

Abstract
Background and purpose:
Changes in extracellular fluid osmolarity, which occur after tissue damage and disease, cause inflammation and maintain chronic inflammatory states by unknown mechanisms. Here, we investigated whether the osmosensitive channel, transient receptor potential vanilloid 4 (TRPV4), mediates inflammation to hypotonic stimuli by a neurogenic mechanism.
Experimental approach:
TRPV4 was localized in dorsal root ganglia (DRG) by immunofluorescence. The effects of TRPV4 agonists on release of pro-inflammatory neuropeptides from peripheral tissues and on inflammation were examined.
Key results:
Immunoreactive TRPV4 was detected in DRG neurones innervating the mouse hindpaw, where it was co-expressed in some neurones with CGRP and substance P, mediators of neurogenic inflammation. Hypotonic solutions and 4α-phorbol 12,13-didecanoate, which activate TRPV4, stimulated neuropeptide release in urinary bladder and airways, sites of neurogenic inflammation. Intraplantar injection of hypotonic solutions and 4α-phorbol 12,13-didecanoate caused oedema and granulocyte recruitment. These effects were inhibited by a desensitizing dose of the neurotoxin capsaicin, antagonists of CGRP and substance P receptors, and TRPV4 gene knockdown or deletion. In contrast, antagonism of neuropeptide receptors and disruption of TRPV4 did not prevent this oedema. TRPV4 gene knockdown or deletion also markedly reduced oedema and granulocyte infiltration induced by intraplantar injection of formalin.
Conclusions and implications:
Activation of TRPV4 stimulates neuropeptide release from afferent nerves and induces neurogenic inflammation. This mechanism may mediate the generation and maintenance of inflammation after injury and during diseases, in which there are changes in extracellular osmolarity. Antagonism of TRPV4 may offer a therapeutic approach for inflammatory hyperalgesia and chronic inflammation.
Keywords: Transient receptor potential vanilloid 4, neurogenic inflammation, pain, substance P, calcitonin gene-related peptide
Introduction
Primary afferent neurones of the dorsal root, trigeminal and vagal ganglia that have small cell bodies and unmyelinated (C) or thinly myelinated (A-δ) fibres contain substance P (SP) and calcitonin gene-related peptide (CGRP). Noxious thermal, mechanical and chemical stimuli activate ion channels and receptors on afferent fibres in many tissues. These stimuli induce SP and CGRP release from afferent fibres in the central nervous system, resulting in nociceptive transmission, and in peripheral tissues, causing neurogenic inflammation (McDonald, 1988; Brain and Williams, 1989; Baluk et al., 1995; 1999; Figini et al., 1997; Grant et al., 2002). Neurogenic inflammation includes SP-stimulated extravasation of plasma proteins and adhesion of leukocytes in postcapillary venules, and CGRP-stimulated arteriolar dilatation. Neurogenic mechanisms also control contraction of smooth muscle in the airway and urinary bladder (Groneberg et al., 2004; Candenas et al., 2005), fluid and electrolyte secretion in the airway and intestinal epithelium (Karaki and Kuwahara, 2004), and activation of cells of the immune system (O'Connor et al., 2004). These processes contribute to major human diseases, including asthma, arthritis, migraine and inflammatory bowel disease, and drugs that disrupt neurogenic inflammation, such as CGRP antagonists, are effective therapies for migraine (Olesen et al., 2004).
An understanding of the mechanisms that cause neurogenic inflammation may lead to new treatments for inflammation and pain. The mechanisms by which chemical agents, such as the pro-inflammatory peptide bradykinin and capsaicin, an agonist of transient receptor potential vanilloid (TRPV) 1, cause neurogenic inflammation and pain have been thoroughly investigated. Far less is known about how physical forces cause neurogenic inflammation and pain. Tissue damage is accompanied by altered hydrostatic pressure of interstitial fluid and changes in the osmolarity of extracellular fluid, which causes cells to shrink or swell, occur in diseases such as diabetes (Puliyel and Bhambhani, 2003; Sorensen et al., 2006), alcoholism (Vamvakas et al., 1998), aquadynia (Misery et al., 2003) and syndromes of inappropriate secretion of antidiuretic hormone (Sorensen et al., 1995), which are associated with altered pain perception. Changes in extracellular osmolarity induce inflammation and pain (Jensen and Norup, 1992; Graven-Nielsen et al., 1997; Drewes et al., 2003; Weerakkody et al., 2003; Bennell et al., 2004), and osmotic challenges (e.g. inhalation of nebulized hypertonic saline and water) provoke asthma (Baba et al., 1989; Choi et al., 2003) and cause inflammation of the airway (Umeno et al., 1990) and bladder (Maggi et al., 1990) by neurogenic mechanisms. Hypertonic solutions stimulate release of SP (Mandahl et al., 1984; Baraniuk et al., 1999) and CGRP (Tramontana et al., 1991; Del Bianco et al., 1992). However, it is not known how changes in extracellular osmolarity induce neuropeptide release and neurogenic inflammation.
We investigated the role of TRPV4 (Alexander et al., 2008) in neurogenic inflammation induced by osmotic stimuli. TRPV channels permit afferent nerves to detect thermal, mechanical and chemical stimuli, and thereby regulate neurogenic inflammation and nociception (reviewed in Pedersen et al., 2005). TRPV4, the mammalian homologue of the C. elegans gene Osmosensory-9, is gated by small reductions in tonicity and by temperatures >27°C (Liedtke et al., 2000; Guler et al., 2002). TRPV4 is expressed by neurosensory structures, including circumventricular organs that detect changes in systemic osmolarity, inner ear hair cells, Merkel cells and sensory neurones. TRPV4−/− mice show abnormal osmotic regulation and decreased nociceptive responses to pressure (Liedtke and Friedman, 2003; Suzuki et al., 2003), and TRPV4 knockdown or deletion reduces nociceptive responses to hypotonic and mildly hypertonic stimuli (Alessandri-Haber et al., 2003; 2005;). In common with other TRPV channels, notably TRPV1, inflammatory agents can sensitize TRPV4 (Alessandri-Haber et al., 2003; 2006; Grant et al., 2007), suggesting that this channel may amplify inflammatory responses to many agonists. Thus, TRPV4 functions to detect osmotic and mechanical stimuli. However, the role of TRPV4 in neurogenic inflammation is completely unknown. We examined the hypothesis that osmotic stimuli activate TRPV4 on sensory nerves to induce neuropeptide release and neurogenic inflammation.
Methods
Animals
C57BL6 mice (male, 6–8 weeks) were from Charles River Laboratories (Wilmington, MA, USA). TRPV4−/− and TRPV4+/+ mice (male and female, 6–8 weeks) have been described previously (Liedtke and Friedman, 2003). All mice were housed under constant humidity and temperature, under a 12-h light and dark cycle. Institutional Animal Care and Use Committees approved all procedures. At the end of procedures, mice were humanely killed using sodium pentobarbitone (200 mg·kg−1 i.p.), followed by bilateral thoracotomy.
Paw inflammation
Control and experimental groups were studied on the same day. Mice were briefly anaesthetized with halothane (5%). Isotonic saline (0.9% NaCl, 283 mOsm), hypotonic solution (deionised water, 17 mOsm), hypertonic saline (10% NaCl, 3250 mOsm), 4α-phorbol 12,13-didecanoate (4αPDD) (500 µM in 6.7% DMSO and 0.9% NaCl) or DMSO (6.7% DMSO and 0.9% NaCl) was injected into the plantar surface of one hind paw (5 µL), using a 30-gauge needle with a 20 µL Hamilton syringe as previously described (Vergnolle et al., 2001). All injected solutions were pH 7.4, corrected with NaOH. The site of injection was marked. To induce neurogenic inflammation, 1% formalin in 10 µL 0.9% NaCl was injected into the paw. Paw diameter was measured before and after injections as an index of oedema using a digital calliper with a resolution of 10 µM (Fisher Scientific, Hampton, NH, USA) (Houle et al., 2005). Paw diameter was measured at the site of injection between the plantar and the dorsal surfaces of the paw by investigators unaware of the experimental treatment. Minimal or no compression was exerted onto the paw at the time of the measurement. After 4 h, myeloperoxidase (MPO) activity was measured in paws as an index of granulocyte recruitment. Tissues were homogenized in 0.5% hexadecyltrimethylammonium bromide phosphate buffered solution (pH 6.0), and supernatants of homogenates were used for enzymatic assay in a solution of 3, 3′-dimethoxybenzidine and 1% hydrogen peroxide. Optical density was measured at 450 nm, and MPO activity was expressed in units of enzyme mg−1 tissue compared with pure enzyme. Paw tissue was also collected at 4 h after agonist injection for histological examination. The injected paws were fixed by immersion in formalin, washed in 50% ethanol in distilled water and embedded in paraffin wax. Sections (5 µM) were mounted on glass slides and stained with haematoxylin and eosin as described previously (Vergnolle et al., 1999).
