

Abstract
Enzyme engineering by directed evolution presents a powerful strategy for tailoring the function and physicochemical properties of biocatalysts to therapeutic and industrial applications. Our laboratory’s research focuses on developing novel molecular tools for protein engineering, as well as on utilizing these methods to customize enzymes and to study fundamental aspects of their structure and function. Specifically, we are interested in nucleoside and nucleotide kinases which are responsible for the intracellular phosphorylation of nucleoside analog (NA) prodrugs to their biologically active triphosphates. The high substrate specificity of the cellular kinases often interferes with prodrug activation and consequently lowers the potency of NAs as antiviral and cancer therapeutics. A working solution to the problem is the co-adminstration of a promiscuous kinase from viruses, bacteria, and other mammals. However, further therapeutic enhancements of NAs depend on the selective and efficient prodrug phosphorylation. In the absence of true NA kinases in nature, we are pursuing laboratory evolution strategies to generate efficient phosphoryl-transfer catalysts. This review summarizes some of our recent work in the field and outlines future challenges.
Keywords: Deoxynucleoside kinase, Directed evolution, Enzyme catalysis, Nucleoside analogs, Protein engineering
Introduction
Enzymes are remarkable catalysts, capable of accelerating chemical reactions by up to 13 orders of magnitude while exhibiting generally high selectivity and specificity for their substrates under environmentally benign reaction conditions. One of the first attempts to rationalize the observed functional capabilities of enzymes was Emil Fischer’s ‘lock-and-key’ model[1] which, despite being overly simplistic, captures some of the basic ideas of enzyme catalysis. It also helps to account for some of the limitations encountered by scientists and engineers as they try to capitalize on the distinct performance of enzymes for applications in the laboratory and industry. Many of the synthetic applications envisioned by researchers involve modified and unnatural substrates, as well as reaction conditions that significantly diverge from the physiological environment in regards to pH, temperature, and solvent composition. In practice, these novel requirements often translate into compromised catalyst performance as the new conditions interfere with the complementarity of enzyme and substrate. In terms of Fischer’s model, the key no longer fits the lock.
A perfect example for such a lock-key mismatch is the application of nucleoside analog (NA) prodrugs in antiviral and cancer therapy. As shown in Fig. 1, NAs closely resemble the natural ribo and 2′-deoxyribonucleosides, the building blocks of RNA and DNA, yet they carry distinct chemical substitutions in the ribose and nucleobase moieties. To exhibit their biological function, NAs depend on intracellular phosphorylation to their corresponding triphosphates via the host’s nucleoside salvage pathway (Fig. 2). At the core of the salvage pathway is a cascade of three kinases, deoxynucleoside kinases (dNK, e.g. thymidine kinase), deoxynucleoside monophosphate kinases (dNMPK, e.g. thymidylate kinase), and nucleoside diphosphate kinase (NDPK), to recycle scavenged 2′-deoxynucleosides and to activate intermediates of the de novo nucleotide biosynthetic pathway. Following cellular uptake via membrane transporters, the prodrugs have to ‘borrow’ these kinases to become activated. Once in their triphosphate state, the NA anabolites then turn into competitive substrates for low-fidelity polymerases and reverse transcriptases found in cancer cells and viruses, respectively. The incorporation of NA-triphosphates results in termination of the DNA replication process, preventing further disease proliferation. The replication machinery of normal, healthy cells on the contrary has higher fidelity, protecting the host from the lethal effects of these suicide substrates.
Fig. 1.

Overview of natural 2′-deoxynucleosides and selected nucleoside analogs. Thymidine (T, 1), 2′-deoxycytidine (dC, 2), 2′-deoxyadenosine (dA, 3), 2′-deoxyguanosine (dG, 4), 3′-azido-3′-deoxythymidine (AZT, 5), 2′,3′-dideoxy-5-fluoro-3′-thiacytidine (FTC, 6), ganciclovir (7), 3-deoxythymidine (ddT, 8). Grey-shaded areas are marking structural and functional modifications in the prodrugs.
Fig. 2.

Nucleoside salvage pathway for the production of 2′-deoxynucleoside triphosphates. Scavenged 2′-deoxynucleosides are imported by designated membrane transporter proteins and serve as substrate for dNK. Alternatively, cleavage of the nucleobase by phosphorylase feds into other pyrimidine and purine metabolism pathways. Separately, thymidylate synthase and ribonucleotide reductase catalyze the final reactions of the nucleotide de novo pathways, producing TMP and dNDPs which are converted to their triphosphates by dNMPK and NDPK. dNK = 2′-deoxynucleoside kinase, dNMPK = 2′-deoxynucleoside monophosphate kinase, NDPK = nucleoside diphosphate kinase
Problems with the concept arise as the intracellular activation of NAs is often compromised by the high substrate specificity of the host’s endogenous kinases. The inefficient phosphorylation of NAs not only reduces the potency of existing pro-drugs but can result in the accumulation of cytotoxic reaction intermediates.[2–4] More importantly, it is responsible for the failure of a large number of potential NA pro-drug candidates in vivo.[5] One promising solution to overcome the shortcomings of the phosphorylation cascade has been the co-administration of prodrugs with exogenous, broad-specificity kinases.[6–9] While biochemical and pre-clinical experiments have demonstrated the effectiveness of the strategy in principle, the studies also uncovered limitations arising from the dNKs’ significantly lower activity for NAs compared to their performance with the native substrates. In addition, the broad substrate specificity of exogenous kinases raised concerns as it interferes with the tightly regulated 2′-deoxynucleoside metabolism.[10] More recent studies have hence focused on identifying orthogonal NA kinases for the selective and efficient activation of these prodrugs.
