

Abstract
Background and purpose:
Although both microsomal prostaglandin E synthase (mPGES)-1 and cyclooxygenase (COX)-2 are critical factors in stroke injury, but the interactions between these enzymes in the ischaemic brain is still obscure. This study examines the hypothesis that mPGES-1 activity is required for COX-2 to cause neuronal damage in ischaemic injury.
Experimental approach:
We used a glutamate-induced excitotoxicity model in cultures of rat or mouse hippocampal slices and a mouse middle cerebral artery occlusion-reperfusion model in vivo. The effect of a COX-2 inhibitor on neuronal damage in mPGES-1 knockout (KO) mice was compared with that in wild-type (WT) mice.
Key results:
In rat hippocampal slices, glutamate-induced excitotoxicity, as well as prostaglandin (PG) E2 production and PGES activation, was significantly attenuated by either MK-886 or NS-398, inhibitors of mPGES-1 and COX-2 respectively; however, co-application of these inhibitors had neither an additive nor a synergistic effect. The protective effect of NS-398 on the excitotoxicity observed in WT slices was completely abolished in mPGES-1 KO slices, which showed less excitotoxicity than WT slices. In the transient focal ischaemia model, mPGES-1 and COX-2 were co-localized in the infarct region of the cortex. Injection of NS-398 reduced not only ischaemic PGE2 production, but also ischaemic injuries in WT mice, but not in mPGES-1 KO mice, which showed less dysfunction than WT mice.
Conclusion and implications:
Microsomal prostaglandin E synthase-1 and COX-2 are co-induced by excess glutamate in ischaemic brain. These enzymes are co-localized and act together to exacerbate stroke injury, by excessive PGE2 production.
Keywords: prostaglandin E2, prostaglandin E synthase, cyclooxygenase, ischaemia, excitotoxicity, glutamate, inflammation, stroke
Introduction
Stroke remains a major cause of death and neuronal disability worldwide. In the early stages of cerebral ischaemia, activation of glutamate receptors initiates the ischaemic cascade that causes the damage (Butcher et al., 1990; Lee et al., 1999). At later times after ischaemia, inflammation is a major factor in the progression of the injury (Dirnagl et al., 1999; Barone and Feuerstein, 1999).
The prostaglandin (PG) E2, one of the most likely candidates for propagation of inflammation, is known to be produced and accumulated at the lesion sites of the ischaemic brain (Kempski et al., 1987; Iadecola et al., 2001; Ikeda-Matsuo et al., 2006). PGE2 is sequentially synthesized from arachidonic acid in two enzymatic steps: cyclooxygenase (COX) and PGE synthase (PGES). Between the two COX isoforms, COX-1 and COX-2, COX-2 is the more highly inducible form (Kaufmann et al., 1997; Chandrasekharan et al., 2002). Although COX-2 has been immunohistochemically detected specifically in neurons in the normal brain (Kaufmann et al., 1997), it has also been shown to be up-regulated after transient ischaemia (Nogawa et al., 1997; Iadecola et al., 1999; Yokota et al., 2004) and has recently been identified in non-neuronal cells as well at some lesion sites, for example, in microglia in the brains of patients with multiple sclerosis and chronic cerebral ischaemia (Tomimoto et al., 2000; Rose et al., 2004). The genetic disruption and chemical inhibition of COX-2 have been shown to ameliorate neuronal death after transient forebrain ischaemia in mice, suggesting that COX-2 is involved in the pathogenic events occurring in cerebral ischaemia (Nogawa et al., 1997; Iadecola et al., 2001; Sasaki et al., 2004). Because deletion and inhibition of COX-2 attenuated the elevation in PGE2 in the post-ischaemic brain, it was suggested that the accumulated PGE2 may mediate the toxic effects of COX-2 in the brain (Nogawa et al., 1997; Iadecola et al., 2001; Kunz et al., 2007). COX-2-derived PGH2 is a precursor of not only PGE2 but also thromboxane A2 (TXA2), PGI2 and PGD2 (Govoni et al., 2001; Sunose et al., 2001; McAdam et al., 2005). PGI2 and PGD2 are known to have protective effects on ischaemic injury (Cui et al., 2006; Saleem et al., 2007; Taniguchi et al., 2007; Wei et al., 2008), while an inhibitor of TXA2 synthase and antagonist of TXA2 TP receptors have been reported to ameliorate brain ischaemic injury (Matsuo et al., 1993; Iijima et al., 1996). Thus, TXA2 may also mediate the toxic effects of COX-2 in the brain. In addition to prostanoids, COX-2 also generates free radical species (Kontos et al., 1980; Armstead et al., 1988; Torres et al., 2004). Previous studies have shown that one of the primary sources of reactive oxygen species in the ischaemic brain is the metabolism of arachidonic acid by COX (Traystman et al., 1991; Nelson et al., 1992; Chan, 2001; Domoki et al., 2001). Thus, COX inhibitors have been shown to reduce free radical production in global cerebral ischaemia (Hall et al., 1993; Candelario-Jalil et al., 2003) and in traumatic brain injury (Tyurin et al., 2000). Therefore, the prostanoids and/or reactive oxygen species have been suggested to mediate the toxic effects of COX-2 in brain ischaemia, but the major COX-2 reaction products mediating the neurotoxicity of COX-2 have not yet been defined.
Three major isozymes of PGES were recently isolated: cytosolic PGES (cPGES), microsomal PGES (mPGES)-1 and mPGES-2. While cPGES and mPGES-2 are constitutively expressed in various cells and tissues, mPGES-1 is induced by pro-inflammatory stimuli and in various models of inflammation (Jakobsson et al., 1999; Murakami et al., 2002). We have demonstrated that the activation of microglia by lipopolysaccharide (LPS) contributes to PGE2 production through the induction of mPGES-1 expression at sites of inflammation of the brain parenchyma (Ikeda-Matsuo et al., 2005). Recently, using models of focal cerebral ischaemia in mPGES-1 knockout (KO) mice, we demonstrated that induction of mPGES-1 contributes to the exacerbation of stroke injury through PGE2 production following ischaemia (Ikeda-Matsuo et al., 2006).
Although mPGES-1 has been shown to act in concert with COX-2 in in vitro transfection models (Murakami et al., 2000) and there is increasing evidence that mPGES-1 is co-induced with COX-2 in some injury models (Samuelsson et al., 2007), the interaction between these enzymes in the ischaemic brain is still obscure. Thus, contrary to a proposed model of exclusive COX-2/mPGES-1 coordination, it has been observed that COX-2 could coordinate with not only mPGES-1 but also cPGES in the brain (Vazquez-Tello et al., 2004). In addition, un-coupled regulation of COX-2 and mPGES-1 has also been observed in rat microglia (de Oliveira et al., 2008). Furthermore, up-regulation of cPGES after peritoneal injection of LPS in rats has also been reported (Tanioka et al., 2000). Considering that the contribution of mPGES-1 and PGE2 in mediating the neurotoxicity of COX-2 has not yet been defined, a study of the relation of mPGES-1 and COX-2 in stroke injury and neuronal death could provide a considerable amount of information that would help in the therapeutic targeting of these enzymes in patients with stroke injury.
Here we report on the co-induction and co-localization of mPGES-1 and COX-2 in the mouse cerebral cortex after transient focal ischaemia in vivo. Using inhibitors of mPGES-1 and COX-2 and mPGES-1 KO mice, we demonstrated that mPGES-1 activity is required for COX-2 to exert enhancement of excitotoxicity, and that mPGES-1 activity exacerbates stroke injury through PGE2 production.
Methods
Animals
All animal care and experimental procedures complied with the guidelines of the Japanese Pharmacological Society. mPGES-1 KO mice and wild-type (WT) mice (C57BL/6J x 129/SvJ background) back-crossed to C57BL/6J mice for >8 generations to avoid artifactual differences caused by genetic background were used (Uematsu et al., 2002). Our preliminary data showed no significant gender differences in infarct volume, degree of oedema, neurological score and motor activity 24 h after ischaemia (data not shown). Therefore, both male and female mice were studied at weight 23–25 g; and data from both sexes were pooled. Wistar rat pups were purchased from Japan SLC, Inc. (Shizuoka, Japan).
Organotypic hippocampal cultures
Brains were rapidly removed from 7-day-old pups, and 300-µm-thick horizontal entorhino-hippocampal slices were placed on transparent membranes (Millicell-CM, Millipore, Billerica, MA, USA) in 6-well culture plates and cultured with 700 µL of culture medium consisting of 50% minimal essential medium, 25% horse serum, and 25% Hanks’ balanced salt solution supplemented with 3.0 mg·mL−1 glucose, 2 mM L-glutamine, 100 U·mL−1 penicillin G and 125 µg·mL−1 streptomycin in a humidified incubator at 37°C in 5% CO2. After 7 days in vitro, slice cultures were exposed to 1 mM glutamate for 15 min for rats and 30 min for mice with or without MK-886 and/or NS-398. An identical treatment was performed with the vehicle as a control group. Then medium was changed to normal culture medium containing 5 µg·mL−1 propidium iodide (PI) with or without MK-886 and/or NS-398 and cultured for 24 h. After that, cells were killed by 24-h incubation with 0.3% Triton X-100 at a low temperature (4°C). PI fluorescence images were obtained with the LSM510 confocal imaging system (Zeiss). The fluorescence intensity at CA1 pyramidale was obtained by measuring averaged gray-scale values of the desired area using a graphic software (Photoshop version 7.0. Adobe systems).