Antagonists
Mice were treated with antagonists previously reported to inhibit neurogenic inflammation (Steinhoff et al., 2000). The CGRP receptor antagonist CGRP8-37 was administered 5 min before intraplantar injections (10 µg·kg−1 i.v. plus 10 µg·kg−1 s.c., dissolved in 0.9% NaCl) and then hourly (10 µg·kg−1 i.v.) (Steinhoff et al., 2000). The SP neurokinin 1 receptor (NK1R) antagonist SR140333 was administered 30 min before intraplantar injections (1 mg·kg−1 s.c., dissolved in 0.1% DMSO plus 0.9% NaCl) (Steinhoff et al., 2000). These antagonists were administered at different times because they have distinctly different half-lives and pharmacokinetic properties. Capsaicin was administered to deplete sensory neurones of neuropeptides. Under halothane anaesthesia, capsaicin was administered three times (one dose of 25 and two doses of 50 mg·kg−1 s.c., dissolved in 10% ethanol, 10% Tween 80 and 80% saline) at 0, 6 and 32 h (Nguyen et al., 2003). Experiments were started 10 days after the last dose of capsaicin, and the effectiveness of capsaicin treatment was verified by instilling one drop of 0.1 mg·mL−1 capsaicin into the eye and counting wiping movements.
siRNA treatments
Mice were anaesthetized with halothane (5%). The following siRNA duplexes were injected intrathecally (10 µL of DEPC water) at 0, 12 and 24 h: TRPV4 siRNA labelled with 6-carboxyfluorescein (6-FAM): 5′-ucuaccaguacuauggcuud(tt)-3′, 3′-d(tt)agauggucaugauaccgaa-5′, control siRNA with the same percentage of GC but targeting no known sequence: 6-FAM 5′-caugcuagguuaguacuugd(tt)-3′, 3′-d(tt)guacgauccaaucaugaac-5′. Other control mice were injected intrathecally with DEPC water. Experiments began 4 days after the first injection, as described previously(Cenac et al., 2008).
Retrograde tracing and immunofluorescence
Mice (n = 3) were anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.), and 1,1′-dioctadecyl-3,3,3′,3-tetramethyl-indocarbocyanine perchlorate (DiI, 17 µg·mL−1, 50% DMSO) was injected into the plantar surface of one hind paw (20 µL). At 10 days after injection, mice were killed and dorsal root ganglia (DRG) (L4-L5) were removed and incubated in 4% paraformaldehyde (100 mM PBS, pH 7.4, 2 h, room temperature) and then 30% sucrose (overnight, 4°C). DRG were embedded in OCT compound, and frozen sections (10 µM) were prepared. Sections were fixed in 4% paraformaldehyde (3 min) and then washed with PBS containing 5% normal goat serum and 3% Triton X-100. Sections were incubated in this buffer with the following primary antibodies: rabbit anti-TRPV4 (1:500) and guinea-pig anti-substance P (1:250) or guinea pig anti-CGRP (1:250) (all overnight, 4°C). Washed slides were incubated with a goat anti-rabbit IgG labelled with FITC (1:200) and goat anti-guinea pig IgG labelled with Alexa-647 (1:1000). As a control for TRPV4 specificity, the primary antiserum was pre-incubated with the peptide used for immunization (10 µM) for 24 h at 4°C before staining.
Confocal microscopy
Single images of sections (1024 × 1024 pixels) were acquired with a Zeiss LSM510 Meta confocal microscope using a 40X EC Plan-Neofluor objective (1.3 n.a.). The 488 line of the Argon laser was used to excite FITC, and the 543 and 633 line of the HeNe lasers were used to excite DiI and Alexa-647, respectively. The total cellular pixel intensity of the DiI fluorescence in individual cells was determined using the LSM510 Meta software, and cells with a total intensity of >800 pixels were chosen as DiI positive neurones.
Neuropeptide release
Slices (∼0.4 mm, 50–70 mg) were prepared from the urinary bladder and airways (trachea and bronchi) of mice at 4°C and were transferred to 2 mL chambers and superfused at 0.4 mL·min−1 with a Krebs solution (mM: NaCl 119, NaHCO3 25, KH2PO4 1.2, MgSO4 1.5, CaCl2 2.5, KCl 4.7 and D-glucose 11) containing 0.1% BSA, 1 µM phosphoramidon and 1 µM captopril (37°C, 96% O2, 4% CO2). After a 60 min stabilization period, 10 min fractions (4 mL) were collected into acetic acid (final concentration 2N): two fractions prior, one fraction during and one fraction after administration of the stimulus. Tissues were stimulated with the TRPV4 agonist 4αPDD (100 µM) (Watanabe et al., 2002a), or with hypotonic solution (228 or 17 mOsm). In some experiments, Ca2+ ions were omitted, or sensory nerves were depleted of neuropeptides using capsaicin (pre-incubation with 10 µM capsaicin for 20 min before stimulation). Freeze-dried fractions were reconstituted with assay buffer and analysed by enzyme immunoassays for CGRP and SP (Trevisani et al., 2004). None of the antibodies cross-reacted with 4αPDD (100 µM). The level of release of CGRP-LI and SP-LI were calculated by subtracting the mean pre-stimulus value from those values obtained during and after stimulation.
Statistical analyses
Results are presented as mean ± SE. Data were tested for normality and then analysed using both anova for repeated measures and one-way anova followed by a Tukey–Kramer multiple-comparison post-test for in vivo data, or a Dunnett's test for in vitro data.
Materials
4αPDD, DNSO, NaCl, pure enzyme, CGRP8-37 and capsaicin were obtained from Sigma (St. Louis, MO, USA), SR140333 from Sanofi Montpellier (Montpellier, France; a generous gift from Dr X. Edmonds-Alt). The siRNA duplexes were purchased from Nucleotide Synthesis Core facilities (University of Calgary, Calgary, Alberta, Canada), DiI was obtained from Invitrogen (Carlsbad, CA, USA), OCT compound from Sakura Finetek (Torrance, CA, USA). Rabbit anti-TRPV4 was purchased from Alomone (Tel Aviv, Israel), guinea-pig anti-substance P from Chemicon (Temecula, CA, USA), guinea pig anti-CGRP from Research Diagnostic Inc (Flanders, NJ, USA), goat anti-rabbit IgG labelled with FITC from Jackson ImmunoResearch (West Grove, PA, USA), and goat anti-guinea-pig IgG labelled with Alexa-647 from Invitrogen.
Results
TRPV4 agonists cause inflammation of peripheral tissues
To assess the role of TRPV4 in inflammation, mice received intraplantar injections of the TRPV4 agonists 4αPDD (Watanabe et al., 2002a) or hypotonic solution (Liedtke et al., 2000; Guler et al., 2002). 4αPDD (5 µL, 500 µM) or hypotonic solution (5 µL, 17 mOsm distilled water) increased paw diameter by three- to fourfold from 30 min to 4 h compared with isotonic saline or DMSO vehicle, indicative of oedema (Figure 1A). 4αPDD and hypotonic solution also increased tissue MPO activity after 4 h, indicative of granulocyte recruitment (Figure 1B). Although hypertonic solution (5 µL, 10% NaCl, 3250 mOsm) caused severe oedema, it did not cause significant granulocyte recruitment (Figure 1B). The structure of the tissue was disrupted in paw tissues that were injected with 4αPDD, hypotonic solution or hypertonic solution, but not those injected with isotonic saline (Figure 1C). Thus, the established activators of TRPV4, 4αPDD and hypotonic solution, caused oedema, granulocyte recruitment and tissue disruption, three of the main features of inflammation.
Figure 1.
