT-PCR. Results were normalized to GAPDH. Levels in WT group were arbitrarily assigned a value of 1. Values are means ± SEM (n = 6−7). *P < 0.05. HYD −: control, without HYD; HYD +, with HYD.
Deletion of AT1 receptors decreased the extent of van Giessen-staining in cardiac interstitial sections from more than 5% (Figure 3A) to less than 2% (Figure 5D) and also decreased collagen I mRNA expression (Figure 6B) in GCA KO mice, compared with basal conditions. Hydralazine treatment did not affect residual cardiac fibrosis, collagen I mRNA expression or these parameters in AT1 KO mice (Figures 5D and 6B).
HR, SBP, cardiac mass, collagen accumulation and cardiac gene expression of hypertrophic and fibrogenic markers in AT2 receptor KO and GCA and in AT2 DKO mice
The potential role of AT2 receptors in hydralazine-induced exacerbation of the cardiac hypertrophy observed in GCA KO mice was also assessed in the present study. As we had observed in WT, GCA KO, AT1 KO and GCA/AT1 DKO mice, there was no difference in basal HR in four groups of AT2 receptor KO and GCA/AT2 DKO mice (data not shown). Hydralazine significantly increased HR in AT2 KO and GCA/AT2 DKO mice over the course of the experiment, but there was no difference between the treated groups (data not shown). Deletion of AT2 receptors did not affect SBP (Figure 7A), compared with corresponding animals that expressed AT2 (Figure 1A). Hydralazine decreased SBP in both AT2 KO and AT2/GCA DKO mice (Figure 7A) but was without effect on BW (AT2 KO control: 34.0 ± 1.3 g; AT2 KO + hydralazine: 36.2 ± 1.5 g; GCA/AT2 DKO control: 35.1 ± 1.1 g; GCA/AT2 DKO + hydralazine: 36.0 ± 1.3 g). Unlike the situation in GCA KO and GCA/ AT1 DKO mice, hydralazine treatment had no effect on the HW/BW (Figure 7B) and LVW/BW (Figure 7C) ratios. The cardiac ANP mRNA expression (Figure 8A) was not changed by hydralazine in AT2 receptor KO but suppressed in GCA/AT2 DKO mice.
Figure 7.
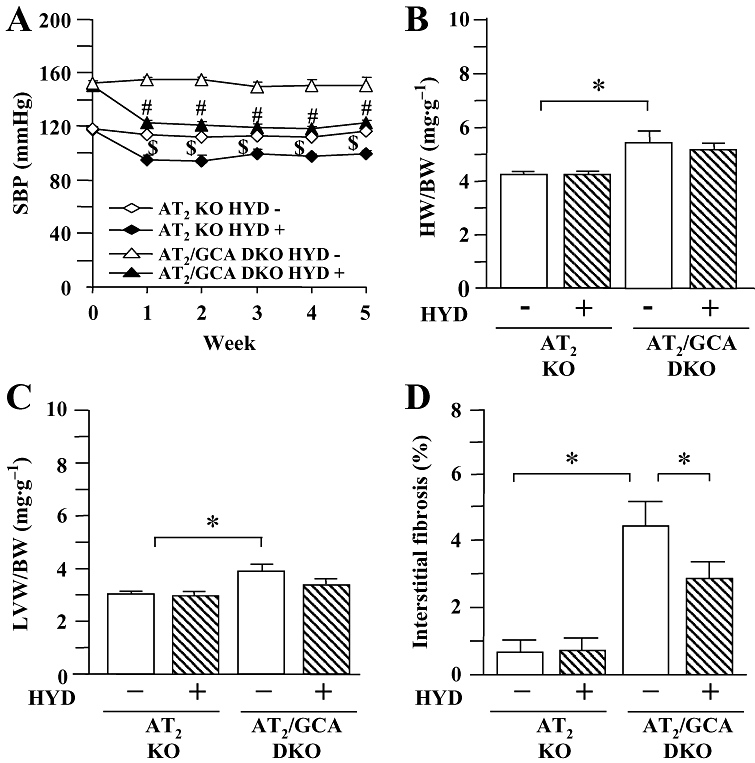
Systolic blood pressure (SBP, A), the ratios of heart weight to body weight (HW/BW, B) and left ventricular weight to body weight (LVW/BW, C) and cardiac interstitial fibrosis (D) in angiotensin II type 2 (AT2) receptor knockout (KO) and guanylyl cyclase-A (GCA)/AT2 double KO (DKO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. SBP was measured in conscious mice prior to, and at weekly intervals after, the commencement of treatment using a computerized tail-cuff method. Hearts and left ventricles were weighed at the conclusion of the experiments (week 5), and HW/BW and LVW/BW were calculated. After van Giessen-staining the area of collagen deposition on the interstitial region and the total left ventricular area were quantified using an image analysing system. Values are means ± SEM (n = 7–8). *P < 0.05; $different from AT2 KO (HYD −), or #different from GCA/AT2 DKO (HYD −) at corresponding time-points (P < 0.05). HYD −: control, without HYD; HYD +, with HYD.