Western blot analysis
The cerebral cortex, striatum and hippocampus were dissected from the brain obtained as described above and lysed by homogenization in 10 mM HEPES-buffered solution (pH 7.4) containing 5 mM EDTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 µg·mL−1 leupeptin, 1 µg·mL−1 pepstatin and 50% glycerol, followed by sonication (3 × 10 s) and centrifugation at 12 000× g for 10 min at 4°C. The protein concentration of the supernatants was measured by the method of Bradford. A total of 15 µg protein of each sample was denatured by boiling for 5 min in sample buffer (62.5 mM Tris-HCl, pH 6.7, 1% SDS, 10% glycerol, 2% 2-mercaptoethanol, 0.025% bromophenol blue), separated by electrophoresis on 15% SDS-polyacrylamide gels, and transferred electrophoretically onto immobilon-P polyvinylidene difluoride membranes (Millipore). The membranes were blocked overnight in Tris-buffered saline-Tween 20 (TBS-T; 10 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 0.1% Tween 20), and 5% skim milk. The membranes were incubated with the appropriate primary antibodies against mPGES-1 (1:500 dilution), mPGES-2 (1:1000), cPGES (1:250), COX-1 (1:250) and COX-2 (1:1000) in TBS-T for 1.5 h. After washing the membranes with TBS-T, horseradish peroxidase-conjugated secondary antibodies were added at a 1:10 000 dilution in TBS-T and incubated for 1 h. After washes with TBS-T, the protein bands were visualized with ECL Western blot detection reagents (Amersham Biosciences, Piscataway, NJ, USA).
RNA extraction and real-time PCR analysis
RNA extraction was performed as described (Ikeda et al., 2000). Briefly, total RNA was extracted with Trizol® (Invitrogen) and then treated with RNase-free DNase I for 15 min at room temperature. Samples of total RNA (2µg) were reverse-transcribed using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA, USA). Expression of COX-2 and mPGES-1 mRNAs was quantified by real-time PCR with the ABI PRISM® (Applied Biosystems) technology and the SYBR® Premix Ex Taq™ (Takara). After amplification (1 cycle at 95°C for 1 min and 40 cycles at 95°C for 15 s, 60°C for 1 min), a melting curve was constructed to determine the melting temperature of each PCR product; their sizes were checked on a 2% agarose gel stained with ethidium bromide (0.1 µg·mL−1). The mRNA levels of each gene of interest and of GAPDH, chosen as a housekeeping gene, were determined in parallel for each sample. Results are expressed as the normalized ratio of mRNA level of each gene of interest to the GAPDH gene. The gene-specific primer pairs used for real-time PCR were as follows: mPGES-1, sense 5’-ATCAAGATGTACGCGGTGGC-3’, antisense 5’-GAGGAAATGTATCCAGGCGA-3’; COX-2, sense 5’-TACAAGCAGTGGCAAAGGCC-3’, antisense 5’-CAGTATTGAGGAGAACAGATGGG-3’; GAPDH, sense 5’-ACCACCAACTGCTTA GCC-3’, antisense 5’-ATCACGCCACAGCTTTCC-3’.
PGE2 assay
The concentration of the PGE2 in dissected brain tissues or culture medium was determined using an enzyme immunoassay (EIA) kit (Cayman Chemicals). The brain tissues were quickly frozen in liquid nitrogen and weighed to determine the wet weight. Prostanoids were extracted by homogenization of the tissues in 70% methanol solution containing 10 µM indomethacin and centrifugation at 15 000× g for 20 min at 4°C. The supernatant was evaporated and dissolved and diluted with the assay buffer. The hippocampal culture medium was also diluted with the assay buffer. The PGE2 concentration was determined according to the instructions provided with the kit.
Assay of enzymatic activity of PGES
Prostaglandin E synthase activities in cell lysates were measured by assessment of the conversion of PGH2 to PGE2. The slices were disrupted by sonication (3 × 10 s, at 1-min intervals) in 100 µL of phosphate-buffered saline (PBS) supplemented with 2.5 mM DTT, 1 mM PMSF, 10 µg·mL−1 leupeptin and 30 µg·mL−1 pepstatin A. After centrifugation of the sonicates at 10 000× g for 5 min at 4°C, the supernatants were further centrifuged at 100 000× g for 1 h at 4°C, and the microsomal membranes (pellets which were resuspended in 100 µL of PBS supplemented with 2.5 mM DTT, 1 mM PMSF, 10 µg·mL−1 leupeptin and 30 µg·mL−1 pepstatin A) were used to measure PGES activity. An aliquot of each lysate (30-µg protein equivalents) was incubated with 0.5 µg of PGH2 for 30 s at 24°C in 100 µL of 0.2 M Tris-HCl, pH 8.0, containing 2 mM glutathione and 14 µM indomethacin. After terminating the reaction by the addition of 100 mM FeCl2, PGE2 contents in the supernatants were quantified by use of an EIA kit (Cayman Chemical).
Induction of transient focal ischaemia
Middle cerebral artery (MCA) occlusion was carried out under halothane anesthesia (5.0% for induction, 1.0% for maintenance) as described previously (Ikeda-Matsuo et al., 2006). Briefly, the right common carotid artery was exposed through a midline incision, and occlusion of the MCA was achieved by inserting a 6-0 nylon monofilament with a heat-blunted tip coated with silicon thread through the proximal external carotid artery into the internal carotid artery and up to the MCA (9 mm from the internal carotid/pterygopalatine artery bifurcation). The occlusion of the MCA was maintained for 1.5 h, followed by reperfusion for 1∼3 days. Body temperature was monitored by a rectal thermometer and maintained at 37°C by using a warm pad during the operation and until the animals were awake. In sham-operated animals, an incision was made over the MCA but the artery was not occluded. The COX-2 inhibitor NS-398 was diluted in saline at pH 8.5 and vehicle consisted of saline (pH 8.5). NS-398 (10 mg·kg−1; i.p.) or vehicle was administered twice daily starting 10 min after reperfusion.
Immunocytochemistry
Animals were anesthetized with sodium pentobarbital and then perfused transcardially with saline, followed by 4% paraformaldehyde in PBS. The brains were removed, post-fixed overnight in a solution containing 4% paraformaldehyde and 4% sucrose in PBS, and then cryoprotected in solutions containing 10%, 15% and 20% sucrose in PBS for 1 day each. The brains were then frozen in dry-ice powder, and coronal sections (20 µm) were cut using a cryostat. The sliced tissues were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.3% Triton-X for 10 min, and then treated with 3% bovine serum albumin (BSA) for 30 min in PBS to block nonspecific binding. The preparations were incubated with appropriate primary antibodies against mPGES-1 (1:250 dilution), COX-2 1:250), CD-11b (1:50) and Neu-N (1:1000) in PBS containing 3% BSA at 4°C overnight, and washed in PBS. Staining was performed according to protocol of the avidin-biotin complex kit (Vector Laboratories Inc., Burlingame, CA, USA). For fluoroimmunostaining, the Cy3- or FITC-conjugated secondary antibodies (1:100) were used. The slices were mounted with Vectashield Mounting Medium (Vector Laboratories Inc.) and examined using a confocal laser scanning system (LSM510) on an Axiovert200M inverted microscope (Carl Zeiss, Germany).
Quantification of infarct volume
After reperfusion for 72 h, the animals were killed and the brains removed. Brains were sectioned coronally into five 2 mm sections and incubated with 2% triphenyltetrazolium chloride (TTC) in saline for 10 min at 37°C. The area of infarct, identified by the lack of TTC staining, was measured on the rostral and caudal surfaces of each slice using Scion image software (Scion Corp., Frederick, MD, USA) and numerically integrated across the thickness of the slice to obtain an estimate of the infarct volume in each slice. The volumes from all slices were summed to calculate the total infarct volume over the entire infarcted hemisphere. The infarct volume was measured separately in the cerebral cortex, striatum and hemisphere, and corrected for swelling by comparing the volume of the neocortex in the infarcted hemisphere with that in the noninfarcted hemisphere. The swelling rates were calculated as the rates of change in the volumes.