Besides searching for orthogonal NA kinases in nature, enzyme engineering by rational design and directed evolution presents a promising alternative. Directed evolution in particular has given scientists and engineers a powerful tool to explore the inner workings of proteins and enzymes, as well as to tailor native biocatalysts to novel substrates and function.[11–16] The process mimics Darwinian evolution, applying iterative cycles of diversification and selection to stepwise modify an existing protein towards progeny with the desired properties. In practice, one or more ‘parental’ genes that encode for target proteins are forced to diversify by saturation and random mutagenesis, as well as in vitro recombination methods, creating gene libraries of typically 103–1010 members. Following overexpression in a suitable host organism or in vitro, the corresponding protein libraries are evaluated for the desired property (e.g. substrate specificity, thermostability etc.) by selection or screening assays.[17] Probably the most common selection strategy is genetic complementation of auxotrophic host strains which can process libraries of 106–107 members. Alternatively, screening protocols are traditionally based on LB-agar or microtiter plate assays and can typically be used to analyze up to 104 library members. Extending the limit on library size, newer screening methods utilizing fluorescence-activated cell sorting (FACS) and in vitro compartmentalization have been developed for protein libraries with up to 108 members.[18,19] Candidates that excel under the chosen conditions are isolated and can either serve as ‘parents’ for the next round of evolution or undergo detailed characterization.
For the creation of effective NA kinases from natural nucleoside and nucleotide kinases by directed evolution, our laboratory is pursuing a highly interdisciplinary approach, applying the tools of molecular biology, biochemistry and molecular evolution in combination with synthetic organic chemistry and computational protein design. This strategy has formed the foundation for the creation of unique kinase libraries and the development of a novel screening protocol to identify promising kinase candidates.
Cellular Kinases, Exogenous Kinases and Substrate Specificity
The enzyme engineering of kinases in the salvage pathway has largely focused on the first phosphorylation step catalyzed by dNKs (Fig. 2). These enzymes exhibit generally high specificity towards natural 2′-deoxyribonucleosides and possess distinct yet complementary preferences for the individual nucleobases (Fig. 3). In humans, two of the dNKs (2′-deoxycytidine kinase (dCK) and thymidine kinase 1(TK1)) are cytoplasmic kinases while thymidine kinase 2 (TK2) and 2′-deoxyguanosine kinase (dGK) are expressed in the mitochondria. Although all four enzymes have distinct substrate preferences, the two enzyme pairs complement each other to ensure phosphorylation of the four natural DNA building blocks.
Fig. 3.

Summary of structure and substrate specificity of the 2′-deoxynucleoside kinase family. Members are divided into type-I and type-II subfamilies based on their distinct structural features. Representative protein structures, shown as grey ribbons, are based on crystal structure coordinates except for the homology-based model of human TK2. Where available, substrate occupying the phosphoryl-acceptor binding site is shown as black spheres while substrate binding in the phosphoryl-donor site is marked by white spheres. Substrate profiles are displayed beneath the structures, reflecting each enzyme’s catalytic efficiency for the four natural 2′-deoxynucleosides (T, dA, dG, dC) by their font sizes.
Crystal structures of several dNKs show these enzymes to belong to two subfamilies; type-I dNKs with a parallel β-sheet in the core, flanked by multiple helices on both sides and a short LID region over the active site, as well as the smaller type-II dNKs with a central parallel β-sheet, flanked by three helices. An extended lasso region, which serves as lid over the active site, is held in place by a ligated zinc atom. We have explored some of the fundamental functional features on the type-II thymidine kinase from the hyperthermophilic eubacterium Thermotoga maritima (TmTK).[20–22] The Thermotoga enzyme is a close structural and functional homolog of the human TK1 and, due to its thermal stability, serves as a (literally) robust model for the latter. Biochemical studies, as well as a series of crystal structures of TmTK with bound substrate(s) and products revealed a large conformational change in the native homotetramer associated with ATP binding in the phosphoryl donor site. This 5Å-lateral expansion of the protein complex from its ‘close’ to ‘open’ conformation is critical to function as the tetramer locked in the ‘close’ state is catalytically inactive. Although it remains unclear whether the conformational transition is forced by ATP binding or occurs spontaneously due to protein dynamics, hence creating a suitable binding site for ATP, our activity measurements as a function of temperature support the latter hypothesis. A discontinuity in the corresponding Arrhenius plot, as well as characteristic changes in the turnover rates are consistent with an ATP-independent conformational change. Furthermore, our results support the idea that the observed conformational changes in TmTK are physiologically relevant and could be involved in regulating its enzyme activity. Based on TmTK’s considerable structural similarity with other type-II dNKs, we anticipate similar behavior across the entire subfamily including human TK1. In regards to NA activation, type-II dNKs play an important role in phosphorylating the two anti-HIV prodrugs 3′-azido-3′-deoxythymidine (AZT; 5) and 2′,3′-dideoxy-2′,3′-didehydro-thymidine (d4T), as well as various thymidine derivatives used for boron neutron-capture therapy.[23,24] Nevertheless, the subfamily’s strict specificity for thymine and uracil-nucleosides, as well as the substrate’s direct interactions with the protein backbone, has complicated enzyme engineering and so far limited its potential in therapeutic applications.
Members of the type-I dNK subfamily on the contrary show great versatility in regards to substrate specificity, even though they all share a common protein scaffold[2] (Fig. 3). For example, human dGK exclusively recognizes purine nucleosides as phosphoryl acceptors while TK2 phosphorylates only pyrimidine nucleosides. Even broader specificity can be found in dCK which activates 2′-deoxyribose carrying cytidine, adenine and guanine moieties while 2′-deoxynucleoside kinase from Drosophila melanogaster (DmdNK) effectively phosphorylates all four natural 2′-deoxynucleosides, although with a preference for pyrimidines.[25] The substrate adaptability of type-I dNKs has made members of this subfamily the target of the majority of past and present enzyme engineering studies.