Figure 8.
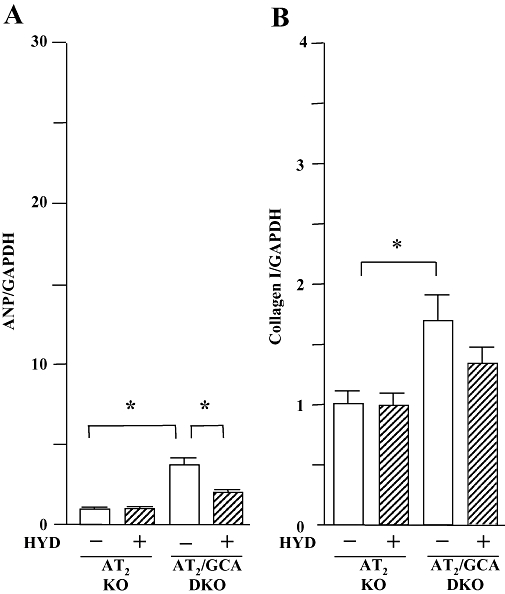
Left ventricular expression of mRNA for atrial natriuretic peptide (ANP, A) and collagen I (B) in angiotensin II type 2 (AT2) receptor and in guanylyl cyclase-A (GCA) and AT2 double knockout (DKO) mice. Hydralazine (HYD) was administered in drinking water (≈24 mg·kg−1·day−1) for 5 weeks, while the mice in control groups received drinking water only. Total RNA was extracted from the left ventricular tissues using TRIzol. The relative levels of specific mRNAs were assessed by real-time RT-PCR. Results were normalized to GAPDH. Levels in wild-type group were arbitrarily assigned a value of 1. Values are means ± SEM (n = 7–8). *P < 0.05. HYD −: control, without HYD; HYD +, with HYD.
In contrast to the effects in GCA KO mice (Figure 3A), cardiac van Giessen-staining was decreased in GCA/AT2 DKO animals (Figure 7D). Hydralazine treatment further decreased cardiac interstitial fibrosis in GCA/AT2 DKO mice, but did not affect collagen deposition in AT2 receptor KO mouse heart (Figure 7D) or collagen I mRNA expression in both genotypes (P > 0.05) (Figure 8B).
Discussion
The present results clearly demonstrated that hydralazine increased the HW/BW and LVW/BW ratios, cardiomyocyte cross-sectional area and cardiac ANP mRNA in mice lacking GCA, but not in WT mice, despite its antihypertensive actions. Thus, vasodilator therapy augmented cardiac hypertrophy under conditions of GCA deficiency. Long-term treatment with hydralazine in rats or humans is known to increase plasma renin activity (Tsoporis and Leenen, 1986; Leenen et al., 1987) and induces cardiac sympathetic hyperactivity (Tsoporis and Leenen, 1988), as well as cardiac volume overload (Tsoporis et al., 1989). However, the molecular mechanisms underlying the pro-hypertrophic actions of hydralazine are presently unclear.