Behaviuoral experimenst
The animals were scored for neurological deficits at 0 h or 1∼3 days after reperfusion. Ischaemic neurological deficits were confirmed and scored as follows: 0, no deficit; 1, reduced outstretching of the contralateral forelimb when lifted by the tail; 2, impaired forepaw walking when made to walk on the forelimbs while being held by the tail; 3, flexion of the torso and contralateral forelimb (>3 s) when lifted by the tail; 4, contralateral forelimb weakness upon application of pressure to the side of the body; 5, circling to the affected side; and 6, no spontaneous locomotor activity. The highest score of the observed defects in each animal (min: 0; max: 6) was taken as the neurological score.
Blood–brain barrier (BBB) permeability
The integrity of the BBB was investigated using Evans Blue. Evans Blue (2% in saline; 200 µL) was injected into mice via a tail vein 1 day after 1.5 h-MCA occlusion and mice were killed 24 h after the injection (2 days after MCA occlusion). Animals were anesthetized with pentobarbital (50 mg·kg−1, i.p.) and perfused transcardially with saline (3.2 mL·min−1) for 10 min until blue color was absent from the effluent. Mice were decapitated and brains were immediately removed and placed on plates, on ice. The cortices were weighed and homogenized in 50% trichloroacetic acid solution to extract the dye in the supernatant. The tissue content of Evans Blue was estimated from the absorbance of 620 nm.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical significance was evaluated with one-way analysis of variance followed by Tukey's test. Values of P < 0.05 were considered to indicate statistical significance.
Materials
MK-886, NS-398, indomethacin and TTC were purchased from Sigma-Aldrich (Deisenhofen, Germany). The IC50 values of MK-886 obtained by suppression of enzyme activity in vitro are 2.4 and 58 µM for mPGES-1 and COX-2 respectively (Claveau et al., 2003; Koeberle et al., 2009). The IC50 values of NS-398 obtained by suppression of enzyme activity in vitro are 20 and 0.04–4 µM for mPGES-1 and COX-2 respectively (Ouellet and Percival, 1995; Warner et al., 1999; Thorén and Jakobsson, 2000). Therefore, we used MK-886 and NS-398 at the concentrations of 1–10 µM and 1 µM, respectively, in the in vitro system. The rabbit anti-human mPGES-1, mPGES-2 and cPGES polyclonal antibodies, anti-ovine COX-1 monoclonal antibody, and PGH2 were from Cayman Chemical (Ann Arbor, MI, USA). Other materials and their sources were as follows: goat anti-human COX-2 polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-neuron-specific nuclear protein (Neu-N) monoclonal antibody (Chemicon, Temecula, CA, USA); anti-CD11b monoclonal antibody (Serotec Inc., Oxford, UK); and multiple-labeling grade secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Other reagents were obtained from Wako Pure Chemical Industries (Osaka, Japan). Anaesthetics were obtained as follows: halothane (Takeda Pharmaceutical, Osaka, Japan) and pentobarbital (Abbott Laboratories, IL, USA).
Results
mPGES-1 and COX-2 are co-induced in hippocampal slices exposed to glutamate
To determine whether mPGES-1 is induced by excitotoxic glutamate stimulation, we first investigated the mPGES-1 protein expression in rat hippocampal slices by Western blot analysis. The slices were stimulated with 1 mM glutamate for 15 min and then cultured with normal medium for 24 h. mPGES-1 protein was slightly expressed in the vehicle-treated control slices (Figure 1A). Glutamate exposure up-regulated the expression to about 3-fold that in the controls (Figure 1B). Although COX-1, COX-2, cPGES and mPGES-2 protein were constitutively expressed in hippocampal slices (Figure 1A), only COX-2 appeared to be up-regulated by glutamate stimulation. However, this up-regulation was not statistically significant (n = 3). To determine whether or not the induction of mPGES-1 and COX-2 was regulated at the pre-translational level, we carried out a real-time PCR analysis. The transcript encoding mPGES-1 was substantially increased 3 h after glutamate exposure (Figure 1C). The mRNA levels of mPGES-1 peaked after 6 h at a value at least 10-fold above the initial levels. The levels decreased after 12 h, and returned to the basal levels by 24 h. COX-2 mRNA was appeared to be increased 3 h after glutamate exposure, but this increase was not significant (Figure 1D). Between 6 and 24 h, the levels of COX-2 decreased gradually to 25% of the basal level, possibly because of the decrease in COX-2-expressing neurons due to the neurotoxic effects of glutamate.
Figure 1.
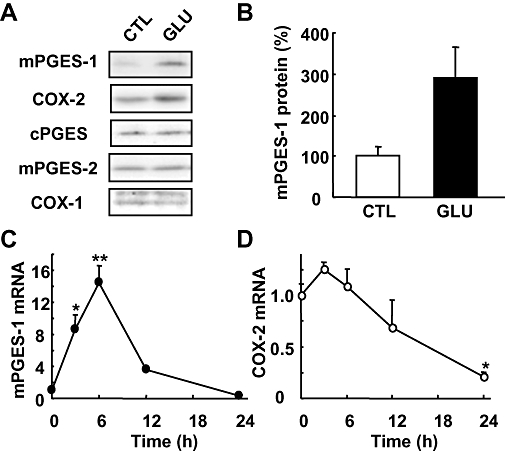
mPGES-1 and COX-2 induction after glutamate exposure in cultures of rat hippocampal slices. (A) Western blot analysis for mPGES-1, COX-2, cPGES, mPGES-2 and COX-1 in the cultures of hippocampal slices 24 h after exposure to 1 mM glutamate (GLU) or vehicle (CTL) for 15 min. Representative data from three separate experiments are presented. (B) Summary data from immunoblotting with mPGES-1 antibody were scaled to a percentage of the control response (n = 3). (C and D) Time course of the expression of mPGES-1 and COX-2 mRNA after glutamate exposure. Quantitated data from real-time PCR analysis with the mPGES-1 (C) and COX-2 (D) primers were normalized to GAPDH (n = 3). **P < 0.01, *P < 0.05 versus the basal level (0 h). COX, cyclooxygenase; cPGES, mPGES, cytosolic/microsomal prostaglandin E synthase.
mPGES-1 is involved in the PGE2 production, PGES activation and excitotoxicity induced by glutamate in hippocampal slices
To elucidate the roles of mPGES-1, we first investigated changes in PGE2 production and PGES enzymatic activity induced by glutamate exposure in the presence or absence of MK-886, an inhibitor of mPGES-1. In hippocampal slice cultures, glutamate elicited an increase in PGE2 to a level 2.5-fold higher than the control level (Figure 2A). This glutamate-induced PGE2 production was suppressed almost completely by MK-886 at a concentration of 3 µM or higher. For the PGES activity assays, we measured the conversion of exogenous PGH2 to PGE2 using membrane fractions of glutamate-exposed cultures. As shown in Figure 2B, the membrane-associated PGES activity was substantially increased 24 h after glutamate exposure. The increased PGES activity was also inhibited by MK-886 in a concentration-dependent manner. These results indicate that glutamate-induced PGE2 production may be mediated by an increase of PGES activity through the induction of mPGES-1 protein.
Figure 2.
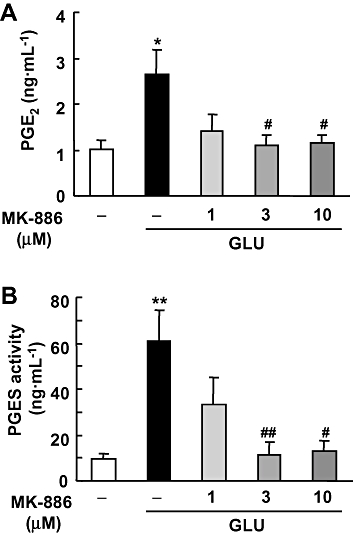
Effect of MK-886 on glutamate-induced PGE2 synthesis and PGES activity in cultures of rat hippocampal slices. MK-886 (1, 3 and 10 µM) was applied during and after the 1 mM glutamate (GLU) exposure for 15 min. (A) The amount of PGE2 in the culture medium 24 h after glutamate exposure was measured using an EIA kit (n = 4). (B) PGES activity in membrane fraction was measured as the conversion of exogenous PGH2 (0.5 µg) to PGE2 for 30 s by the lysate of these cells (30 µg protein). The amount of PGE2 was measured by the same protocol as in (A) (n = 4–5). **P < 0.01, *P < 0.05 versus the control; ##P < 0.01, #P < 0.05 versus glutamate alone. EIA, enzyme immunoassay; PGES, prostaglandin E synthase.
To determine whether or not mPGES-1 induction affects the excitotoxicity induced by glutamate exposure, the cellular damage to hippocampal slices was assessed by fluorescent image analysis of the PI uptake. The exposure of slices to glutamate resulted in neuronal death, with a selective uptake of PI in the CA1 region of the hippocampus (Figure 3A). The increased PI uptake induced by glutamate exposure was inhibited significantly by MK-886 in a concentration-dependent manner (Figure 3B). Taken together, these results suggest that the PGE2 produced by induction of mPGES-1 expression exacerbates excitotoxicity in the hippocampal slice cultures.