Probing Substrate Specificity in Kinases by Mutagenesis
The diverse substrate profiles of type-I kinase family members in respect to individual nucleobases presented an initial focus for our enzyme engineering studies. While crystal structures of multiple family members with different pyrimidine and purine nucleosides, as well as nucleoside analogs have clearly established that all substrates bind with superimposable orientation in the active site, an assessment of the individual enzyme–substrate interactions has been complicated by the generally low protein sequence identity in the subfamily (30–50%). Nevertheless, earlier random mutagenesis experiments with DmdNK, as well as crystallographic studies of human dCK had identified the same three residues in both enzymes, namely Ala100/Arg104/Asp133 in dCK (corresponding to Val84/Met88/Ala110 in DmdNK), as being critical for thymidine discrimination in the human kinase[26,27] (Fig. 4). The elaborate hydrogen-bonding network in dCK allows for favorable binding interaction between the exocyclic amino group of the substrate and Asp133, a contact that would be lost upon binding of thymidine. Furthermore, the network also preorients the Arg104 side chain in a conformation which creates a steric clash with the 5-methyl group of thymine. In contrast, Val84/Met88/Ala110 in DmdNK lack these hydrogen-bonding interactions and instead create a large, generic hydrophobic binding pocket, capable of accommodating pyrimidines and purines alike.
Fig. 4.

Active site comparison of A) human 2′-deoxycytidine kinase (PDB code: 1P6O)[27] and B) 2′-deoxynucleoside kinase from Drosophila melanogaster (PDB code: 1J90).[26] Both enzymes carry 2′-deoxycytidine in the phosphoryl acceptor binding site. Across the type-I kinase family, Arg128 (Arg105) and Glu53 (Glu52) are highly conserved as they form critical hydrogen bonding interactions with the 5′-hydroxyl group of the substrate. In dCK, the hydrogen bonding network is extended to include Arg104 and Asp133 which in turn interacts with the exocyclic amino group on the substrate’s nucleobase. In contrast, the same residues in DmdNK form a large binding pocket which lacks specific contacts and could explain the broader substrate specificity of the fruitfly enzyme. Figures adapted from ref. [28]; created in PyMol (DeLano, Palo Alto, CA).
Based on preliminary data by Lavie and coworkers,[27] we built rTK3, a variant of dCK with the substitutions at Ala100Val/Arg104Met/Glu133Ala by site-directed mutagenesis, thereby eliminating the enzyme’s discrimination against thymidine as reflected by a raise in catalytic efficiency (kcat/KM) by over 1000-fold (Table 1).[28] The mutations also benefit the phosphorylation of 2′-deoxycytidine (dC) while the activation of purine nucleosides shows a substantial drop. These findings prompted the question whether i) Ala100Val, Arg-104Met and Glu133Ala were the only mutations leading to thymidine kinase activity, and ii) there are other positions throughout the protein which, upon mutagenesis, could result in a similar change in substrate specificity. To answer these questions, we used site-saturation and whole-gene random mutagenesis of dCK in combination with functional selection of library members in the thymidine kinase-deficient E. coli auxotroph KY895.[29] Random mutagenesis yielded two unique kinase variants, epTK6 and epTK16, with five and three mutations, respectively. More importantly, both candidates carried an Arg104Gln substitution and changes in Asp133 to either Gly or Asn, supporting the functional relevance of these two positions for thymidine kinase activity. Subsequent reverse engineering further probed the role of position 100, identifying it as a lesser contributor to activity gains, and determined the other mutations to be irrelevant for catalytic performance. Interestingly, data from the kinetic analysis of the reverse engineered epTK6A indicated a much more balanced substrate profile compared to previously characterized rTK3, showing improved thymidine phosphorylation by >400-fold but wild type-like activity for the remaining DNA precursors (Table 1). The broader specificity of epTK6A was shown to extend beyond the natural substrates as reflected in substantially increased activity for several NAs. The ease to create such a broad-specificity dNK is intriguing, especially if we consider recent hypotheses for enzyme evolution via ‘generalist’ intermediates.[30]
Table 1.
Catalytic performance of dCK variants
| WT dCK | rTK3 | epTK6A | ssTK1A | ssTK2A | |
|---|---|---|---|---|---|
| Ala100 | Val100 | … | Val100 | Val100 | |
| Arg104 | Met104 | Gln104 | Met104 | Met104 | |
| Asp133 | Ala133 | Gly133 | Ser133 | Thr133 | |
| T | 0.095 | 105 (+1105) | 39 (+410) | 342 (+3600) | 406 (+4273) |
| dC | 63 | 777 (+12) | 356 (+6) | 400 (+6.3) | 50 (−1.2) |
| dA | 38 | 6 (−6) | 8 (−5) | 4 (−10) | 2 (−20) |
| dG | 21 | 0.23 (−100) | 19 (1) | 2 (−10) | 2 (−10) |
The kcat/KM values of the parental enzyme, as well as individual variants are listed. The fold change relative to dCK is shown in parentheses. Data were adapted from ref. [28].
Given the strong evidence for the central role of amino acids 104 and 133 in thymidine kinase activity, we next performed simultaneous site-saturation mutagenesis in the two positions in search of alternate amino acid pairs. While selection of the library in our auxotrophic E. coli strain yielded over a dozen functional variants, DNA sequence analysis showed that substitutions in position 104 were dominated by Met with Gln being the less frequent alternate. Mutations in position 133 were more diverse, showing Thr, Ser, and Asn as the most common substitutions. In combination with Ala100Val, the kinetic analysis of the two most frequent mutants Arg104Met/Asp133Ser (ssTK1A) and Arg104Met/Asp133Thr (ssTK2A) revealed a ~4000-fold enhanced thymidine kinase activity (Table 1). The additional gain in performance over rTK3 likely results from favorable hydrogen-bonding interactions between the Ser/Thr side chains with the carbonyl oxygen in the 4-position of thymine. More so, the 8-fold decline in dC activity as a consequence of the extra methyl group in Thr133 (ssTK2A) was rationalized by unfavorable steric interactions between the side chain and the substrate’s exocyclic amino group. In summary, we successfully applied a variety of enzyme engineering methods to identify and verify two key positions in human dCK, responsible for the phosphorylation of thymidine. Exploring a number of amino acids pairs in these positions, the resulting kinetic data further confirmed our ability to exert control over the pyrimidine nucleobase preferences in dCK.