It is well established that angiotensin II plays an important role in the development of cardiac hypertrophy, and that AT1 receptors mediate most of the known physiological effects of angiotensin II (Hunyady and Turu, 2004). Hydralazine has been reported to increase plasma renin activity in spontaneously hypertensive rats (Tsoporis and Leenen, 1986; 1988;). We have demonstrated previously that genetic or pharmacological blockade of AT1 receptors attenuated cardiac hypertrophy in GCA KO mice (Li et al., 2002) and that AT1 receptors were responsible for hypertrophy induced in GCA KO mouse heart by androgens (Li et al., 2004). We initially speculated that angiotensin II/AT1 receptor signalling might participate in hydralazine-induced cardiac growth. Somewhat unexpectedly, however, hydralazine treatment increased cardiac mass and enhanced cardiac expression of the hypertrophic marker ANP in GCA/AT1 DKO mice, but not in mice lacking AT1 receptors alone. Thus, the present findings suggest that hydralazine-induced pro-hypertrophic signalling was independent of AT1 receptors. On the other hand, the persistence of the antihypertensive actions of hydralazine in the GCA/AT1 receptor DKO mice supports the contention that cardiac hypertrophy was independent of blood pressure in these animals.
The AT2 receptor is the other major angiotensin II receptor subtype. Its expression is up-regulated in cardiovascular pathologies, including cardiac hypertrophy (Suzuki et al., 1993; Lopez et al., 1994) and heart failure (Tsutsumi et al., 1998). Although the role of AT2 receptors in cardiac remodelling remains controversial, accumulating lines of evidence appear to support the view that AT2 receptors can promote cardiac growth in certain pathological situations. Indeed, at least in some tissues, AT1 and AT2 receptors share common signalling pathways that stimulate cell and tissue proliferation (Mifune et al., 2000; Senbonmatsu et al., 2000; Ichihara et al., 2001; Yan et al., 2003; D'Amore et al., 2005). We have recently demonstrated that expression of AT2 receptors at the mRNA and protein level is up-regulated in hearts of GCA KO mice and that both genetic and pharmacological blockade of AT2 receptors ameliorated the cardiac hypertrophy induced by GCA deficiency (Li et al., 2009). The pro-hypertrophic effect of hydralazine observed in GCA KO and GCA/AT1 DKO mice was completely abolished in GCA/AT2 DKO animals, and hydralazine treatment also decreased cardiac mass and ANP gene expression. These results suggest that hydralazine-induced pro-hypertrophic signalling in the heart was dependent on AT2 receptors.
The other finding in the present study is that hydralazine treatment attenuates cardiac fibrosis in GCA KO mice. It is generally considered that there are two types of cardiac fibrosis: reparative and reactive fibrosis. Reparative fibrosis appears to be a reaction to the loss of myocardial material (due to necrosis or apoptosis, after myocardial ischemia or senescence) and is primarily interstitial in location. In contrast, reactive fibrosis is observed in the absence of cell loss and appears to be a reaction to inflammation; it is primarily located perivascularly (Swynghedauw, 1999). Hydralazine therapy has previously been found to be ineffective in the modification of reactive fibrosis (Mukherjee and Sen, 1993; Norton et al., 1997). In contrast, hydralazine treatment prevented the development of reparative fibrosis in spontaneously hypertensive rats (Tsotetsi et al., 2001). In addition, hydralazine has been shown to inhibit prolyl hydroxylase activity (Bhatnagar et al., 1972; Knowles et al., 2004), which prevents the post-translational modification of collagen prolyl residues essential for the formation of stable collagen fibres (Murad et al., 1985). In the present study, even though hydralazine promoted the growth of cardiomyocytes, the same treatment diminished cardiac interstitial fibrosis in GCA KO mice. This suggests that there may be different mechanisms by which the growth of cardiomyocytes and cardiac fibroblasts is regulated in GCA-deficient mice. Future studies are required to address whether myocardial ischaemia and/or prolyl hydroxylase overactivity are associated with the excessive cardiac collagen accumulation in GCA deficiency.
In conclusion, the present findings indicate that vasodilator therapy with hydralazine induces AT2 receptor-dependent, but AT1 receptor-independent, growth of cardiomyocytes in mice lacking GCA. Thus, the possibility that exacerbation of cardiac hypertrophy may occur during the use of hydralazine in patients with decreased GCA activity should now be considered. On the other hand, attenuation of cardiac fibrosis by hydralazine treatment could be beneficial in the management of cardiac disease. The precise mechanisms underlying the anti-fibrotic action of hydralazine should now be addressed further.