Figure 3.
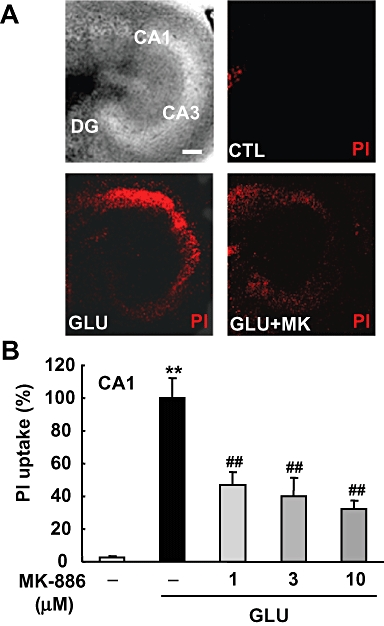
Effect of MK-886 on glutamate-induced excitotoxicity in cultures of rat hippocampal slices. MK-886 (1, 3 and 10 µM) was applied during and after the 1 mM glutamate exposure for 15 min. Twenty-four h later, PI uptake was analysed. (A) A representative differential interference microscopic image of a cultured hippocampal slice and confocal images of PI fluorescence of a control slice (CTL), a slice that received glutamate exposure (GLU) and a slice that received glutamate exposure with 10 µM of MK-886 (GLU+MK) are shown (scale bar: 200 µm). (B) Summary data from PI uptake analysis in the CA1 region with or without glutamate and/or MK-886 were scaled to a percentage of the response of glutamate alone (n = 11–13 slices per group). **P < 0.01 versus the control; ##P < 0.01 versus glutamate alone. PI, propidium iodide.
mPGES-1 and COX-2 are coordinately involved in the PGE2 production, PGES activity and excitotoxicity induced by glutamate in hippocampal slices
To address the question of whether COX-2 and mPGES-1 are functionally coupled to produce PGE2 and thereby enhance glutamate-induced toxicity, we investigated the synergistic effect of MK-886 and NS-398 on PGE2 production, PGES activity and excitotoxicity induced by glutamate. The glutamate-induced PGE2 production was potently inhibited by both NS-398 and MK-886, but co-application of NS-398 and MK-886 had neither an additive nor a synergistic effect (Figure 4A). NS-398 decreased PGE2 production to below the basal level, indicating the necessity of constitutive COX-2 for basal PGE2 production. We next examined the effects of these inhibitors on PGES activity. Treatment with MK-886, but not NS-398, inhibited the glutamate-induced PGES activity (Figure 4B), indicating that NS-398 specifically inhibits COX-2 but not mPGES-1. Under the same conditions, the excitotoxicity induced by glutamate was significantly, but not completely, inhibited by either NS-398 or MK-886; however, co-application of NS-398 and MK-886 had neither an additive nor a synergistic effect: the inhibition by the combination of the two agents was no greater than that by either agent singly (Figure 4C). These results suggest that COX-2 and mPGES-1 are sequentially involved in the PGE2 production and excitotoxicity induced by glutamate in the rat hippocampal slice culture.
Figure 4.
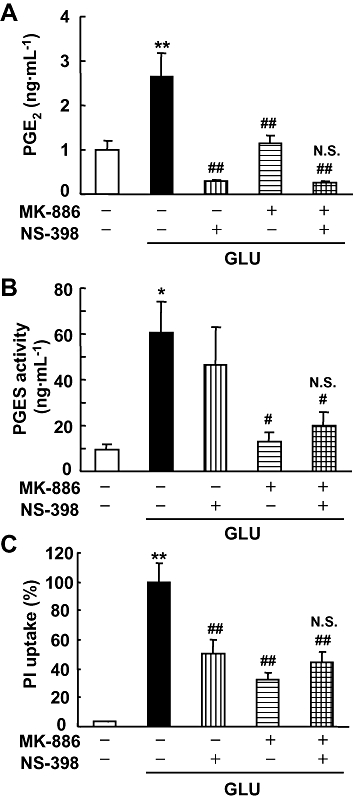
Effect of co-application of MK-886 and NS-398 on glutamate-induced PGE2 synthesis, PGES activity and excitotoxicity in cultures of rat hippocampal slices. MK-886 (10 µM) and NS-398 (1 µM) were applied during and after the 1 mM glutamate (GLU) exposure for 15 min. (A) The amount of PGE2 in the culture medium 24 h after glutamate exposure was measured using an EIA kit (n = 4). (B) PGES activity in the membrane fraction was measured as the conversion of exogenous PGH2 (0.5 µg) to PGE2 for 30 s by the lysate of these cells (30 µg protein). The amount of PGE2 was measured by the same protocol as in (A) (n = 4–5). (C) PI uptake was analysed 24 h after glutamate exposure. Summary data from PI uptake analysis in the CA1 region with or without glutamate and/or MK-886 and/or NS-398 were scaled to a percentage of the response of glutamate alone (n = 11–13 slices per group). **P < 0.01, *P < 0.05 versus the control; ##P < 0.01, #P < 0.05, N.S. (not significant) versus glutamate alone. EIA, enzyme immunoassay; PGES, prostaglandin E synthase; PI, propidium iodide.
To further examine the roles of mPGES-1 and functional coupling of mPGES-1 to COX-2 in glutamate-induced excitotoxicity, we next examined the effect of glutamate and NS-398 on excitotoxicity using hippocampal slices from mPGES-1 KO and WT mice. Glutamate-induced PGE2 production was completely abolished in hippocampal slices from mPGES-1KO mice (Figure 5A), indicating a crucial role of mPGES-1 in glutamate-induced PGE2 production. The expression levels of the other related enzymes, mPGES-2, cPGES, COX-1 and COX-2, were similar between WT- and KO-derived slices (Figure 5B). The glutamate-induced excitotoxicity in slices from WT mice was significantly higher than that in slices from mPGES-1 KO mice (Figure 6). As observed in rat slices, both indomethacin and NS-398 inhibited the glutamate-induced toxicity in slices from WT mice to almost the same level as the toxicity in slices from mPGES-1-KO mice without inhibitors. On the other hand, neither indomethacin treatment nor NS-398 treatment inhibited glutamate-induced excitotoxicity in slices from mPGES-1 KO mice (Figure 6B). Taken together, these results suggest that mPGES-1 itself plays a role in the excitotoxicity induced by glutamate and that mPGES-1 functionally coupled to COX-2 exacerbates this excitotoxicity.
Figure 5.
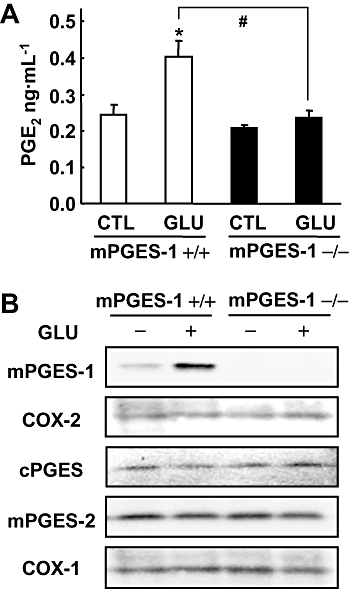
mPGES-1 is an essential component for PGE2 production after glutamate exposure in cultures of mice hippocampal slices. (A) The production of PGE2 in the culture medium of slices from mPGES-1 KO (−/−) and WT (+/+) mice 24 h after 1 mM glutamate (GLU) exposure for 30 min and in control hippocampal slices (CTL) (n = 3). (B) Western blot analysis for mPGES-1, COX-2, cPGES, mPGES-2 and COX-1 in cultures of hippocampal slices 24 h after glutamate exposure. Representative data from three separate experiments are presented. *P < 0.05 versus the control; #P < 0.05 versus glutamate alone in slices from WT mice. COX, cyclooxygenase; cPGES, mPGES, cytosolic/microsomal prostaglandin E synthase; KO, knockout; WT, wild-type.
Figure 6.