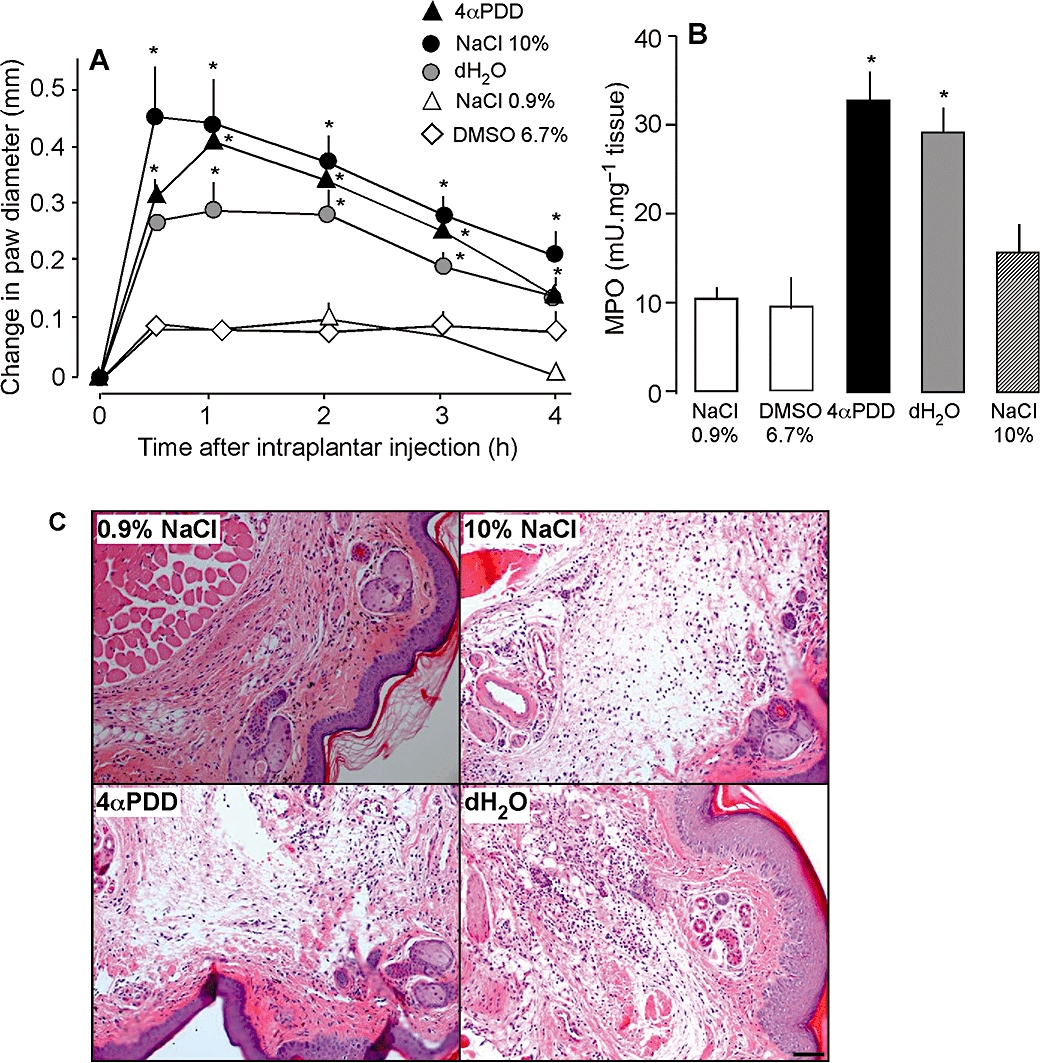
Effects of intraplantar injections of 4α PDD, distilled water (dH2O), 10% NaCl and 0.9% NaCl on paw diameter (A), MPO (B) and histology of paw tissues (C). Scale bar = 200 µm. 4αPDD, dH2O and 10% NaCl increased paw diameter and paw MPO activity, and disrupted the structure of the tissue compared with 0.9% NaCl. *P < 0.05 compared with 0.9% NaCl, n = 16 per group for all except for histology, n = 5 per group. 4αPDD, 4α-phorbol 12,13-didecanoate; MPO, myeloperoxidase.
TRPV4 agonists cause inflammation by a neurogenic mechanism
To determine the role of sensory nerves in TRPV4-induced inflammation, we chronically treated mice with capsaicin, which depletes C and Aδ fibres of neuropeptides. Capsaicin treatment did not affect paw diameter (not shown) or MPO activity (Figure 2D) in mice receiving intraplantar isotonic saline. However, capsaicin strongly inhibited the effects of 4αPDD and hypotonic solution on paw diameter and MPO activity at most times (Figure 2A, B and D). Capsaicin also inhibited the effects of hypertonic solution on paw diameter from 2–4 h, but not on MPO activity (Figure 2C and D).
Figure 2.
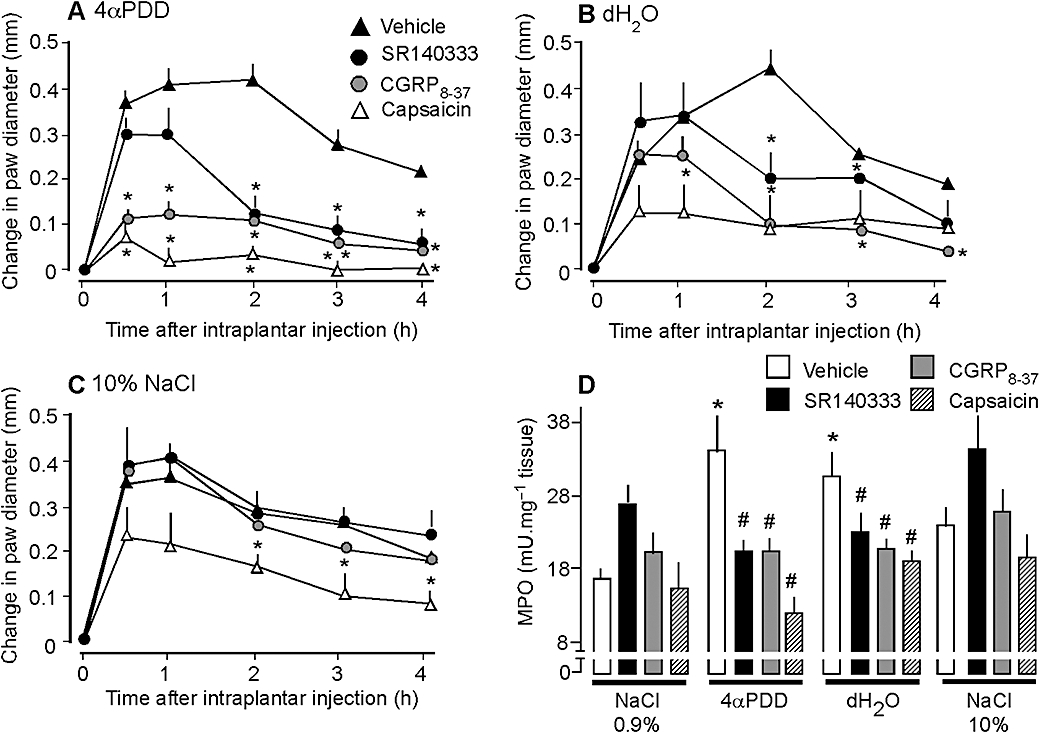
Effects of capsaicin, CGRP8-37, SR140333 and vehicle (control) on paw diameter (A–C) and MPO (D) of mice after intraplantar injections of 4αPDD (A), distilled water (dH2O, B), 10% NaCl (C) and 0.9% NaCl (D). Capsaicin, CGRP8-37 and SR140333 inhibited the effects of 4αPDD and dH2O on diameter and MPO activity, but had little or no effect on responses to 10% NaCl. *P < 0.05 compared with vehicle, n = 8 per group. 4αPDD, 4α-phorbol 12,13-didecanoate; CGRP, calcitonin gene-related peptide; MPO, myeloperoxidase.
SP and CGRP released from C and Aδ fibres promote neurogenic inflammation of peripheral tissues (McDonald, 1988; Brain and Williams, 1989; Baluk et al., 1995; 1999; Figini et al., 1997; Grant et al., 2002). To determine whether SP and CGRP mediate the effects of TRPV4 agonists on inflammation, we treated mice with antagonists of the CGRP and SP receptors. The CGRP antagonist CGRP8-37 and the NK1R antagonist SR140333 inhibited the effects of 4αPDD and hypotonic solution on paw diameter and MPO activity (Figure 2A, B and D). Although both antagonists similarly inhibited granulocyte recruitment, the CGRP receptor antagonist more effectively reduced oedema, inhibiting 4αPDD-induced oedema from 30 min to 4 h, and inhibiting hypotonic solution-induced oedema from 2–4 h. The NK1 receptor antagonist reduced 4αPDD-induced oedema from 2–4 h, and inhibited hypotonic solution-induced oedema only at 2 h. Neither CGRP8-37 nor SR140333 affected oedema or MPO activity in mice receiving hypertonic saline (Figure 2C and D). Thus, 4αPDD and hypotonic solution cause paw inflammation by a mechanism that depends on the release of CGRP and SP from capsaicin-sensitive nerves. Hypertonic solution induces inflammation by a different mechanism, involving capsaicin-sensitive fibres, but not CGRP or SP receptors.