Nevertheless, we have also come to realize that there are limits to what extent primary shell residues, amino acids that directly contact substrate in the enzyme active site, can alter the overall performance of an enzyme. In a separate study, we analyzed and compared the active sites of DmdNK and human TK2.[31] While the two enzymes share similar preferences for pyrimidine substrates, their catalytic performance differs by almost two orders of magnitude. We found this difference to be particularly interesting in light of the fact that the two enzymes carry identical amino acids in 27 out of 29 positions within 6 Å of the phosphoryl acceptor binding site. While individual, as well as combined mutations of the two positions (L78F/L116M) in TK2 did raise enzyme activity in an additive fashion, the gains were comparably moderate. These findings strongly suggested that more distant portions of the enzyme also contribute to its overall catalytic performance and encouraged us to expand our kinase engineering efforts beyond the immediate vicinity of the active site.
Chimeragenesis in Kinase Engineering
The functional role of protein regions distant to the active site can be explored by random mutagenesis and in vitro recombination. The latter approach, better known as DNA shuffling, exchanges entire protein fragments and domains between two or more parental structures, a strategy that has been shown more efficient in generating progeny with improved properties than mutagenesis.[32] However, traditional DNA shuffling is based on homologous gene recombination, requiring the parental DNA sequences to share at least 70% identity. For the shuffling of our type-I dNKs, this method is not suitable as the sequence identities of various family members do not exceed 50%. To overcome this limitation, we applied SCRATCHY, an alternative in vitro recombination protocol that does not depend on sequence identity of the parental genes.[33,34]
The application of SCRATCHY enabled us to create extensive libraries of chimeric proteins between the human TK2 and DmdNK. Subsequently, chimeras with thymidine kinase activity were identified with the help of our auxotrophic E. coli strain. From among the functional hybrid kinases, candidates HD-16 and HDHD-12 were selected for in-depth characterization. HD-16 is a single crossover chimera, carrying the N-terminus of TK2 (amino acids 1–175) and the C-terminus of DmdNK (amino acids 176–236). In contrast, HDHD-12 contains three crossovers, starting with the Nterminal TK2 sequence (amino acids 1–11), switching to DmdNK from residues 12–88 and back to TK2 from position 89–172, and finally completing the chimera with the C-terminal portion from DmdNK (amino acids 173–233). When tested with natural 2′-deoxynucleosides, the kinetic analysis showed both chimeras to exhibit intermediary catalytic performances relative to their parents (Table 2). However, experiments with 2′,3′-dideoxycytidine (ddC) and d4T showed increased rates of phosphorylation for both NAs. More so, in the case of d4T neither parent had measurable activity for the analog while both chimeras successfully phosphorylated the prodrug. Although the activities themselves are very low and offer little immediate value for practical applications, our experiments were able to identify another hotspot in the type-I dNK scaffold, the C-terminal region of the protein. Ongoing experiments in our group and by others in the field are now focusing on the functional role of this region which, despite being approximately 20 Å away from the phosphoryl acceptor binding site, appears to influence enzyme activity and substrate specificity. Our working hypothesis argues for communication between the two sites to be transmitted via conformational changes of the LID region, located between the two segments in the kinase structure.
Table 2.
Catalytic performance of chimeric kinases
| WT TK2 | WT DmdNK | HD-16 | HDHD-12 | |
|---|---|---|---|---|
| TK: 1–175 DmdNK: 176–236 | TK: 1–11 DmdNK: 12–88 TK: 89–172 DmdNK: 173–233 | |||
| T | 2,700 | 91,000 (+33) | 13,000(+5) | 14,000(+5) |
| dC | 1,200 | 90,000(+75) | 2,300(+2) | 9,100 (+8) |
| dA | nd | 830(1) | 9.3 (−100) | 22(−33) |
| dG | nd | 180(1) | 1.5 (−100) | 4.5 (−33) |
| ddC | nd | 39 (1) | 3.4 (−11) | 110 (+3) |
| d4T | nd | nd | 11 | 19 |
The kcat/KM values (× 102 s−1 M−1) of the parental enzymes, as well as individual chimera are listed. The fold change relative to TK2 (for pyrimidines) and DmdNK (for purines and nucleoside analogs) are shown in parentheses. nd = not detectable. Data were adapted from ref. [33].
New Methods for More Effective Directed Evolution of Kinases
Over the course of our directed evolution experiments on dNKs, one reoccurring issue has been the restrictions posed by library analysis with the thymidine kinase-deficient E. coli strain KY895. In directed protein evolution, the golden rule of ‘you get what you select for’ has been upheld many times over the last decade. In our particular case, library selection with the E. coli auxotroph would inadvertently identify the thymidine kinases among our library members, not, as originally planned, variants with NA kinase activity. An alternative is in vivo screening on replica plates, testing for cytotoxicity of NAs.[35] Replica plating, while directly evaluating library members for NA activation, is very laborious and ultimately depends on the toxicity of the phosphorylated NA to the host, a criterion that in our tests in E. coli could not be extended beyond AZT. In light of these biases and functional deficiencies of existing assays, we sought the design and implementation of a new, more adequate selection or screening technique for orthogonal NA kinases.