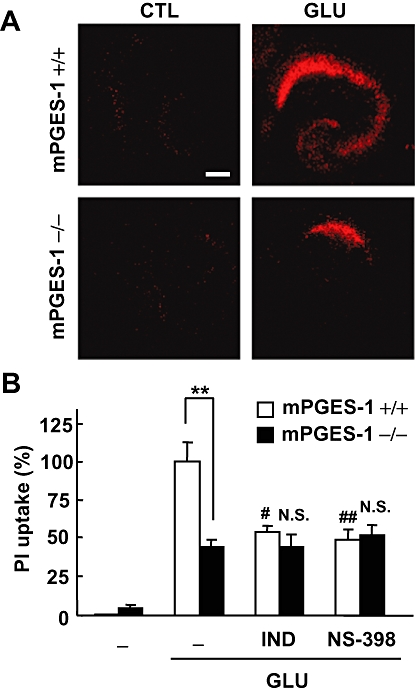
Effects of COX inhibitors on the glutamate-induced excitotoxicity in an mPGES-1 KO and a WT mouse hippocampal slice culture. PI uptake was analysed 24 h after 1 mM glutamate exposure for 30 min. (A) Representative confocal images of PI fluorescence of a control slice (CTL) and a slice that received glutamate (GLU) exposure in WT (+/+) mice and mPGES-1 KO (−/−) mice are shown (scale bar: 200 µm). (B) Summary data from PI uptake analysis in the CA1 region with or without glutamate and indomethacin (IND, 1 µM) or NS-398 (1 µM) were scaled to a percentage of the glutamate response in slices from WT mice. Indomethacin and NS-398 were applied during and after the glutamate exposure (n = 6–11 slices per group). **P < 0.01, ##P < 0.01, #P < 0.05, versus glutamate alone in a slice from a WT mouse; N.S. (not significant) versus glutamate alone in a slice from a mPGES-1 KO mouse. COX, cyclooxygenase; mPGES, microsomal prostaglandin E synthase; KO, knockout; PI, propidium iodide; WT, wild-type.
mPGES-1 and COX-2 are co-induced after focal cerebral ischaemia
To extend the observations from the slice cultures to in vivo models of ischaemic injury, we used the mouse MCA occlusion-reperfusion model which showed an increase in PGE2 production in the post-ischaemic cortex (Ikeda-Matsuo et al., 2006). We have reported previously that mPGES-1 and COX-2 protein were up-regulated in a rat ischaemia model with 2h occlusion (Ikeda-Matsuo et al., 2006). In the present study, to examine the effect of giving repeated NS-398 injections for 3 days after ischaemia (Sugimoto and Iadecola, 2003), we used a less severe occlusion model in order to avoid the death of mice after reperfusion. Occlusion of the MCA for 1.5 h and reperfusion resulted in an expansion of infarction up to 3 days after ischaemia; the infarct volumes of the whole brain after 24 h, 48 h and 72 h were 4.8 ± 0.6%, 13.1 ± 2.4% and 13.5 ± 2.1% respectively (n = 5–9). Because the PGE2 content in the ipsilateral cortex peaked at 1 day after ischaemia and returned to the basal level by 3 days (data not shown), we investigated the effect of NS-398 on post-ischaemic PGE2 production at 1 day after ischaemia. Injection of 10 mg·kg−1 NS-398 significantly inhibited the post-ischaemic PGE2 production in WT mice (Figure 7A). In mPGES-1 KO, whose basal PGE2 level in the cerebral cortex was significantly lower than that in WT mice (P < 0.01), post-ischaemic PGE2 production was completely absent, indicating that mPGES-1 contributes to the post-ischaemic PGE2 production, and also, at least in part, to the basal PGE2 production in the cerebral cortex. The constant level of 6-keto-PGF1α (the stable end product of PGI2) and the decreased level of thromboxane B2 (the stable end product of thromboxane A2) in the cerebral cortex after ischaemia were similar in vehicle-injected and NS-398-injected WT mice and also in mPGES-1 KO mice (data not shown). The inducible expression of mPGES-1 in both the cerebral cortex and striatum was decreased and almost disappeared in NS-398-injected mice, while the expressions of mPGES-2, cPGES, and COX-1 and the inducible expression of COX-2 were similar in vehicle-injected mice and NS-398-injected mice (Figure 7B). The expression levels of mPGES-2, cPGES and COX-1 were similar in WT and mPGES-1 KO mice (data not shown). The inducible expression of COX-2 was observed in WT and mPGES-1 KO mice; the expression levels in the ipsilateral cortex and striatum of WT mice were 130 ± 4% (n = 3, P < 0.05) and 192 ± 158% (n = 3, P < 0.05) respectively, versus those in the contralateral hemisphere, while those in KO mice were 113 ± 16% (n = 3, not significant) and 138 ± 3% (n = 3, P < 0.01) respectively. Immunostaining of mouse brain slices for mPGES-1 and COX-2 revealed co-induction and co-localization of these proteins in the ipsilateral cortex (Figure 7C). As we have reported in the rat 2h occlusion model (Ikeda-Matsuo et al., 2006), immunostaining of mouse brain slices for mPGES-1, as well as COX-2, revealed induction of these proteins in the Neu-N-positive cells in the peri-infarct region and CD11b-positive cells in the core region of the post-ischaemic cortex (Figure 7D).
Figure 7.
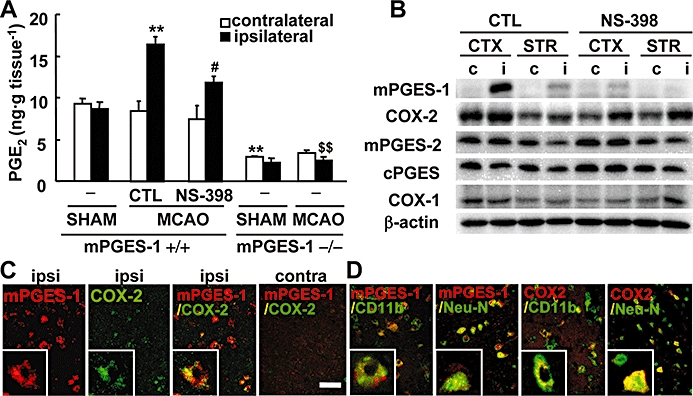
Co-induction of mPGES-1 and COX-2 contributes to post-ischaemic PGE2 production. (A) The production of PGE2 in the ipsilateral or contralateral cortex of mPGES-1 KO (−/−) or WT (+/+) mice injected with NS-398 or vehicle (CTL), 24 h after MCA occlusion (MCAO) and sham operation (SHAM) (n = 4–6 mice per group). (B) Western blot analysis for mPGES-1, COX-2, cPGES, mPGES-2, COX-1 and β-actin in the ipsilateral (i) or contralateral (c) cerebral cortex (CTX) and striatum (STR) of WT mice injected with NS-398 or vehicle (CTL). Representative data from three separate experiments are presented. (C) The double-immunostaining of mPGES-1 (red) and COX-2 (green) in the ischaemic regions of the ipsilateral (ipsi) and contralateral (contra) cortex. Insets show high magnification of staining. (D) The double-immunostaining of mPGES-1 (red) or COX-2 (red) and CD11b (green) in the ischaemic core region or Neu-N (green) in the peri-infarct region of the ipsilateral cortex. Insets show high magnification of staining. The photos shown here are representative examples from three separate experiments (scale bar, 20 µm; insets, 5 µm). **P < 0.01 versus the ipsilateral cortex of sham-operated WT mice or contralateral cortex of WT mice after MCA occlusion; #P < 0.05 versus the ipsilateral cortex of CTL-injected WT mice after MCA occlusion; $$P < 0.01 versus the ipsilateral cortex of WT mice after MCA occlusion. COX, cyclooxygenase; cPGES, mPGES, cytosolic/microsomal prostaglandin E synthase; KO, knockout; MCA, middle cerebral artery; Neu-N, neuron-specific nuclear protein; WT, wild-type.
mPGES-1 and COX-2 were coordinately involved in the exacerbation of ischaemic injury in vivo
To elucidate the functional coupling of mPGES-1 and COX-2 in the ischaemic brain in vivo, we investigated the effect of NS-398 on ischaemic brain injury in both WT and mPGES-1 KO mice. Injection of NS-398 (10 mg·kg−1, i.p.) after reperfusion significantly decreased infarct volume in the whole brain and cerebral cortex of WT mice (Figure 8A,B). As we have reported previously, the infarct volume of mPGES-1 KO mice was significantly smaller than that of WT mice. NS-398 did not attenuate the infarction in mPGES-1 KO mice, suggesting that mPGES-1 is required for the effects of COX-2, which contribute to the expansion of infarction. The injection of NS-398 ameliorated the neurological dysfunctions by 48 h after ischaemia in WT mice, while it showed no effect in mPGES-1 KO mice by 72 h after ischaemia (Figure 8C). Furthermore, NS-398 significantly attenuated oedema in the WT mice but not in the mPGES-1 KO mice (Figure 9A).
Figure 8.