TRPV4 is present in DRG neurones containing CGRP and SP
We have previously reported that TRPV4 colocalizes with CGRP and SP in DRG neurones of the rat (Grant et al., 2007). The colocalization of TRPV4, CGRP and SP in primary spinal afferent neurones innervating the mouse paw would be consistent with a direct effect of TRPV4 agonists on peptide release in this tissue. To identify DRG neurones innervating the paw, we injected the retrograde tracer DiI into the plantar surface, and then localized TRPV4, CGRP and SP using immunofluorescence. TRPV4-like immunoreactivity (LI) was detected in a large number of neurones of variable diameter where it was localized in the cytosol and nucleus (Figure 3). TRPV4 has also been detected in the nuclei of neurones of the subventricular organ of mice (Liedtke and Friedman, 2003). Of the retrogradely labelled DRG neurones containing DiI, 51% (23 of 45 neurones with total DiI pixel intensity >800) expressed TRPV4-LI. Some neurones expressing TRPV4-LI also contained CGRP-LI and SP-LI (Figure 3, arrows). However, TRPV4-LI was also found in DiI-labelled neurones that did not contain detectable CGRP-LI or SP-LI (Figure 3, arrow heads). Pre-adsorption of the TRPV4 antiserum with the peptide used for immunization prevented TRPV4 staining. Thus, TRPV4 is present in a large proportion of DRG neurones innervating the paw, and some of these neurones also contain the pro-inflammatory neuropeptides SP and CGRP.
Figure 3.
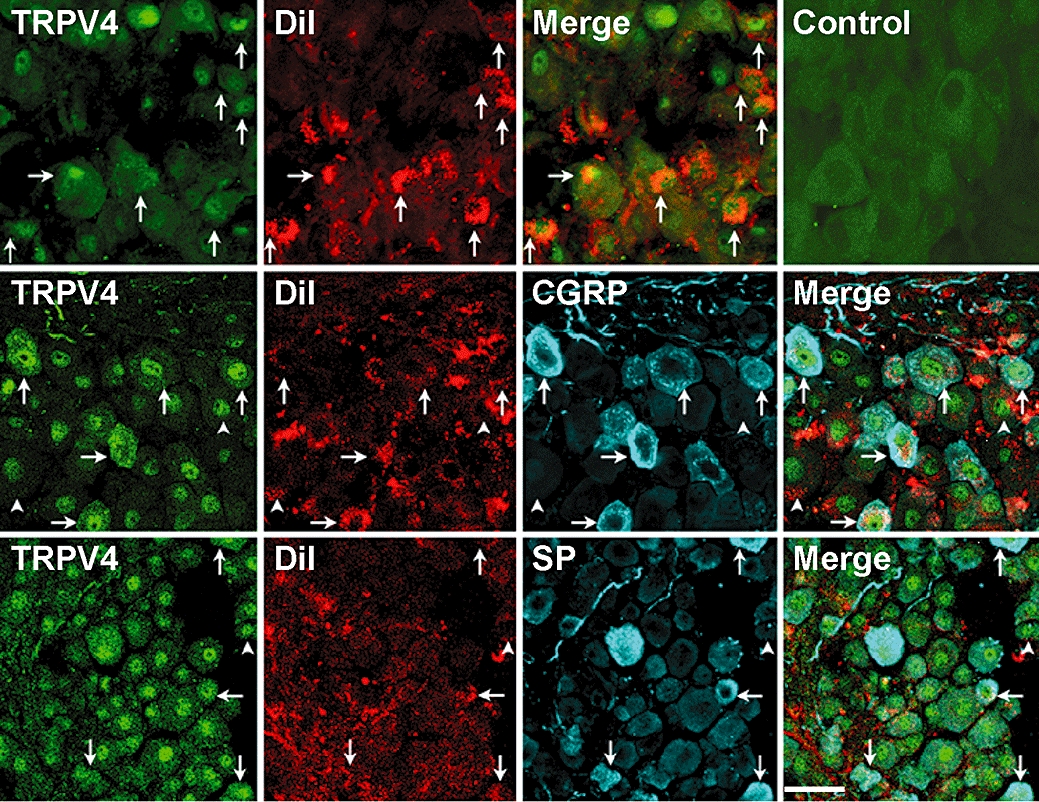
Localization of TRPV4, CGRP and SP in DiI-labelled DRG (L4-L5) innervating the mouse paw. DiI was injected into the plantar surface of the hind paw to retrogradely label neurones innervating this tissue. TRPV4-LI was detected in some DiI-labelled neurones containing CGRP-LI and SP-LI (arrows). However, some neurones expressing TRPV4-LI did not contain detectable DiI, CGRP-LI or SP-LI (arrow heads). Pre-absorption of the antibody abolished staining (control). Scale bar = 40 µM in top and middle rows and 50 µM in bottom row. CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglia; LI, like-immunoreactivity; SP, substance P; TRPV, transient receptor potential vanilloid 4 (TRPV).
TRPV4 agonists stimulate the release of SP and CGRP
To directly determine whether TRPV4 agonists promote secretion of pro-inflammatory neuropeptides in peripheral tissues, we measured the release of SP-LI and CGRP-LI from superfused tissues. We examined peptide release from the urinary bladder and airways because these tissues are richly innervated by primary spinal afferent neurones containing CGRP and SP, and are established sites of neurogenic inflammation in mice (Figini et al., 1997; Baluk et al., 1999). We were unable to study peptide release from the skin since this tissue is more sparsely innervated by peptidergic neurones and even powerful stimulants, such as capsaicin release only small amounts of CGRP and SP that are difficult to measure (Geppetti, unpublished observations). We found that 4αPDD (100 µM) stimulated a >sevenfold increase in SP-LI release, and a 12-fold increase in CGRP-LI release from both tissues (Figure 4A–D). Hypotonic solution (228 mOsm) stimulated a five- to eightfold increase in SP-LI release, and a 6- to 11-fold increase in CGRP-LI release from both tissues (Figure 4A–D). Removal of extracellular Ca2+ ions or capsaicin-desensitization of tissues inhibited the stimulating effects of 4αPDD and hypotonic solution. More hypotonic solutions (17 mOsm) stimulated an even larger peptide release (not shown). Thus, 17 mOsm solution induced a 21-fold increase in SP-LI release from bladder (maximal 125 ± 25), a 20-fold increase in SP-LI release from airway (maximal 98 ± 12), a 30-fold increase in CGRP-LI release from bladder (maximal 357 ± 62), and a 57-fold increase in CGRP-LI release from airway (maximal 452 ± 51) (all values fmol g−1 20 min−1). Thus, known agonists of TRPV4 stimulate the release of pro-inflammatory peptides from sensory nerve endings in peripheral tissues.
Figure 4.
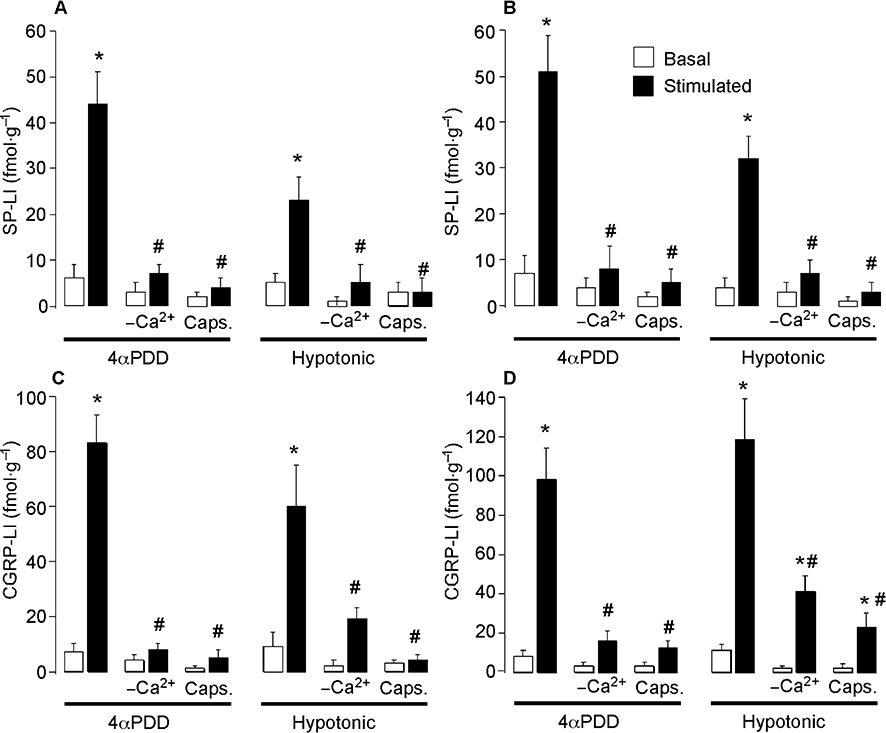
Effects of 4αPDD and hypotonic solution on the release of SP (A, B) and CGRP (C, D) from mouse urinary bladder (left) and airways (right). 4αPDD and hypotonic solution stimulated release of CGRP and SP, and the effects were abolished by removal of extracellular Ca2+ ions (-Ca2+) and by capsaicin (Caps) treatment. Neuropeptide release was measured over 20 min. *P < 0.05 compared with basal; #P < 0.05 compared with 100 µM 4αPDD and hypotonic solution. 4αPDD, 4α-phorbol 12,13-didecanoate; CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglia; SP, substance P.