Inspired by new library screening methods for directed evolution libraries based on FACS,[36,37] we developed a similar strategy for identifying enzymes that can phosphorylate NAs. Conceptionally, our idea was based on the notion that i) nucleosides and NAs are efficiently transported across the cell membrane while their corresponding monophosphates, the product of the kinase-catalyzed phosphoryl transfer, become trapped in the host cytoplasm and ii) fluorescent nucleoside analogs (fNAs) with an excitation maxima of >300 nm are substrates for type-I dNKs. The spectral shift is necessary as proteins and other small-molecule metabolites such as ATP possess intrinsic fluorescence properties under physiological conditions and otherwise interfere with NA detection due to the overlapping absorption maxima. Fortunately, earlier DNA structure studies, utilizing fluorescent 2′-deoxyribonucleosides including etheno-adenine (11),[38] as well as furano (9) and pyrrolo-pyrimidines (10) and pterines (12,13)[39,40] (Fig. 5) have already demonstrated that relatively small synthetic modifications of a nucleoside’s pyrimidine or purine moiety could red-shift its absorption spectrum, enabling detection of these fNAs with high sensitivity in complex mixtures such as the cytoplasm. We argued that the combination of these modified nucleobases with sugar derivatives found in NA prodrugs would create suitable substrates for dNK library screening.
Fig. 5.
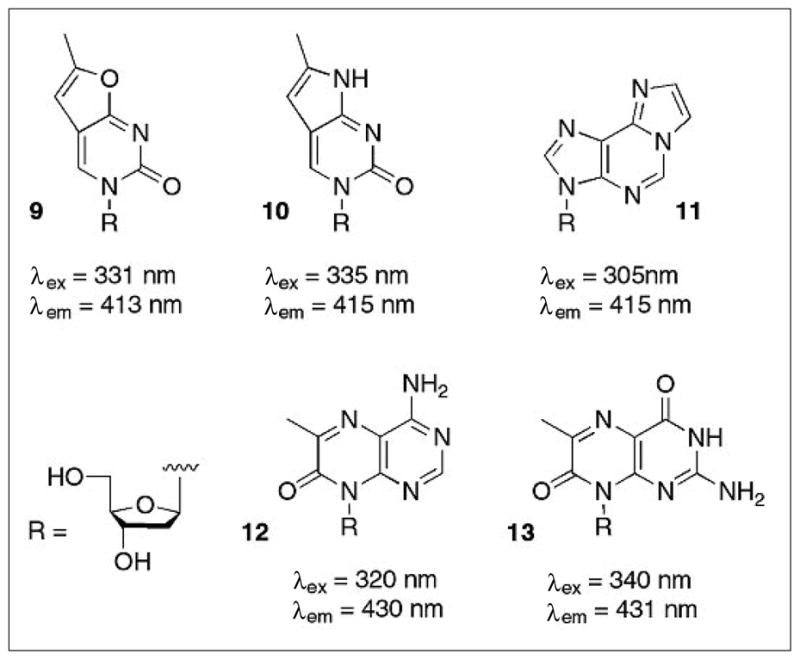
Structures and spectral properties of fluorescent analogs of pyrimidines and purines. Furano and pyrrolo-pyrimidines (8,9) can substitute for thymine and cytosine while etheno-adenine (10) and pterines (11,12) are replacements for purines. Fluorescent nucleoside analogs carry the appropriate sugar derivative instead of the 2′-deoxyribosyl moiety as the R-group.
For our specific purposes, we incubated bacteria that express individual dNK library members with a target fNA, relying on broad-specificity nucleoside transporters to efficiently translocate the fluorophor across the membrane and produce the corresponding fNA-monophosphate in the presence of a functional fNA kinase (Fig. 6). Upon accumulation of the fluorophor in the cytoplasm, FACS was employed to identify and isolate hosts with the highest fluorescence intensity.[37,41]
Fig. 6.

Schematic overview of our new kinase screening assay based on fluorescence activated cell sorting (FACS). Accumulation of the fluorescent nucleoside (or nucleoside analog) monophosphate in bacteria that express a kinase with the desired substrate specificity raises the fluorescence intensity of the host cell. FACS can rapidly detect and separate this subpopulation of the cell culture for subsequent characterization or additional rounds of directed evolution. The assay was validated with E. coli cells, expressing either DmdNK with thymidine kinase activity (+ kinase) or the thymidine kinase activity-deficient human dCK (− kinase). Upon incubation with furano-thymidine, the fluorescence intensity increase 20 to 30-fold in host cells expressing DmdNK. Figure adapted from ref. [42].
The new FACS-based assay was first put to the test over four rounds of directed evolution of DmdNK to identify enzymes for the phosphorylation of 3′-deoxythymidine (ddT; 8).[42] Even though the isolated kinases had as few as four mutations, their kinetic characterization showed a dramatic change in substrate specificity. For example in variant R4.V3 (Thr85Met/Glu172Val/Tyr179Phe/His193Tyr), the catalytic efficiency for natural pyrimidine 2′-deoxyribonucleosides was lowered by up to 48,000-fold while activity for the fddT and ddT was largely preserved (Table 3). Reverse engineering studies revealed that these functional changes were largely accomplished by two amino acid substitutions in the phosphoryl-acceptor binding site, responsible for interacting with the substrate’s ribose moiety (Fig. 7). The substitution of Val172 back to wild type Glu172 in R4.V3-[172] reestablished significant activity levels of the natural 2′-deoxynucleosides (Table 3), verifying the residue’s major role in determining substrate specificity. The amino acid reversion at position 179 resulted in less dramatic changes but nevertheless confirmed this residue’s contribution to the overall specificity switch. Further analysis also showed that the new assay was faithful to the golden rule; using a nucleoside analog with modifications in the nucleobase moiety (fddT) did bias the outcome of the experiment. In addition to the two active site mutations, a third mutation at Thr85 was linked to enlarging the nucleobase binding pocket to better accommodate the extra furnao moiety. Upon reversion of Met85 to its native Thr (R4.V3-[85]), we observed a 10-fold increase in the catalytic efficiency for ddT. In comparison with the parental DmdNK, R4.V3-[85] showed only residual activity for the natural DNA building blocks but phosphorylated ddT with 6-fold greater efficiency, overall making it a truly orthogonal NA kinase.
Table 3.