Acknowledgments
This work was supported in partly by research grants from the Japanese Ministry of Education, Science and Culture, the Japanese Ministry of Health and Welfare and the Japanese Society for the Promotion of Science, Japan.
Glossary
Abbreviations:
- ANP
atrial natriuretic peptide
- AT1
angiotensin II type 1 receptor
- BW
body weight
- DKO
double knockout
- GCA
guanylyl cyclase-A
- HR
heart rate
- HW
heart weight
- KO
knockout
- LVW
left ventricular weight
- SBP
systolic blood pressure
- WT
wild-type
Statement of conflicts of interest
None.
References
- Bauer JA, Fung HL. Concurrent hydralazine administration prevents nitroglycerin-induced hemodynamic tolerance in experimental heart failure. Circulation. 1991;84:35–39. doi: 10.1161/01.cir.84.1.35. [DOI] [PubMed] [Google Scholar]
- Bhatnagar RS, Rapaka SSR, Liu TZ, Wolfe SM. Hydralazine-induced disturbances in collagen biosynthesis. Biochim Biophys Acta. 1972;271:125–132. doi: 10.1016/0005-2795(72)90140-7. [DOI] [PubMed] [Google Scholar]
- D'Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension. 2005;46:1347–1354. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- Fenje P, Leenen FH. Effects of minoxidil on blood pressure and cardiac hypertrophy in two-kidney, one-clip hypertensive rats. Can J Physiol Pharmacol. 1985;63:161–164. doi: 10.1139/y85-029. [DOI] [PubMed] [Google Scholar]
- Gerber JG, Nies AS. Ahtihypertensive agents and the drug therapy of hypertension. In: Gilman AG, Rall TW, Nies AS, Taylor P, editors. The Pharmacological Basis of Therapeutics. New York: Pergamon Press; 1990. pp. 784–813. [Google Scholar]
- Gogia H, Mehra A, Parikh S, Raman M, Ajit-Uppal J, Johnson JV, et al. Prevention of tolerance to hemodynamic effects of nitrates with concomitant use of hydralazine in patients with chronic heart failure. J Am Coll Cardiol. 1995;26:1575–1580. doi: 10.1016/0735-1097(95)00368-1. [DOI] [PubMed] [Google Scholar]
- Heart Failure Society of America. Executive summary: HFSA 2006 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2006;12:10–38. doi: 10.1016/j.cardfail.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult. Circulation. 2005;112:e154–e235. doi: 10.1161/CIRCULATIONAHA.105.167586. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009a;119:e391–e479. doi: 10.1161/CIRCULATIONAHA.109.192065. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009b;53:e1–e90. doi: 10.1016/j.jacc.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Turu G. The role of the AT1 angiotensin receptor in cardiac hypertrophy: angiotensin II receptor or stretch sensor? Trends Endocrinol Metab. 2004;15:405–408. doi: 10.1016/j.tem.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Ichihara S, Senbonmatsu T, Price E, Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin type 2 receptor is essential for ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104:346–351. doi: 10.1161/01.cir.104.3.346. [DOI] [PubMed] [Google Scholar]
- Kleaveland JP, Reichek N, McCarthy DM, Chandler T, Priest C, Muhammed A, et al. Effects of six-month afterload reduction therapy with hydralazine in chronic aortic regurgitation. Am J Cardiol. 1986;57:1109–1116. doi: 10.1016/0002-9149(86)90684-3. [DOI] [PubMed] [Google Scholar]
- Knowles HJ, Tian YM, Mole DR, Harris AL. Novel mechanism of action for hydralazine: induction of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases. Circ Res. 2004;95:162–169. doi: 10.1161/01.RES.0000134924.89412.70. [DOI] [PubMed] [Google Scholar]
- Leenen FH, Prowse S. Time-course of changes in cardiac hypertrophy and pressor mechanisms in two-kidney, one-clip hypertensive rats during treatment with minoxidil, enalapril or after uninephrectomy. J Hypertens. 1987;5:73–83. doi: 10.1097/00004872-198702000-00011. [DOI] [PubMed] [Google Scholar]