To determine whether 4αPDD and hypotonic solution stimulate neuropeptide release by activating TRPV4, we measured neuropeptide release from the urinary bladder and airways of TRPV4+/+ and TRPV4−/− mice. 4αPDD and hypotonic solution both induced a >sixfold increase in SP-LI release, and a >10-fold increase in CGRP-LI release from both tissues from TRPV4+/+ mice (n = 6, not shown). In contrast, 4αPDD and hypotonic solution did not stimulate release of SP-LI or CGRP-LI above baseline from urinary bladder or airways from TRPV4−/− mice (n = 4). Thus, 4αPDD and hypotonic solution stimulate neuropeptide release by activating TRPV4.
TRPV4 mediates the inflammatory actions of 4αPDD and hypotonic solution, but not those of hypertonic solution
To evaluate directly the contribution of TRPV4 to inflammation, we used the complimentary approaches of gene-knockdown (siRNA) and gene-deletion (TRPV4−/− mice). TRPV4-targeted siRNA or control siRNA, with fluorescent labels to detect cellular uptake, were injected intrathecally into mice. After 4 days, fluorescent siRNA had accumulated in a large proportion of DRG neurones (Figure 5A), to a similar extent whether animals were injected with siRNA control (92.7 ± 7.0%) or TRPV4 siRNA (94.7 ± 3.5%). Injection of TRPV4 siRNA resulted in a marked reduction in the intensity of TRPV4-LI (Figure 5A) and in the number of neurones with detectable TRPV4-LI (Figure 5B), when compared with mice treated with control siRNA or vehicle (DEPC water). TRPV4 siRNA inhibited the stimulant effects of 4αPDD and hypotonic solution on paw diameter by ∼ 50% from 30 min – 2 h, and prevented the increase in MPO activity at 4 h when compared with control siRNA (Figure 6B, C and E). In contrast, TRPV4 siRNA did not affect paw diameter and MPO activity in mice receiving hypertonic solution (Figure 6D and E) or isotonic saline (Figure 6A and E). In TRPV4+/+ mice, 4αPDD or hypotonic solution increased paw diameter and MPO activity, but these effects were significantly reduced in TRPV4−/− mice (Figure 7A–D). Thus, knockdown or deletion of TRPV4 inhibits the pro-inflammatory actions of 4αPDD and hypotonic solution, suggesting that TRPV4 mediates these effects. In contrast, hypertonic solutions induce inflammation by a TRPV4-independent mechanism.
Figure 5.
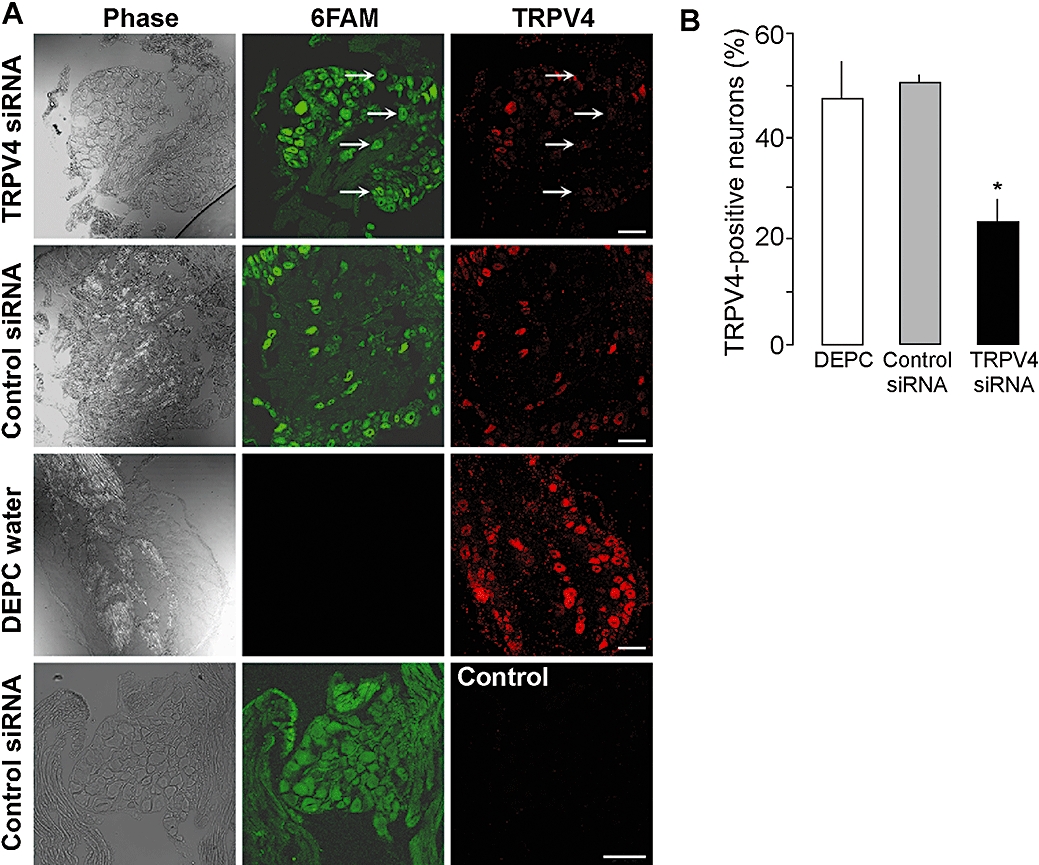
Down-regulation of TRPV4 with siRNA. Vehicle (DEPC water), control siRNA or TRPV4 siRNA was injected intrathecally and DRG were collected after 4 days to verify uptake of fluorescent 6FAM-siRNA and to localize TRPV4-LI (A). Both control and TRPV4 6FAM-siRNA were detected in many cells of the DRG. TRPV4-LI was detected in DRG after control siRNA, and this signal was prevented by pre-adsorpion of antibody (control). Arrows show neurones that have taken up siRNA (6FAM positive) and that did not express TRPV4-LI. Scale bar = 50 µM. Injection of TRPV4 siRNA down-regulated TRPV4-LI. TRPV4 siRNA also reduced the number of neurones with detectable TRPV4-LI (B). *P < 0.05 compared with DEPC water. DRG, dorsal root ganglia; LI, like-immunoreactivity; TRPV, transient receptor potential vanilloid 4 (TRPV).
Figure 6.