Catalytic performance of ddT-kinases
| WT DmdNK | R4.V3 | R4.V3-[85] | R4.V3-[172] | |
|---|---|---|---|---|
| Thr85Met | … Glu172Val | Thr85Met … | ||
| Glu172Val | Tyr179Phe | Tyr179Phe | ||
| Tyr179Phe | His193Tyr | His193Tyr | ||
| His193Tyr | ||||
| T | 4813 | 0.1 (−48,000) | 1.4 (−3,400) | 326 (−14) |
| dC | 5850 | 0.36 (−16,000) | 0.39 (−15,000) | 2925 (−2) |
| dA | 161 | nd | nd | 57 (−2.5) |
| dG | 28 | nd | nd | 6 (−4.5) |
| ddT | 4.6 | 3 (−1.5) | 28 (+6) | 2.6 (−1.8) |
| fddT | 1.3 | 1.9 (+1.5) | 0.4 (−3) | 7.5 (+6) |
The kcat/KM values (× 103 s−1 M−1) of the parental enzyme, as well as evolved variants are listed. The fold change relative to DmdNK are shown in parentheses. nd = not detectable. Data were adapted from ref. [42].
Fig. 7.

Structure model of active sites in DmdNK (A) and R4.V3 (B). The location of the three relevant mutations Glu172Val, Tyr179Phe, and Thr85Met are highlighted in DmdNK with thymidine, bound in the phosphoryl acceptor site (PDB code: 1OT3).[43] A) Glu172 in the wild-type enzyme forms a direct hydrogen-bonding interaction with the 3′-hydroxyl group of the substrate while Tyr179 contacts the same position via hydrogen bonding to a water molecule (red sphere) in the active site. B) Directed evolution of DmdNK for ddT phosphorylation introduces two hydrophobic residues in position 172 and 179, better accommodating the binding of the nucleoside analog. Separately, the mutation of Thr85 to Met is believed to slightly enlarge the binding pocket for the nucleobase portion by inducing a conformational shift of the helical region.
Although we anticipated a larger increase in the catalytic activity of R4.V3-[85] for ddT, based on the selection pressure during FACS analysis, the moderate effect seen in vitro translated into more dramatic improvements in vivo. In bacterial cultures, as well as in more recent mammalian cell culture work, our ddT kinase clearly outperformed the parental DmdNK. We attribute this effect to the ddT kinase’s ability to discriminate against naive 2′-deoxynucleosides which are present at low micromolar concentrations inside the cell. In contrast to the engineered kinase, the ability of DmdNK to activate the NA is compromised by substrate competition for the active site between ddT and natural 2′-deoxynucleosides. Following this initial demonstration of our new FACS-based kinase screening assay to identify NA kinases in directed evolution libraries, we are poised to expand our engineering efforts to find novel kinases with improved properties for existing, as well as new NA prodrugs.
Future Perspectives: Multifunctional Kinases
Another new and exciting research challenge presents itself as a direct consequence of the recent success in engineering 2′-deoxynucleoside kinases. Early experiments suggest that the increased efficiency of initial nucleoside analog phosphorylation could shift the bottleneck of activation to the second step of the reaction cascade, the conversion of nucleoside monophosphate to its diphosphate. While earlier studies have demonstrated that mutagenesis and in vitro recombination can also broaden the substrate specificity of dNMPKs,[44,45] an attractive alternative would be to merge dNK and dNMPK activity by creating a dual-function biocatalyst. A single enzyme capable of performing both phosphoryl-transfer reactions would not only simplify its clinical application but could also minimize the accumulation of cytotoxic intermediates and raise the efficiency of the overall activation process.
From a practical perspective, the idea of creating a bis-phosphorylation kinase for nucleoside analogs is supported by the structural similarities of enzymes in the two subfamilies. As shown in Fig. 8A, the crystal structures from representatives of both subfamilies, DmdNK and the E. coli thymidylate kinase (EcTMPK), show striking similarities between the two enzyme scaffolds. Furthermore, the same structures show superimposable binding sites for phosphoryl-donor and -acceptor, respectively, while results from mechanistic studies are consistent with direct phosphoryl-transfer in both cases (in contrast to nucleoside diphosphate kinase, the third enzyme in the salvage pathway, which proceeds via a phospho-histidine intermediate[48]). Probably the strongest argument in favor of developing a dual-function kinase is the existence of precedence in nature. Generally, dNKs and dNMPKs are strictly limited to their respective reactions, yet two promiscuous enzymes have been reported: the thymidine kinases from Herpes Simplex and Equine herpes virus.[47,49,50] In both cases, the homology of their core structures clearly identifies them as members of the 2′-deoxynucleoside kinase family (Fig. 8B). Furthermore, phylogenetic analysis places them closer to the dNK subgroup, a finding that is consistent with kinetic experiments which confirm the herpes enzymes’ primary function as thymidine kinases with ~10% secondary activity as thymidylate kinases.[49,50]
Fig. 8.

Structure comparison of dNK, dNMPK and dual-function kinase. A) Superposition of DmdNK (PDB code: 1OT3)[43] and thymidylate kinase from E. coli (PDB code: 4TMK).[46] Despite overall low sequence identity and strict specificity for either 2′-deoxynucleosides or thymidine monophosphate, the two enzymes have matching functional elements (LID region highlighted in yellow/orange, P-loop shown in green) and striking overall structural similarity. The bisubstrate analog Tp5A (P1-(5′-adenosyl)P5-(5′-thymidyl)pentaphosphate) bound in the active site of the E. coli kinase is shown in sticks B) The thymidine kinase from Herpes Simplex virus (PDB code: 1P7C)[47] is primarly a thymidine kinase, yet can also perform the second phosphorylation step from TMP to TDP. The enzyme carries N and C-terminal extensions but otherwise shares the same core structure with dNKs and dNMPKs including the central β-sheet, the LID region (orange), and the P-loop (green). The binding conformation of the co-crystallized Tp5A analog also closely resembles the orientation seen in the above E. coli structure.