- Leenen FH, Smith DL, Farkas RM, Reeves RA, Marquez-Julio A. Vasodilators and regression of left ventricular hypertrophy. Hydralazine versus prazosin in hypertensive humans. Am J Med. 1987;82:969–978. doi: 10.1016/0002-9343(87)90160-4. [DOI] [PubMed] [Google Scholar]
- Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, et al. Guanylyl cyclase A inhibits angiotensin II type 1a receptor-mediated cardiac remodeling, an endogenous protective mechanism in the heart. Circulation. 2002;106:1722–1728. doi: 10.1161/01.cir.0000029923.57048.61. [DOI] [PubMed] [Google Scholar]
- Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, et al. Androgen contributes to gender-related cardiac hypertrophy and fibrosis in mice lacking the gene encoding guanylyl cyclase-A. Endocrinology. 2004;145:951–958. doi: 10.1210/en.2003-0816. [DOI] [PubMed] [Google Scholar]
- Li Y, Saito Y, Kuwahara K, Rong X, Kishimoto I, Harada M, et al. Guanylyl cyclase-A inhibits angiotensin II type 2 receptor-mediated pro-hypertrophic signaling in the heart. Endocrinology. 2009;150:3759–3765. doi: 10.1210/en.2008-1353. [DOI] [PubMed] [Google Scholar]
- Lopez JJ, Lorell BH, Ingefinger JR, Weinberg EO, Schunkert H, Diamant D, et al. Distribution and function of cardiac angiotensin AT1- and AT2-receptor subtypes in hypertrophied rat hearts. AM J Physiol. 1994;267:H844–H852. doi: 10.1152/ajpheart.1994.267.2.H844. [DOI] [PubMed] [Google Scholar]
- Lopez MJ, Wong SKF, Kishimoto I, Dubois S, Mach V, Friesen J, et al. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995;378:65–68. doi: 10.1038/378065a0. [DOI] [PubMed] [Google Scholar]
- Mifune M, Sasamura H, Shimizu-Hirota R, Miyazaki H, Saruta T. Angiotensin II type 2 receptors stimulate collagen synthesis in cultured vascular smooth muscle cells. Hypertension. 2000;36:845–850. doi: 10.1161/01.hyp.36.5.845. [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Sen S. Alteration of collagen phenotypes in hypertensive hypertrophy: role of blood pressure. J Mol Cell Cardiol. 1993;25:185–196. doi: 10.1006/jmcc.1993.1021. [DOI] [PubMed] [Google Scholar]
- Murad S, Tajima S, Pinnell SR. A paradoxical effect of hydralazine on prolyl and lysyl hydroxylase activities in cultured human skin fibroblasts. Arch Biochem Biophys. 1985;241:356–363. doi: 10.1016/0003-9861(85)90557-0. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, et al. Role of natriuretic peptide receptor GC-A in myocardial infarction evaluated using genetically engineered mice. Hypertension. 2005;46:441–447. doi: 10.1161/01.HYP.0000173420.31354.ef. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Soma M, Takahashi Y, Rehemudula D, Kanmatsuse K, Furuya K. Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ Res. 2000;86:841–845. doi: 10.1161/01.res.86.8.841. [DOI] [PubMed] [Google Scholar]
- Norton GR, Tsotetsi J, Trifunovic B, Hartford C, Candy GP, Woodiwiss AJ. Myocardial stiffness is attributed to alterations in cross-linked rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation. 1997;96:1991–1998. doi: 10.1161/01.cir.96.6.1991. [DOI] [PubMed] [Google Scholar]
- Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci USA. 1997;94:14731–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram BL, Ischise S, Frolich ED. Effect of methyldopa, clonidine, and hydralazine on cardiac mass and haemodynamicsin Wistar-Kyoto and SHR. Cardiovasc Res. 1982;16:40–46. doi: 10.1093/cvr/16.1.40. [DOI] [PubMed] [Google Scholar]