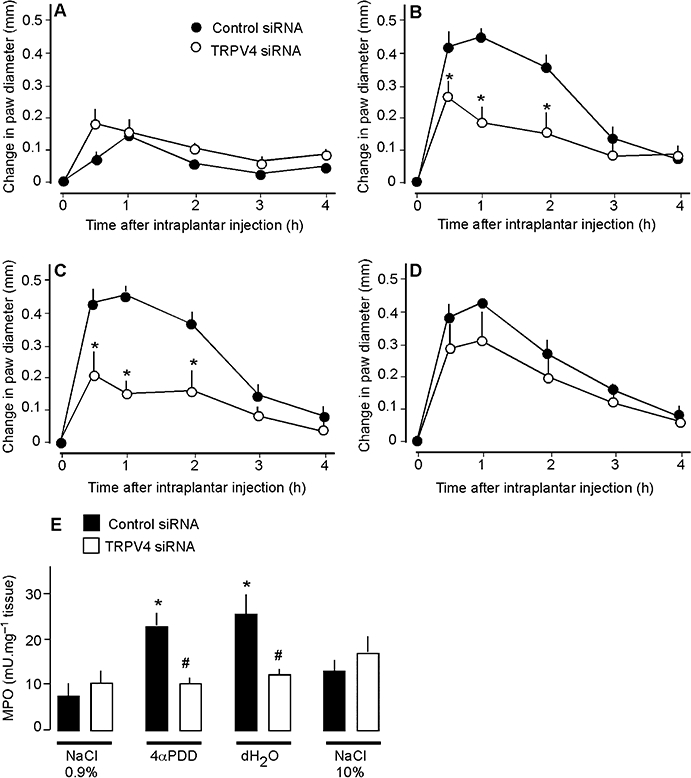
Effects of TRPV4 down-regulation with siRNA on paw diameter (A–D) and MPO (E) of mice after intraplantar injections of 0.9% NaCl (A), 4αPDD (B), distilled water (dH2O, C) or 10% NaCl (D). TRPV4 siRNA inhibited the effects of 4αPDD and distilled water on diameter and MPO activity, but did not affect the responses to 10% NaCl. *P < 0.05 compared with control siRNA (A–D) or 0.9% NaCl (E); #P < 0.05 compared with control siRNA (E), n = 6 in each group. MPO, myeloperoxidase; TRPV, transient receptor potential vanilloid 4 (TRPV).
Figure 7.
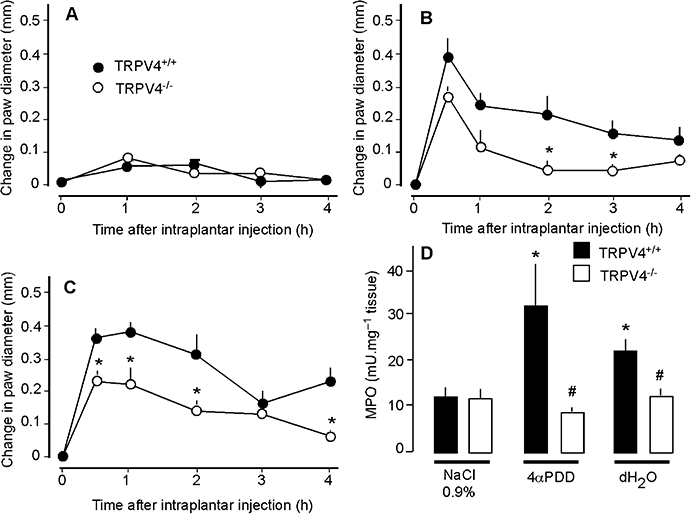
Effects of TRPV4 deletion on paw diameter (A–C) and MPO (D) of mice after intraplantar injections of 0.9% NaCl (A), 4αPDD (B), or distilled water (dH2O, C). TRPV4-deficient mice (TRPV4−/−) showed significantly less oedema and MPO activity than did wild-type littermates (TRPV4+/+). *P < 0.05 compared with TRPV4+/+ (A–C) or compared with 0.9% NaCl (D); #P < 0.05 to TRPV4+/+; n = 5 for each group of mice that received 4αPDD or distilled water, n = 7 for TRPV4+/+ and n = 8 for TRPV4−/− that received 0.9% NaCl. MPO, myeloperoxidase; TRPV, transient receptor potential vanilloid 4 (TRPV).
TRPV4 contributes to formalin-induced inflammation
We used TRPV4 knockdown or deletion to determine the contribution of TRPV4 to the inflammatory action of formalin, which is partially mediated by a neurogenic mechanism (Damas and Liegeois, 1999). Intraplantar injection of formalin caused a large and sustained increase in paw diameter and MPO activity in mice receiving control siRNA and in TRPV4+/+ mice (Figure 8A–C). However, in mice treated with TRPV4 siRNA and in TRPV4−/− mice, the effects of formalin on paw diameter and MPO activity were significantly reduced. Thus, TRPV4 contributes to formalin-induced inflammation, suggesting that TRPV4 is an important endogenous mediator of this neurogenic inflammatory reaction.
Figure 8.
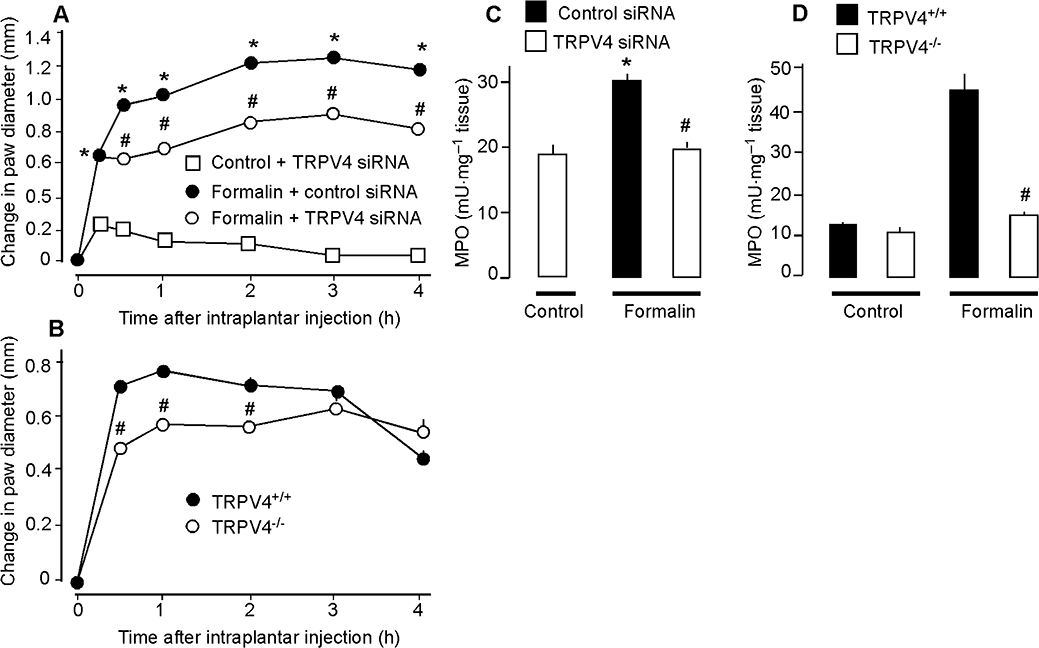
Effects of TRPV4 down-regulation with siRNA (A, C) or TRPV4 deletion (B, D) on control non-inflamed and formalin-induced changes in paw volume (A, B) and MPO (C, D). In (A) and (C), controls received an intraplantar injection of 0.9% NaCl and an intrathecal injection of TRPV4 siRNA. In (D), control TRPV4+/+ and TRPV4–/– mice received an intraplantar injection of 0.9% NaCl. TRPV4 siRNA-treated and TRPV4–/– mice showed a reduced inflammatory response to formalin with a diminished oedema and MPO activity. *P < 0.05 compared with control non-inflamed (A, C); #P < 0.05 compared with control siRNA + formalin (A, C) or TRPV4+/+ (B, D), n = 6 for all mice treated with siRNA, n = 7 for TRPV4+/+ and n = 8 for TRPV4−/− mice that received formalin. MPO, myeloperoxidase; TRPV, transient receptor potential vanilloid 4 (TRPV).
Discussion and conclusions
Inflammation is one of the first defensive responses to injury. However, an exaggerated inflammatory response can produce tissue damage, as is observed in chronic inflammatory diseases such as asthma, arthritis and inflammatory bowel disease. The development of more effective treatments for these diseases depends on a better understanding of the mediators of inflammation. In this study, we have shown for the first time that TRPV4 mediates neurogenic inflammation induced by a decrease in osmolarity of extracellular fluid, and we demonstrate that this mechanism occurs in an experimental model of injury.
Tissue injury generates and releases many well-characterized chemical mediators that can activate afferent nerves to cause neurogenic inflammation. By comparison, far less is known about how physical changes, such as altered hydrostatic or osmotic pressure of interstitial fluid, which also occur during injury and disease (Sorensen et al., 1995; 2006; Vamvakas et al., 1998; Misery et al., 2003; Puliyel and Bhambhani, 2003), affect the function of afferent nerves. Our results show that TRPV4, a channel that responds to small reductions in extracellular osmolarity (Liedtke et al., 2000; Guler et al., 2002), and which participates in systemic osmotic regulation (Liedtke and Friedman, 2003), plays an important role in tonicity-induced inflammation. We found that hypotonic solutions that can activate TRPV4 (Guler et al., 2002; Liedtke et al., 2000) and the TRPV4 agonist 4αPDD (Watanabe et al., 2002a) cause oedema and granulocyte recruitment, two of the main features of inflammation. These effects were markedly diminished by TRPV4 gene knockdown or deletion, and thus depend, in large part, upon TRPV4 or a product of the TRPV4 gene.