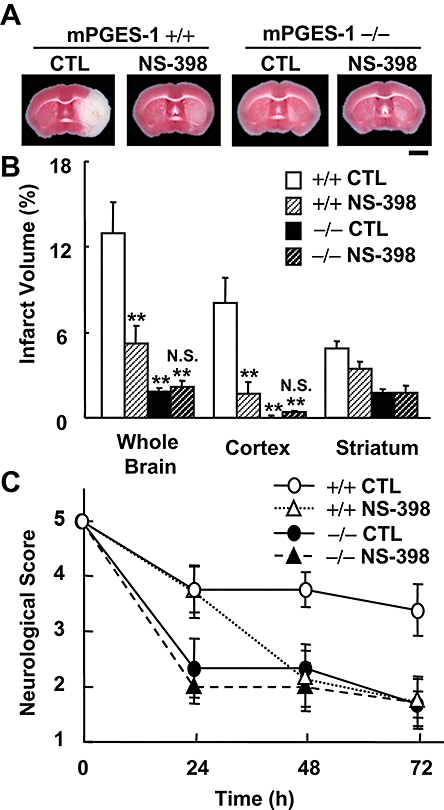
Protective effect of NS-398 on post-ischaemic symptoms in WT mice but not in mPGES-1 KO mice. NS-398 (10 mg·kg−1; i.p.) or vehicle was administered twice daily starting 10 min after MCA occlusion. (A) Representative TTC-stained coronal sections 72 h after MCA occlusion of the WT (+/+) mice and mPGES-1 KO (−/−) mice injected with vehicle (CTL) or NS-398 (scale bar: 2 mm). (B) The volume of infarcted cortex 72 h after ischaemia was estimated and expressed as a percentage of the corrected tissue volume (n = 7–9 mice per group). (C) Neurological dysfunction in the NS-398-injected WT and mPGES-1 KO mice 24, 48 and 72 h after ischaemia (n = 7–9 mice per group); **P < 0.01 versus WT control mice; N.S. (not significant) versus mPGSE-1 KO control mice. KO, knockout; MCA, middle cerebral artery; mPGES, microsomal prostaglandin E synthase; WT, wild-type.
Figure 9.
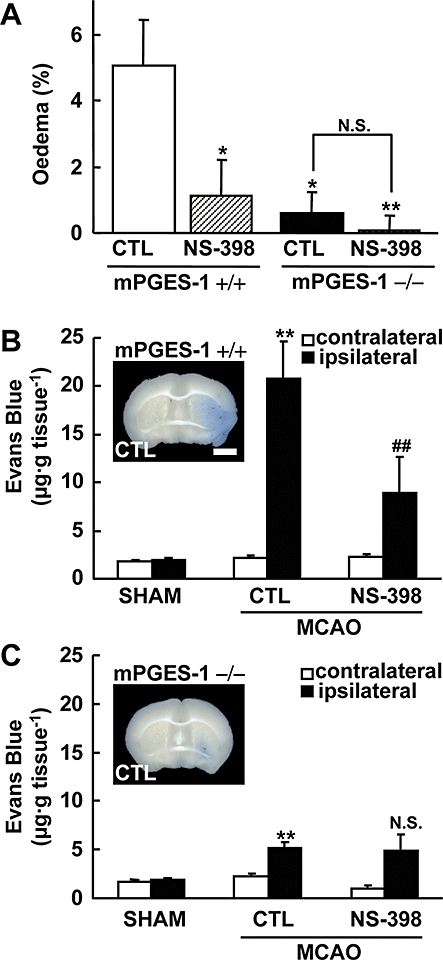
Protective effect of NS-398 on oedema and Evans Blue extravasation after ischaemia in WT mice but not in mPGES-1 KO mice. NS-398 (10 mg·kg−1; i.p.) or vehicle was administered twice daily starting 10 min after MCA occlusion. (A) The corrected oedema percentage in the NS-398 or vehicle (CTL)-injected WT (+/+) and mPGES-1 KO (−/−) mice (n = 7–8 mice per group). **P < 0.01, *P < 0.05 versus the vehicle-injected WT mice. (B and C) The Evans Blue contents of the ipsilateral and contralateral cortex after MCA occlusion (MCAO) or sham-operation (SHAM) in WT (B) and mPGES-1 KO (C) mice, injected with NS-398 or vehicle were measured 48 h after transient ischaemia (n = 6–7 mice per group). **P < 0.01 versus the contralateral cortex of mice after MCA occlusion or the ipsilateral cortex of sham-operated mice; ##P < 0.01, N.S. (not significant) versus the ipsilateral cortex of vehicle-treated mice after MCA occlusion The insets in (B) and (C) show representative results of Evans Blue-stained brain slices of a vehicle-injected WT (+/+) and mPGES-1 KO (−/−) mouse respectively (scale bar: 2 mm). KO, knockout; MCA, middle cerebral artery; mPGES, microsomal prostaglandin E synthase; WT, wild-type.
The permeability of BBB was quantified by leakage of Evans Blue into the ischaemic cortex. Evans Blue content in the brain of sham-operated mice was very low (Figure 9B). Forty-eight h after MCA occlusion, Evans Blue leakage into the ischaemic cortex of WT mice was markedly increased compared with that in sham-operated mice. NS-398 significantly reduced the Evans Blue level in the ischaemic cortex in WT mice. In mPGES-1 KO mice, slight but significant Evans Blue leakage was observed after ischaemia; however, NS-398 did not attenuate the leakage in the mPGES-1 KO mice (Figure 9C). Taken together, these results suggest that mPGES-1 is needed for COX-2 to exert its effects, which contribute to the oedema, BBB disruption and neuronal dysfunctions observed after ischaemia, and that mPGES-1 is functionally coupled with COX-2 to exacerbate stroke injury through PGE2 production.
Discussion
Here we have shown that mPGES-1 and COX-2 are co-induced and co-localized after ischaemic stimuli and coordinately contribute to the exacerbation of ischaemic excitotoxicity. Using mPGES-1 KO mice and inhibitors of mPGES-1 and COX-2, we provide clear evidence that mPGES-1 activity is necessary for the neurotoxicity consequent on COX-2 induction in both in vitro and in vivo ischaemic models.
We have recently demonstrated that up-regulation of mPGES-1 exacerbates the stroke injury observed after ischaemia (Ikeda-Matsuo et al., 2006). In this study, cultures of hippocampal slices from mPGES-1 KO mice showed lower glutamate-induced excitotoxicity compared with those of WT mice, indicating that mPGES-1 also exacerbates the excitotoxicity in vitro. Thus, this excitotoxicity model of cultured hippocampal slices is a useful in vitro tool for investigation of the mPGES-1-related neurotoxicity observed in transient focal ischaemia in vivo. Using this model, we showed that mPGES-1 activity was required for the neurotoxicity of COX-2.
Both mRNA and protein expression of mPGES-1 were up-regulated by glutamate exposure, indicating that induction of mPGES-1 is regulated, at least in part, at the pre-translational level. Considering that an excess amount of extracellular glutamate is present in the post-ischaemic cortex (Dávalos et al., 2000), glutamate may be one of the most potent inducers of mPGES-1 in the post-ischaemic cortex. Indeed, two transcription factors, NF-κB and Egr-1, which have been demonstrated to be involved in transcriptional regulation of mPGES-1 (Naraba et al., 2002; Barakat et al., 2009), are known to be activated by glutamate and the glutamate receptor agonist N-methyl-D-aspartate, as well as by MCA occlusion (Bading et al., 1995; Huang et al., 2003; Lu et al., 2003; Fan and Cooper, 2009). On the other hand, the mRNA and protein of COX-2 were not significantly up-regulated in the present study. In a similar focal ischaemia model, Miettinen et al. (1997) have also demonstrated moderate induction of the COX-2 protein. Because COX-2 is constitutively expressed in neurons (Kaufmann et al., 1997; Quan et al., 1998), the expression level of COX-2 analysed by Western blot could be attributed to both glutamate/MCA-occlusion-induced up-regulation and the neurotoxic effect, which decreases COX-2-expressing neurons. On the other hand, the immunostaining for COX-2 showed the induction of COX-2 in the ipsilateral cortex of mice after MCA occlusion, possibly because the induction of COX-2 was much higher than the basal COX-2 expression, which was undetectable by this method. The double immunostaining for mPGES-1 and COX-2 showed that mPGES-1 and COX-2 were co-induced and co-localized in Neu-N-positive neurons and CD11b-positive neutrophil/macrophage/microglia in the peri-infarct region and ischaemic core region respectively. Although we found no apparent changes in the expression of cPGES, mPGES-2 or COX-1 after glutamate exposure or transient ischaemia, the possibility that other types of PGES or COX contribute to post-ischaemic PGE2 production could not be excluded. However, our results using mPGES-1-KO mice and NS-398 indicate the predominant roles of mPGES-1 and COX-2 in post-ischaemic PGE2 production, as well as the potentiation of ischaemic injury, in the brain. On the other hand, basal PGE2 levels in the brain were reduced but not abolished by genetic deletion of mPGES-1 and were not affected by inhibition of COX-2, suggesting that the contribution of not only mPGES-1, but also other cPGES and/or mPGES-2 and COX-1 are involved in the basal PGE2 production (Figure 7A). Injection of NS-398 did not affect the expression of COX-1, COX-2, mPGES-2 or cPGES, but reduced the expression of mPGES-1, indicating that a positive-feedback mechanism may be involved in the induction of mPGES-1 expression and thereby escalate PGE2 production and inflammation after cerebral ischaemia, as reported in rheumatoid synovial fibroblasts (Kojima et al., 2003). Therefore, the protective effect of NS-398 on ischaemic injuries may be attributed to inhibition of post-ischaemic PGE2 production by not only the inhibition of COX-2 activity, but also the inhibition of mPGES-1 expression.