While there have been numerous enzyme engineering studies involving Herpes Simplex virus thymidine kinase, a majority of these studies have focused on the initial phosphorylation of ganciclovir (7) and acyclovir, two nucleoside analogs used in cancer treatment and antiherpal therapy, respectively.[51–58] However, as neither prodrug relies on the herpes kinase for its conversion to the diphosphate, few of these investigations have assessed the impact of mutations on the secondary activity, the phosphorylation of TMP to TDP.[59–61] In its current state, the dual functionality of Herpes Simplex virus thymidine kinase is mostly a curiosity but one with potential to offer new insight in the catalytic plasticity of enzymes and to create more effective tools for nucleoside prodrug activation.
Acknowledgments
This work was supported in part by the U.S. National Institutes of Health [GM69958] and a grant to the Emory Center for AIDS Research [AI050409] from the National Institutes of Health and by institutional funding from the Emory University Health Science Center.
Biography

Stefan Lutz holds a BSc degree from the Zurich University of Applied Sciences (Switzerland), and a MSc degree from the University of Teesside (UK). He obtained his PhD from the University of Florida and spent three years as a post-doctoral researcher with Stephen Benkovic at Pennsylvania State University under a fellowship of the Swiss National Science Foundation. Since 2002 he has been a chemistry professor at Emory University in Atlanta, Georgia (USA). The research in the Lutz laboratory focuses on the structure-function relationship of proteins through combinatorial protein engineering.
References
- 1.Fischer E. Ber Dtsch Chem Ges. 1894;27:2984. [Google Scholar]
- 2.Arner ES, Eriksson S. Pharmacol Ther. 1995;67:155. doi: 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson S, Munch-Petersen B, Johansson K, Eklund H. Cell Mol Life Sci. 2002;59:1327. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gentry GA. Pharmacol Ther. 1992;54:319. doi: 10.1016/0163-7258(92)90006-l. [DOI] [PubMed] [Google Scholar]
- 5.Shi J, McAtee JJ, Schlueter Wirtz S, Tharnish P, Juodawlkis A, Liotta DC, Schinazi RF. J Med Chem. 1999;42:859. doi: 10.1021/jm980510s. [DOI] [PubMed] [Google Scholar]
- 6.Moolten FL. Cancer Res. 1986;46:5276. [PubMed] [Google Scholar]
- 7.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. Science. 1992;256:1550. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 8.Jordheim LP, Galmarini CM, Dumontet C. Cancer Chemother Pharmacol. 2006;58:547. doi: 10.1007/s00280-006-0195-8. [DOI] [PubMed] [Google Scholar]
- 9.Solaroli N, Johansson M, Balzarini J, Karlsson A. Gene Ther. 2007;14:86. doi: 10.1038/sj.gt.3302835. [DOI] [PubMed] [Google Scholar]
- 10.Song S, Pursell ZF, Copeland WC, Longley MJ, Kunkel TA, Mathews CK. Proc Natl Acad Sci USA. 2005;102:4990. doi: 10.1073/pnas.0500253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tracewell CA, Arnold FH. Curr Opin Chem Biol. 2009;13:3. doi: 10.1016/j.cbpa.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlt JA, Babbitt PC. Curr Opin Chem Biol. 2009;13:10. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernhardt P, O’Connor SE. Curr Opin Chem Biol. 2009;13:35. doi: 10.1016/j.cbpa.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tao J, Xu JH. Curr Opin Chem Biol. 2009;13:43. doi: 10.1016/j.cbpa.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 15.Turner NJ. Nat Chem Biol. 2009;5:567. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 16.Kazlauskas RJ, Bornscheuer UT. Nat Chem Biol. 2009;5:526. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 17.Lutz S, Patrick WM. Curr Opin Biotechnol. 2004;15:291. doi: 10.1016/j.copbio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Farinas ET. Comb Chem High Throughput Screen. 2006;9:321. doi: 10.2174/138620706776843200. [DOI] [PubMed] [Google Scholar]
- 19.Griffiths AD, Tawfik DS. Trends Biotechnol. 2006;24:395. doi: 10.1016/j.tibtech.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Lutz S, Lichter J, Liu L. J Am Chem Soc. 2007;129:8714. doi: 10.1021/ja0734391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segura-Pena D, Lichter J, Trani M, Konrad M, Lavie A, Lutz S. Structure. 2007;15:1555. doi: 10.1016/j.str.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segura-Pena D, Lutz S, Monnerjahn C, Konrad M, Lavie A. J Mol Biol. 2007;369:129. doi: 10.1016/j.jmb.2007.02.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welin M, Kosinska U, Mikkelsen NE, Carnrot C, Zhu C, Wang L, Eriksson S, Munch-Petersen B, Eklund H. Proc Natl Acad Sci USA. 2004;101:17970. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Madhoun AS, Johnsamuel J, Barth RF, Tjarks W, Eriksson S. Cancer Res. 2004;64:6280. doi: 10.1158/0008-5472.CAN-04-0197. [DOI] [PubMed] [Google Scholar]
- 25.Munch-Petersen B, Knecht W, Lenz C, Sondergaard L, Piskur J. J Biol Chem. 2000;275:6673. doi: 10.1074/jbc.275.9.6673. [DOI] [PubMed] [Google Scholar]