Intraplantar injection of formalin induces neurogenic inflammation and pain, and is commonly used to assess the efficacy of anti-inflammatory and analgesic drugs (Damas and Liegeois, 1999). Because our results suggest that TRPV4 activation provokes neurogenic inflammation, we investigated the contribution of this channel to formalin-induced inflammation. TRPV4 knockdown or deletion inhibited formalin-induced oedema and granulocyte recruitment. Our results indicate that endogenous TRPV4 participates in the development of inflammation and suggest that TRPV4 blockade could be a novel therapy for the treatment of neurogenic inflammation.
TRPV4 knockdown or deletion reduced but did not abolish oedema induced by hypotonic solution and by 4αPDD, suggesting the involvement of additional mechanisms. Other members of the TRP family may be involved, such as TRPA1, TRPM3 and TRPV2, which can also respond to decreased osmolarity and mechanical stimuli (Pedersen et al., 2005). Moreover, TRPV4−/− mice may have developed compensatory mechanisms, such as overexpression and activation of other receptors or channels that respond to osmotic or mechanical stimuli. Such compensatory mechanisms could mask a complete inhibition of oedema. Although intrathecal siRNA acutely diminished expression of TRPV4 in DRG neurones, thus reducing the likelihood of compensation, intraplantar hypotonic solutions and 4αPDD may activate TRPV4 in other cell types, such as endothelial cells or keratinocytes, to induce oedema (Watanabe et al., 2002b; Chung et al., 2003; Fian et al., 2007). TRPV4 knockdown or deletion abolished hypotonic solution- and 4αPDD-induced granulocyte recruitment. Thus, TRPV4 present on afferent neurones could account for most of the effects of hypotonic solution and 4αPDD on the recruitment of inflammatory cells.
Our results show that hypotonic solutions and 4αPDD activate TRPV4 to induce inflammation by a neurogenic mechanism. Hypotonic solutions and 4αPDD stimulated the release of SP and CGRP from the urinary bladder and airway, established sites of neurogenic inflammation in mice (Figini et al., 1997; Baluk et al., 1999). This response was Ca2+-dependent, and is thus dependent on neurosecretion, and prevented by pre-treatment with a desensitizing concentration of capsaicin, indicating that CGRP and SP originate from peripheral terminals of capsaicin-sensitive nociceptive fibres. These results are consistent with the observation that hypotonic solutions and 4αPDD promote the release of SP and CGRP from the central projections of these fibres in the spinal cord (Grant et al., 2007). The finding that immunoreactive TRPV4 colocalized in DRG neurones with CGRP and SP suggests that TRPV4 agonists can directly stimulate neuropeptide release from afferent nerves. Indeed, we detected TRPV4-LI, CGRP-LI and SP-LI in the soma of DRG neurones innervating the skin, identified by retrograde tracing. Furthermore, ablation of sensory nerves using capsaicin, or antagonism of SP, NK1 or CGRP receptors, inhibited oedema and granulocyte recruitment caused by intraplantar injections of hypotonic solution or 4αPDD. Whereas capsaicin abolished TRPV4-induced oedema and granulocyte recruitment, indicating an absolute requirement for capsaicin-sensitive afferent nerves, antagonism of SP NK1R and CGRP receptors individually was less effective, suggesting involvement of both SP and CGRP in the inflammatory response. These results are consistent with the involvement of both SP and CGRP in neurogenic inflammation of mouse skin (Grant et al., 2002), and with the role of TRPV4 in hypotonicity-induced nociception (Alessandri-Haber et al., 2003). When considered together, these results suggest that hypotonic solutions and 4αPDD activate TRPV4 on fibres of primary nociceptive neurones in peripheral tissues, such as the skin, to induce the local release of SP and CGRP. SP activates the NK1 receptor on endothelial cells of postcapillary venules, causing plasma extravasation and granulocyte recruitment, and CGRP induces arteriolar vasodilatation and hyperaemia. The combined effects of SP-stimulated plasma extravasation and CGRP-stimulated vasodilatation may contribute to the measured increase in paw diameter, and SP-stimulated granulocyte recruitment may account for the increased MPO activity in tissues. However, TRPV4 is also expressed by cell types other than neurones, such as endothelial cells (Watanabe et al., 2002b; Chung et al., 2003; Fian et al., 2007). Thus, although our results strongly suggest that TRPV4 agonists promote oedema and granulocyte recruitment by a neurogenic mechanism, we cannot exclude the possibility that hypotonic solution and 4αPDD directly activate TRPV4 on endothelial cells to cause inflammation.
A hypertonic stimulus caused oedema, but, in contrast to a hypotonic stimulus and 4αPDD, it did not cause granulocyte recruitment. Similarly, whereas antagonists of SP, NK1 and CGRP receptors inhibited inflammation induced by hypotonic solution and by 4αPDD, they did not affect the oedema response to a hypertonic stimulus. Moreover, TRPV4 knockdown did not modify hypertonic solution-induced oedema. Thus, hypertonic stimuli induce oedema by a mechanism that does not involve activation of TRPV4 on afferent nerves. Our results are in accordance with a report that a moderately hypertonic stimulus (10% NaCl) induces pain behaviour by a TRPV4-independent process (Alessandri-Haber et al., 2005). However, capsaicin pre-treatment diminished hypertonic oedema, suggesting an involvement of afferent neurones, perhaps through activation of channels other than TRPV4. These findings support the report that hypertonic saline increases vascular permeability in rat trachea by a neurogenic mechanism (Umeno et al., 1990).
Although TRPV4 has been implicated in hypotonic, neuropathic and chemotherapy-induced pain (Alessandri-Haber et al., 2003; 2004;), our study is the first to implicate this channel directly in inflammatory processes. Inflammatory mediators can sensitize TRPV4, thereby enhancing its activation (Alessandri-Haber et al., 2003; 2006; Grant et al., 2007). Thus, our study provides evidence that TRPV4 mediates inflammatory responses to hypoosmotic stimuli and to the amplification of inflammation. Antagonism of this channel may offer a novel therapeutic approach for both inflammatory hyperalgesia and chronic inflammation.
Acknowledgments
Supported by INSERM-Avenir and the ‘Fondation Bettencourt-Schueller’ (NV, NC), the Canadian Institute for Health Research (NV, GWZ), the Alberta Heritage Foundation for Medical Research (NV, GWZ, CA), and NIH grants DK57480, DK39957 (NWB), DK52388 (EFG) and the Crohn's and Colitis Foundation of America (1730, EFG).
Glossary
Abbreviations:
- 4αPDD
4α-phorbol 12,13-didecanoate
- CGRP
calcitonin gene-related peptide
- Dil
1,1′-dioctadecyl-3,3,3′,3-tetramethyl-indocarbocyanine perchlorate
- DRG
dorsal root ganglia
- LI
like-immunoreactivity
- SP
substance P
- TRPV
transient receptor potential vanilloid 4 (TRPV)
Conflicts of interest
None.