There was no compensatory up-regulation of other enzymes involved in PGE2 biosynthesis in mPGES-1 KO mice. By using these mice, we showed that genetic disruption of mPGES-1 ameliorated glutamate-induced excitotoxicity and brain injury. Similar protective effects were observed by COX-2 inhibition; however, inhibition by both COX-2 and mPGES-1 had neither an additive nor a synergistic protective effect, indicating that mPGES-1 activity and PGE2 are required for the COX-2 neurotoxicity observed after ischaemia. In other words, among the COX-2 reaction products, PGE2 made the greatest contribution to mediation of the neurotoxicity of COX-2 in our experimental ischaemia models. Kunz et al. (2007) have recently reported that the COX-2 inhibitor NS-398 attenuated PGE2 production and brain injury, but did not affect production of reactive oxygen species, suggesting that COX-2 is not a major source of oxygen radicals after cerebral ischaemia. Indeed, the reported phenotype of mPGES-1 KO mice after ischaemic insult resembles that of COX-2 KO mice (Iadecola et al., 2001; Ikeda-Matsuo et al., 2006).
How, then, is the toxicity of post-ischaemic PGE2 mediated? It has been shown that the heightened temperatures induced by intracerebroventricular injection of PGE2 play a significant role in potentiating the neural damage caused by global ischaemia (Thornhill and Asselin, 1999). In our experimental model, the rectal temperature was maintained at 37°C during the operation. Furthermore, as we were able to detect the COX-2/mPGES-1-related neuronal excitotoxicity even in the cultured hippocampal slice system, we speculate that some or all of the stroke injury observed in our in vivo system was the result of direct damage through the induction of these enzymes at the parenchymal lesion site. PGE2 can efflux by simple diffusion after synthesis and activate four receptor subtypes (EP1–4; nomenclature follows Alexander et al., 2008), with quite different signalling cascades. The various roles of the PGE2 receptors in the neuronal death induced by excitotoxicity and ischaemic stroke have been clarified by genetic deletion and selective inhibition of each EP receptor (McCullough et al., 2004; Takadera et al., 2004; Ahmad et al., 2005) – for example, deletion and inhibition of the EP1 receptors partially reduces the neuronal damage caused by excitotoxicity and ischaemic stroke (Ahmad et al., 2006; Kawano et al., 2006). Interestingly, a recent study showed that genetic deletion of the EP3 receptors reduces the neuronal damage caused by oxygen/glucose deprivation or ischaemic stroke (Saleem et al., 2009). We have also confirmed the amelioration of excitotoxicity and stroke injuries by inhibition and deletion of EP3 receptors (unpublished experiments). Thus, EP3 receptors could be one of the important effectors for the neurotoxicity of PGE2. Further studies will be needed to identify the mechanisms by which PGE2 synthesized by COX-2/mPGES-1 affects neurotoxicity.
In summary, we have shown that mPGES-1 contributes crucially to the neurotoxicity of COX-2 through PGE2 production and plays a critical role in excitotoxicity and stroke injuries. Accumulating reports show that COX-2 inhibitors attenuate ischaemia-induced neurotoxicity and injury (Sugimoto and Iadecola, 2003; Candelario-Jalil et al., 2004; Ahmad et al., 2009). These findings have important implications for the therapeutic potential of COX-2 inhibitors in the treatment of stroke. Our study suggests that not only COX-2 inhibitors, but also mPGES-1 inhibitors may have therapeutic potential in the treatment of stroke. Considering that COX-2 inhibitors may non-selectively suppress the production of many types of prostanoids that are essential for normal physiological function of the brain (Kaufmann et al., 1997) and that a large number of epidemiological studies have provided evidence of an increased cardiovascular risk associated with the use of COX-2 inhibitors (McAdam et al., 1999; Minuz, 2008), an mPGES-1 inhibitor may prove to be an injury-selective inhibitor with fewer side effects. Thus, our results suggest that mPGES-1 is a promising novel target for the treatment of human stroke.
Acknowledgments
This study was partially supported by a Grant-in-Aid for Young Scientists (B) (18790063, Y.I.-M) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by The Mochida Memorial Foundation for Medical and Pharmaceutical Research (Y.I.-M) and by The Uehara Memorial Foundation (Y.I.-M).
Glossary
Abbreviations:
- BBB
blood–brain barrier
- COX
cyclooxygenase
- cPGES, mPGES
cytosolic/microsomal prostaglandin E synthase
- EIA
enzyme immunoassay
- KO
knockout
- LPS
lipopolysaccharide
- MCA
middle cerebral artery
- Neu-N
neuron-specific nuclear protein
- PG
prostaglandin
- PI
propidium iodide
- TTC
triphenyltetrazolium chloride
- WT
wild-type
Conflict of interest
The authors state no conflict of interest.
References
- Ahmad AS, Ahmad M, de Brum-Fernandes AJ, Doré S. Prostaglandin EP4 receptor agonist protects against acute neurotoxicity. Brain Res. 2005;1066:71–77. doi: 10.1016/j.brainres.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- Ahmad M, Zhang Y, Liu H, Rose ME, Graham SH. Prolonged opportunity for neuroprotection in experimental stroke with selective blockade of cyclooxygenase-2 activity. Brain Res. 2009;1279:168–173. doi: 10.1016/j.brainres.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM, Mirro R, Busija DW, Leffler CW. post-ischaemic generation of superoxide anion by newborn pig brain. Am J Physiol. 1988;255:H401–H403. doi: 10.1152/ajpheart.1988.255.2.H401. [DOI] [PubMed] [Google Scholar]
- Bading H, Segal MM, Sucher NJ, Dudek H, Lipton SA, Greenberg ME. N-methyl-D-aspartate receptors are critical for mediating the effects of glutamate on intracellular calcium concentration and immediate early gene expression in cultured hippocampal neurons. Neuroscience. 1995;64:653–664. doi: 10.1016/0306-4522(94)00462-e. [DOI] [PubMed] [Google Scholar]
- Barakat W, Herrmann O, Baumann B, Schwaninger M. NF-kappaB induces PGE2-synthesizing enzymes in neurons. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:153–160. doi: 10.1007/s00210-009-0421-0. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Bullock R, Graham DI, McCulloch J. Correlation between amino acid release and neuropathologic outcome in rat brain following middle cerebral artery occlusion. Stroke. 1990;21:1727–1733. doi: 10.1161/01.str.21.12.1727. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, González-Falcón A, García-Cabrera M, Alvarez D, Al-Dalain S, Martínez G, et al. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem. 2003;86:545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, González-Falcón A, García-Cabrera M, León OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA. 2002;99:13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- Cui Y, Takamatsu H, Kakiuchi T, Ohba H, Kataoka Y, Yokoyama C, et al. Neuroprotection by a central nervous system-type prostacyclin receptor ligand demonstrated in monkeys subjected to middle cerebral artery occlusion and reperfusion: a positron emission tomography study. Stroke. 2006;37:2830–2836. doi: 10.1161/01.STR.0000245088.60282.22. [DOI] [PubMed] [Google Scholar]
- Dávalos A, Shuaib A, Wahlgren NG. Neurotransmitters and pathophysiology of stroke: evidence for the release of glutamate and other transmitters/mediators in animals and humans. J Stroke Cerebrovasc Dis. 2000;9:2–8. doi: 10.1053/jscd.2000.18908. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Domoki F, Perciaccante JV, Puskar M, Bari F, Busija DW. Cyclooxygenase-2 inhibitor NS398 preserves neuronal function after hypoxia/ischemia in piglets. Neuroreport. 2001;12:4065–4068. doi: 10.1097/00001756-200112210-00041. [DOI] [PubMed] [Google Scholar]
- Fan W, Cooper NG. Glutamate-induced NFkappaB activation in the retina. Invest Ophthalmol Vis Sci. 2009;50:917–925. doi: 10.1167/iovs.08-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govoni S, Masoero E, Favalli L, Rozza A, Scelsi R, Viappiani S, et al. The Cycloxygenase-2 inhibitor SC58236 is neuroprotective in an in vivo model of focal ischemia in the rat. Neurosci Lett. 2001;303:91–94. doi: 10.1016/s0304-3940(01)01675-5. [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Althaus JS, VonVoigtlander PF. Hydroxyl radical production and lipid peroxidation parallels selective post-ischemic vulnerability in gerbil brain. J Neurosci Res. 1993;34:107–112. doi: 10.1002/jnr.490340111. [DOI] [PubMed] [Google Scholar]