- Rubattu S, Bigatti G, Evangelista A, Lanzani C, Stanzione R, Zagato L, et al. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J Am Coll Cardiol. 2006;48:499–505. doi: 10.1016/j.jacc.2005.12.081. [DOI] [PubMed] [Google Scholar]
- Sanada S, Node K, Minamino T, Takashima S, Ogai A, Asanuma H, et al. Long-acting Ca2+ blockers prevent myocardial remodeling induced by chronic NO inhibition in rats. Hypertension. 2003;41:963–967. doi: 10.1161/01.HYP.0000062881.36813.7A. [DOI] [PubMed] [Google Scholar]
- Sen S, Tarazi RC, Bumpus FM. Cardiac hypertrophy in SHR. Circ Res. 1974;35:775–781. doi: 10.1161/01.res.35.5.775. [DOI] [PubMed] [Google Scholar]
- Sen S, Tarazi RC, Bumpus FM. Cardiac hypertrophy and antihypertensive therapy. Cardiovasc Res. 1977;11:427–433. doi: 10.1093/cvr/11.5.427. [DOI] [PubMed] [Google Scholar]
- Senbonmatsu T, Ichihara S, Price E, Jr, Gaffney FA, Inagami T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest. 2000;106:R25–R29. doi: 10.1172/JCI10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Matsubara H, Urakami M, Inada M. Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Cir Res. 1993;73:439–447. doi: 10.1161/01.res.73.3.439. [DOI] [PubMed] [Google Scholar]
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Taylor AL, Ziesche S, Yancy C, Carson P, D'Agostino R, Jr, Ferdinand K, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351:2049–2057. doi: 10.1056/NEJMoa042934. [DOI] [PubMed] [Google Scholar]
- Tsoporis J, Leenen FH. Effects of hydralazine on blood pressure, pressor mechanisms and cardiac hypertrophy in twokidney, one-clip hypertensive rats. Can J Physiol Pharmacol. 1986;64:1528–1534. doi: 10.1139/y86-257. [DOI] [PubMed] [Google Scholar]
- Tsoporis J, Leenen FH. Effects of arterial vasodilators on cardiac hypertrophy and sympathetic activity in rats. Hypertension. 1988;11:376–386. doi: 10.1161/01.hyp.11.4.376. [DOI] [PubMed] [Google Scholar]
- Tsoporis J, Yuan BX, Leenen FH. Arterial vasodilators, cardiac volume load, and cardiac hypertrophy in normotensive rats. Am J Physiol. 1989;256(3 Pt 2):H876–H880. doi: 10.1152/ajpheart.1989.256.3.H876. [DOI] [PubMed] [Google Scholar]
- Tsotetsi OJ, Woodiwiss AJ, Netjhardt M, Qubu D, Brooksbank R, Norton GR. Attenuation of cardiac failure, dilatation, damage, and detrimental interstitial remodeling without regression of hypertrophy in hypertensive rats. Hypertension. 2001;38:846–851. doi: 10.1161/hy1001.092649. [DOI] [PubMed] [Google Scholar]
- Tsutamoto T, Kanamori T, Wada A, Kinoshita M. Uncoupling of atrial natriuretic peptide extraction and cyclic guanosine monophosphate production in the pulmonary circulation in patients with severe heart failure. J Am Coll Cardiol. 1992;20:541–546. doi: 10.1016/0735-1097(92)90005-8. [DOI] [PubMed] [Google Scholar]
- Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation. 1993;87:70–75. doi: 10.1161/01.cir.87.1.70. [DOI] [PubMed] [Google Scholar]
- Tsutsumi Y, Matsubara H, Ohkubo N, Mori Y, Nozawa Y, Murasawa S, et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Cir Res. 1998;83:1035–1046. doi: 10.1161/01.res.83.10.1035. [DOI] [PubMed] [Google Scholar]
- Usami S, Kishimoto I, Saito Y, Harada M, Kuwahara K, Nakagawa Y, et al. Association of CT dinucleotide repeat polymorphism in the 5′-flanking region of the guanylyl cyclase (GC)-A gene with essential hypertension in the Japanese. Hypertens Res. 2008;31:89–96. doi: 10.1291/hypres.31.89. [DOI] [PubMed] [Google Scholar]
- Yan X, Price RL, Nakayama M, Ito K, Schuldt AJ, Manning WJ, et al. Ventricular-specific expression of angiotensin II type 2 receptors causes dilated cardiomyopathy and heart failure in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;285:H2179–H2187. doi: 10.1152/ajpheart.00361.2003. [DOI] [PubMed] [Google Scholar]