References
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, et al. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444–4452. doi: 10.1523/JNEUROSCI.0242-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain. 2005;118:70–79. doi: 10.1016/j.pain.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci. 2006;26:3864–3874. doi: 10.1523/JNEUROSCI.5385-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. edn. 2008 revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, Hayashida M, Yasunami J, Takamatsu I, Adachi K, Toyoshima K. Bronchoconstrictive effect of ultrasonic nebulized distilled water on asthmatic children. J Asthma. 1989;26:299–307. doi: 10.3109/02770908909073267. [DOI] [PubMed] [Google Scholar]
- Baluk P, Bertrand C, Geppetti P, McDonald DM, Nadel JA. NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am J Physiol. 1995;268:L263–9. doi: 10.1152/ajplung.1995.268.2.L263. [DOI] [PubMed] [Google Scholar]
- Baluk P, Thurston G, Murphy TJ, Bunnett NW, McDonald DM. Neurogenic plasma leakage in mouse airways. Br J Pharmacol. 1999;126:522–528. doi: 10.1038/sj.bjp.0702323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraniuk JN, Ali M, Yuta A, Fang SY, Naranch K. Hypertonic saline nasal provocation stimulates nociceptive nerves, substance P release, and glandular mucous exocytosis in normal humans. Am J Respir Crit Care Med. 1999;160:655–662. doi: 10.1164/ajrccm.160.2.9805081. [DOI] [PubMed] [Google Scholar]
- Bennell K, Hodges P, Mellor R, Bexander C, Souvlis T. The nature of anterior knee pain following injection of hypertonic saline into the infrapatellar fat pad. J Orthop Res. 2004;22:116–121. doi: 10.1016/S0736-0266(03)00162-1. [DOI] [PubMed] [Google Scholar]
- Brain SD, Williams TJ. Interactions between the tachykinins and calcitonin gene-related peptide lead to the modulation of oedema formation and blood flow in rat skin. Br J Pharmacol. 1989;97:77–82. doi: 10.1111/j.1476-5381.1989.tb11926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Altier C, Chapman K, Liedtke W, Zamponi GW, Vergnolle N. Transcient receptor potential vanilloid-4 has a major role in visceral hypersensitivity symptoms. Gastroenterology. 2008;135:937–946. doi: 10.1053/j.gastro.2008.05.024. [DOI] [PubMed] [Google Scholar]
- Candenas L, Lecci A, Pinto FM, Patak E, Maggi CA, Pennefather JN. Tachykinins and tachykinin receptors: effects in the genitourinary tract. Life Sci. 2005;76:835–862. doi: 10.1016/j.lfs.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Choi IS, Hong SN, Lee YK, Koh YI, Jang AS, Lee HC. Asthmatic airway inflammation is more closely related to airway hyperresponsiveness to hypertonic saline than to methacholine. Korean J Intern Med. 2003;18:83–88. doi: 10.3904/kjim.2003.18.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–32046. doi: 10.1074/jbc.M303251200. [DOI] [PubMed] [Google Scholar]
- Damas J, Liegeois JF. The inflammatory reaction induced by formalin in the rat paw. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:220–227. doi: 10.1007/pl00005345. [DOI] [PubMed] [Google Scholar]
- Del Bianco E, Tramontana M, Bertrand C, Geppetti P. Release of sensory CGRP by hypertonic NaCl is not blocked by tetrodotoxin, omega-conotoxin, nifedipine and ruthenium red. Life Sci. 1992;51:PL73–PL76. doi: 10.1016/0024-3205(92)90234-g. [DOI] [PubMed] [Google Scholar]
- Drewes AM, Babenko L, Birket-Smith L, Funch-Jensen P, Arendt-Nielsen L. Induction of non-painful and painful intestinal sensations by hypertonic saline: a new human experimental model. Eur J Pain. 2003;7:81–91. doi: 10.1016/s1090-3801(02)00070-8. [DOI] [PubMed] [Google Scholar]
- Fian R, Grasser E, Treiber F, Schmidt R, Niederl P, Rosker C. The contribution of TRPV4-mediated calcium signaling to calcium homeostasis in endothelial cells. J Recept Signal Transduct Res. 2007;27:113–124. doi: 10.1080/10799890701402446. [DOI] [PubMed] [Google Scholar]
- Figini M, Emanueli C, Grady EF, Kirkwood K, Payan DG, Ansel J, et al. Substance P and bradykinin stimulate plasma extravasation in the mouse gastrointestinal tract and pancreas. Am J Physiol. 1997;272:G785–G793. doi: 10.1152/ajpgi.1997.272.4.G785. [DOI] [PubMed] [Google Scholar]
- Grant AD, Gerard NP, Brain SD. Evidence of a role for NK1 and CGRP receptors in mediating neurogenic vasodilatation in the mouse ear. Br J Pharmacol. 2002;135:356–362. doi: 10.1038/sj.bjp.0704485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graven-Nielsen T, Arendt-Nielsen L, Svensson P, Jensen TS. Quantification of local and referred muscle pain in humans after sequential i.m. injections of hypertonic saline. Pain. 1997;69:111–117. doi: 10.1016/s0304-3959(96)03243-5. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Quarcoo D, Frossard N, Fischer A. Neurogenic mechanisms in bronchial inflammatory diseases. Allergy. 2004;59:1139–1152. doi: 10.1111/j.1398-9995.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle S, Papez MD, Ferazzini M, Hollenberg MD, Vergnolle N. Neutrophils and the kallikrein-kinin system in proteinase-activated receptor 4-mediated inflammation in rodents. Br J Pharmacol. 2005;146:670–678. doi: 10.1038/sj.bjp.0706371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Norup M. Experimental pain in human temporal muscle induced by hypertonic saline, potassium and acidity. Cephalalgia. 1992;12:101–106. doi: 10.1046/j.1468-2982.1992.1202101.x. [DOI] [PubMed] [Google Scholar]
- Karaki SI, Kuwahara A. Regulation of intestinal secretion involved in the interaction between neurotransmitters and prostaglandin E2. Neurogastroenterol Motil. 2004;16(Suppl. 1):96–99. doi: 10.1111/j.1743-3150.2004.00482.x. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DM. Neurogenic inflammation in the rat trachea. I. Changes in venules, leucocytes and epithelial cells. J Neurocytol. 1988;17:583–603. doi: 10.1007/BF01260988. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Abelli L, Giuliani S, Somma V, Furio M, Patacchini R, et al. Motor and inflammatory effect of hyperosmolar solutions on the rat urinary bladder in relation to capsaicin-sensitive sensory nerves. Gen Pharmacol. 1990;21:97–103. doi: 10.1016/0306-3623(90)90602-i. [DOI] [PubMed] [Google Scholar]
- Mandahl A, Brodin E, Bill A. Hypertonic KCI, NaCl and capsaicin intracamerally causes release of substance P-like immunoreactive material into the aqueous humor in rabbits. Acta Physiol Scand. 1984;120:579–584. doi: 10.1111/j.1748-1716.1984.tb07423.x. [DOI] [PubMed] [Google Scholar]
- Misery L, Meyronet D, Pichon M, Brutin JL, Pestre P, Cambazard F. Aquadynia: a role for VIP? Ann Dermatol Venereol. 2003;130:195–198. [PubMed] [Google Scholar]
- Nguyen C, Coelho AM, Grady E, Compton SJ, Wallace JL, Hollenberg MD, et al. Colitis induced by proteinase-activated receptor-2 agonists is mediated by a neurogenic mechanism. Can J Physiol Pharmacol. 2003;81:920–927. doi: 10.1139/y03-080. [DOI] [PubMed] [Google Scholar]
- O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201:167–180. doi: 10.1002/jcp.20061. [DOI] [PubMed] [Google Scholar]
- Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Puliyel JM, Bhambhani V. Ketoacid levels may alter osmotonicity in diabetic ketoacidosis and precipitate cerebral edema. Arch Dis Child. 2003;88:366. doi: 10.1136/adc.88.4.366-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JB, Andersen MK, Hansen HH. Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in malignant disease. J Intern Med. 1995;238:97–110. doi: 10.1111/j.1365-2796.1995.tb00907.x. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Molyneaux L, Yue DK. The relationship among pain, sensory loss, and small nerve fibers in diabetes. Diabetes Care. 2006;29:883–887. doi: 10.2337/diacare.29.04.06.dc05-2180. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- Tramontana M, Cecconi R, Del Bianco E, Santicioli P, Maggi CA, Alessandri M, et al. Hypertonic media produce Ca(2+)-dependent release of calcitonin gene-related peptide from capsaicin-sensitive nerve fibres in the rat urinary bladder. Neurosci Lett. 1991;124:79–82. doi: 10.1016/0304-3940(91)90826-f. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Gazzieri D, Benvenuti F, Campi B, Dinh QT, Groneberg DA, et al. Ethanol causes inflammation in the airways by a neurogenic and TRPV1-dependent mechanism. J Pharmacol Exp Ther. 2004;309:1167–1173. doi: 10.1124/jpet.103.064162. [DOI] [PubMed] [Google Scholar]
- Umeno E, McDonald DM, Nadel JA. Hypertonic saline increases vascular permeability in the rat trachea by producing neurogenic inflammation. J Clin Invest. 1990;85:1905–1908. doi: 10.1172/JCI114652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamvakas S, Teschner M, Bahner U, Heidland A. Alcohol abuse: potential role in electrolyte disturbances and kidney diseases. Clin Nephrol. 1998;49:205–213. [PubMed] [Google Scholar]
- Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol. 1999;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, et al. Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat Med. 2001;7:821–826. doi: 10.1038/89945. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002a;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002b;277:47044–47051. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- Weerakkody NS, Percival P, Hickey MW, Morgan DL, Gregory JE, Canny BJ, et al. Effects of local pressure and vibration on muscle pain from eccentric exercise and hypertonic saline. Pain. 2003;105:425–435. doi: 10.1016/S0304-3959(03)00257-4. [DOI] [PubMed] [Google Scholar]