- 26.Knecht W, Sandrini MP, Johansson K, Eklund H, Munch-Petersen B, Piskur J. EMBO J. 2002;21:1873. doi: 10.1093/emboj/21.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A. Nat Struct Biol. 2003;10:513. doi: 10.1038/nsb942. [DOI] [PubMed] [Google Scholar]
- 28.Iyidogan P, Lutz S. Biochemistry. 2008;47:4711. doi: 10.1021/bi800157e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Igarashi K, Hiraga S, Yura T. Genetics. 1967;57:643. doi: 10.1093/genetics/57.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. Nat Genet. 2005;37:73. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 31.Gerth ML, Lutz S. Biochem Biophys Res Commun. 2007;354:802. doi: 10.1016/j.bbrc.2007.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trani M, Lutz S. In: Protein Engineering Handbook. Lutz S, Bornscheuer UT, editors. Wiley VCH Verlag; Weinheim: 2008. p. 493. [Google Scholar]
- 33.Gerth ML, Lutz S. J Mol Biol. 2007;370:742. doi: 10.1016/j.jmb.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lutz S, Ostermeier M, Moore GL, Maranas CD, Benkovic SJ. Proc Natl Acad Sci USA. 2001;98:11248. doi: 10.1073/pnas.201413698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christians FC, Scapozza L, Crameri A, Folkers G, Stemmer WP. Nat Biotechnol. 1999;17:259. doi: 10.1038/7003. [DOI] [PubMed] [Google Scholar]
- 36.Lairson LL, Watts AG, Wakarchuk WW, Withers SG. Nat Chem Biol. 2006;2:724. doi: 10.1038/nchembio828. [DOI] [PubMed] [Google Scholar]
- 37.Olsen MJ, Stephens D, Griffiths D, Daugherty P, Georgiou G, Iverson BL. Nat Biotechnol. 2000;18:1071. doi: 10.1038/80267. [DOI] [PubMed] [Google Scholar]
- 38.Barrio JR, Secrist JA, 3rd, Leonard NJ. Biochem Biophys Res Commun. 1972;46:597. doi: 10.1016/s0006-291x(72)80181-5. [DOI] [PubMed] [Google Scholar]
- 39.Berry DA, Jung KY, Wise DS, Sercel AD, Pearson WH, Mackie H, Randolph JB, Somers RL. Tetrahedron Lett. 2004;45:2457. [Google Scholar]
- 40.Hawkins ME, Pfleiderer W, Balis FM, Porter D, Knutson JR. Anal Biochem. 1997;244:86. doi: 10.1006/abio.1996.9879. [DOI] [PubMed] [Google Scholar]
- 41.Georgiou G. Adv Protein Chem. 2000;55:293. doi: 10.1016/s0065-3233(01)55007-x. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Li Y, Liotta D, Lutz S. Nucleic Acids Res. 2009;37:4472. doi: 10.1093/nar/gkp400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikkelsen NE, Johansson K, Karlsson A, Knecht W, Andersen G, Piskur J, Munch-Petersen B, Eklund H. Biochemistry. 2003;42:5706. doi: 10.1021/bi0340043. [DOI] [PubMed] [Google Scholar]
- 44.Willmon CL, Krabbenhoft E, Black ME. Gene Ther. 2006;13:1309. doi: 10.1038/sj.gt.3302794. [DOI] [PubMed] [Google Scholar]
- 45.Brundiers R, Lavie A, Veit T, Reinstein J, Schlichting I, Ostermann N, Goody RS, Konrad M. J Biol Chem. 1999;274:35289. doi: 10.1074/jbc.274.50.35289. [DOI] [PubMed] [Google Scholar]
- 46.Lavie A, Ostermann N, Brundiers R, Goody RS, Reinstein J, Konrad M, Schichtling I. Proc Natl Acad Sci USA. 1998;95:14045. doi: 10.1073/pnas.95.24.14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gardberg A, Shuvalova L, Monnerjahn C, Konrad M, Lavie A. Structure. 2003;11:1265. doi: 10.1016/j.str.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 48.Lascu I, Gonin P. J Bioenerg Biomembr. 2000;32:237. doi: 10.1023/a:1005532912212. [DOI] [PubMed] [Google Scholar]
- 49.Chen MS, Walker J, Prusoff WH. J Biol Chem. 1979;254:10747. [PubMed] [Google Scholar]
- 50.Chen MS, Prusoff WH. J Biol Chem. 1978;253:1325. [PubMed] [Google Scholar]
- 51.Dube DK, Parker JD, French DC, Cahill DS, Dube S, Horwitz MS, Munir KM, Loeb LA. Biochemistry. 1991;30:11760. doi: 10.1021/bi00115a004. [DOI] [PubMed] [Google Scholar]
- 52.Munir KM, French DC, Dube DK, Loeb LA. Protein Eng. 1994;7:83. doi: 10.1093/protein/7.1.83. [DOI] [PubMed] [Google Scholar]
- 53.Munir KM, French DC, Dube DK, Loeb LA. J Biol Chem. 1992;267:6584. [PubMed] [Google Scholar]
- 54.Munir KM, French DC, Loeb LA. Proc Natl Acad Sci USA. 1993;90:4012. doi: 10.1073/pnas.90.9.4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Black ME, Hruby DE. J Biol Chem. 1992;267:9743. [PubMed] [Google Scholar]
- 56.Black ME, Loeb LA. Methods Mol Biol. 1996;57:335. doi: 10.1385/0-89603-332-5:335. [DOI] [PubMed] [Google Scholar]
- 57.Black ME, Loeb LA. Biochemistry. 1993;32:11618. doi: 10.1021/bi00094a019. [DOI] [PubMed] [Google Scholar]
- 58.Kokoris MS, Black ME. Protein Sci. 2002;11:2267. doi: 10.1110/ps.2460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Degreve B, Esnouf R, De Clercq E, Balzarini J. Mol Pharmacol. 2001;59:285. doi: 10.1124/mol.59.2.285. [DOI] [PubMed] [Google Scholar]
- 60.Hinds TA, Compadre C, Hurlburt BK, Drake RR. Biochemistry. 2000;39:4105. doi: 10.1021/bi992453q. [DOI] [PubMed] [Google Scholar]
- 61.Mercer KE, Ahn CE, Coke A, Compadre CM, Drake RR. Protein Eng. 2002;15:903. doi: 10.1093/protein/15.11.903. [DOI] [PubMed] [Google Scholar]