- Huang FP, Wang ZQ, Wu DC, Schielke GP, Sun Y, Yang GY. Early NFkappaB activation is inhibited during focal cerebral ischemia in interleukin-1beta-converting enzyme deficient mice. J Neurosci Res. 2003;73:698–707. doi: 10.1002/jnr.10654. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol. 1999;98:9–14. doi: 10.1007/s004010051045. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima T, Sawa H, Shiokawa Y, Saito I, Ishii H, Nakamura Z, et al. Thromboxane synthetase inhibitor ameliorates delayed neuronal death in the CA1 subfield of the hippocampus after transient global ischemia in gerbils. J Neurosurg Anesthesiol. 1996;8:237–242. doi: 10.1097/00008506-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Ueno A, Naraba H, Matsuki N, Oh-ishi S. Intracellular Ca2+ increase in Neuro-2A cells and rat astrocytes following stimulation of bradykinin B2 receptor. Jpn J Pharmacol. 2000;84:140–145. doi: 10.1254/jjp.84.140. [DOI] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Ota A, Fukada T, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglndin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc Natl Acad Sci USA. 2006;103:11790–11795. doi: 10.1073/pnas.0604400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kempski O, Shohami E, von Lubitz D, Hallenbeck JM, Feuerstein G. post-ischaemic production of eicosanoids in gerbil brain. Stroke. 1987;18:111–119. doi: 10.1161/01.str.18.1.111. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Northoff H, Hofmann B, Schneider G, Werz O. MK-886, an inhibitor of the 5-lipoxygenase-activating protein, inhibits cyclooxygenase-1 activity and suppresses platelet aggregation. Eur J Pharmacol. 2009;608:84–90. doi: 10.1016/j.ejphar.2009.02.023. [DOI] [PubMed] [Google Scholar]
- Kojima F, Naraba H, Sasaki Y, Beppu M, Aoki H, Kawai S. Prostaglandin E2 is an enhancer of interleukin-1beta-induced expression of membrane-associated prostaglandin E synthase in rheumatoid synovial fibroblasts. Arthritis Rheum. 2003;48:2819–2828. doi: 10.1002/art.11261. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Povlishock JT, Dietrich WD, Magiera CJ, Ellis EF. Cerebral arteriolar damage by arachidonic acid and prostaglandin G2. Science. 1980;209:1242–1245. doi: 10.1126/science.7403881. [DOI] [PubMed] [Google Scholar]
- Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to post-ischaemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Lu A, Tang Y, Ran R, Clark JF, Aronow BJ, Sharp FR. Genomics of the periinfarction cortex after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:786–810. doi: 10.1097/01.WCB.0000062340.80057.06. [DOI] [PubMed] [Google Scholar]
- McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdam BF, Byrne D, Morrow JD, Oates JA. Contribution of cyclooxygenase-2 to elevated biosynthesis of thromboxane A2 and prostacyclin in cigarette smokers. Circulation. 2005;112:1024–1029. doi: 10.1161/CIRCULATIONAHA.105.542696. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Izumiyama M, Onodera H, Kurosawa A, Kogure K. Effect of a novel thromboxane A2 receptor antagonist, S-1452, on post-ischaemic brain injury in rats. Stroke. 1993;24:2059–2064. doi: 10.1161/01.str.24.12.2059. [DOI] [PubMed] [Google Scholar]
- Miettinen S, Fusco FR, Yrjänheikki J, Keinänen R, Hirvonen T, Roivainen R, et al. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A2. Proc Natl Acad Sci U S A. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minuz P. Nonsteroidal anti-inflammatory drugs and cardiovascular risk: is prostacyclin inhibition the key event? J Am Coll Cardiol. 2008;52:1637–1639. doi: 10.1016/j.jacc.2008.08.042. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68–69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- Naraba H, Yokoyama C, Tago N, Murakami M, Kudo I, Fueki M, et al. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J Biol Chem. 2002;277:28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- Nelson CW, Wei EP, Povlishock JT, Kontos HA, Moskowitz MA. Oxygen radicals in cerebral ischemia. Am J Physiol. 1992;263:H1356–H1362. doi: 10.1152/ajpheart.1992.263.5.H1356. [DOI] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira AC, Candelario-Jalil E, Bhatia HS, Lieb K, Hüll M, Fiebich BL. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia. 2008;56:844–855. doi: 10.1002/glia.20658. [DOI] [PubMed] [Google Scholar]
- Ouellet M, Percival MD. Effect of inhibitor time-dependency on selectivity towards cyclooxygenase isoforms. Biochem J. 1995;306:247–251. doi: 10.1042/bj3060247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Whiteside M, Herkenham M. Cyclooxygenase 2 mRNA expression in rat brain after peripheral injection of lipopolysaccharide. Brain Res. 1998;802:189–197. doi: 10.1016/s0006-8993(98)00402-8. [DOI] [PubMed] [Google Scholar]
- Rose JW, Hill KE, Watt HE, Carlson NG. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J Neuroimmunol. 2004;149:40–49. doi: 10.1016/j.jneuroim.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Saleem S, Zhuang H, de Brum-Fernandes AJ, Maruyama T, Narumiya S, Doré S. PGD(2) DP1 receptor protects brain from ischemia-reperfusion injury. Eur J Neurosci. 2007;26:73–78. doi: 10.1111/j.1460-9568.2007.05627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem S, Kim YT, Maruyama T, Narumiya S, Doré S. Reduced acute brain injury in PGE2 EP3 receptor-deficient mice after cerebral ischemia. J Neuroimmunol. 2009;208:87–93. doi: 10.1016/j.jneuroim.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Yamagata K, Takemiya T, Tanaka S, Omura-Matsuoka E, et al. Amelioration of hippocampal neuronal damage after transient forebrain ischemia in cyclooxygenase-2-deficient mice. J Cereb Blood Flow Metab. 2004;24:107–113. doi: 10.1097/01.WCB.0000100065.36077.4A. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Iadecola C. Delayed effect of administration of COX-2 inhibitor in mice with acute cerebral ischemia. Brain Res. 2003;960:273–276. doi: 10.1016/s0006-8993(02)03805-2. [DOI] [PubMed] [Google Scholar]
- Sunose Y, Takeyoshi I, Ohwada S, Tsutsumi H, Iwazaki S, Kawata K, et al. The effect of cyclooxygenase-2 inhibitor FK3311 on ischemia-reperfusion injury in a canine total hepatic vascular exclusion model. J Am Coll Surg. 2001;192:54–62. doi: 10.1016/s1072-7515(00)00773-0. [DOI] [PubMed] [Google Scholar]
- Takadera T, Shiraishi Y, Ohyashiki T. Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int. 2004;45:713–719. doi: 10.1016/j.neuint.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Mohri I, Okabe-Arahori H, Aritake K, Wada K, Kanekiyo T, et al. Prostaglandin D2 protects neonatal mouse brain from hypoxic ischemic injury. J Neurosci. 2007;27:4303–4312. doi: 10.1523/JNEUROSCI.0321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- Thorén S, Jakobsson PJ. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur J Biochem. 2000;267:6428–6434. doi: 10.1046/j.1432-1327.2000.01735.x. [DOI] [PubMed] [Google Scholar]
- Thornhill J, Asselin J. The effect of head cooling on the physiological responses and resultant neural damage to global hemispheric hypoxic ischemia in prostaglandin E2 treated rats. Brain Res. 1999;825:36–45. doi: 10.1016/s0006-8993(99)01210-x. [DOI] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Lin JX, Budka H. Cyclooxygenase-2 is induced in microglia during chronic cerebral ischemia in humans. Acta Neuropathol. 2000;99:26–30. doi: 10.1007/pl00007402. [DOI] [PubMed] [Google Scholar]
- Torres L, Anderson C, Marro P, Mishra OP, Delivoria-Papadopoulos M. Cyclooxygenase-mediated generation of free radicals during hypoxia in the cerebral cortex of newborn piglets. Neurochem Res. 2004;29:1825–1830. doi: 10.1023/b:nere.0000042208.20730.23. [DOI] [PubMed] [Google Scholar]
- Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol. 1991;71:1185–1195. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, et al. Oxidative stress following traumatic brain injury in rats: quantitation of biomarkers and detection of free radical intermediates. J Neurochem. 2000;75:2178–2189. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- Vazquez-Tello A, Fan L, Hou X, Joyal JS, Mancini JA, Quiniou C, et al. Intracellular-specific colocalization of prostaglandin E2 synthases and cyclooxygenases in the brain. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1155–R1163. doi: 10.1152/ajpregu.00077.2004. [DOI] [PubMed] [Google Scholar]
- Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Kibler KK, Koehler RC, Maruyama T, Narumiya S, Doré S. Prostacyclin receptor deletion aggravates hippocampal neuronal loss after bilateral common carotid artery occlusion in mouse. Neuroscience. 2008;156:1111–1117. doi: 10.1016/j.neuroscience.2008.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota C, Kaji T, Kuge Y, Inoue H, Tamaki N, Minematsu K. Temporal and topographic profiles of cyclooxygenase-2 expression during 24 h of focal brain ishemia in rats. Neurosci Lett. 2004;357:219–222. doi: 10.1016/j.neulet.2003.12.109. [DOI] [PubMed] [Google Scholar]