

Abstract
Pathogens transmitted by arthropod vectors are common in human populations, agricultural systems and natural communities. Transmission of these vector-borne pathogens depends on the population dynamics of the vector species as well as its interactions with other species within the community. In particular, predation may be sufficient to control pathogen prevalence indirectly via the vector. To examine the indirect effect of predators on vectored-pathogen dynamics, we developed a theoretical model that integrates predator–prey and host–pathogen theory. We used this model to determine whether predation can prevent pathogen persistence or alter the stability of host–pathogen dynamics. We found that, in the absence of predation, pathogen prevalence in the host increases with vector fecundity, whereas predation on the vector causes pathogen prevalence to decline, or even become extinct, with increasing vector fecundity. We also found that predation on a vector may drastically slow the initial spread of a pathogen. The predator can increase host abundance indirectly by reducing or eliminating infection in the host population. These results highlight the importance of studying interactions that, within the greater community, may alter our predictions when studying disease dynamics. From an applied perspective, these results also suggest situations where an introduced predator or the natural enemies of a vector may slow the rate of spread of an emerging vector-borne pathogen.
Keywords: host–pathogen, predation, disease ecology, biological control, vector
1. Introduction
Pathogens are a critical component of many ecological communities, often regulating host populations and influencing community structure. In recent years there has been an increasing appreciation of the importance of studying diseases within the context of the larger ecological community (Grenfell et al. 1995; Hudson et al. 2002; Ostfeld & Holt 2004; Collinge & Ray 2006; Keesing et al. 2006). In particular, diseases that are transmitted by a vector are dependent on the population dynamics of the vector species as well as the interactions of the vector and host populations with other species within the community (Ostfeld & Keesing 2000; Zavaleta & Rossignol 2004; Keesing et al. 2006). Diseases transmitted by arthropod vectors are common in wildlife and in agricultural and human communities (Anderson & May 1991). Zoonotic diseases such as Lyme disease and West Nile virus are transmitted to humans and animals by arthropod vectors, and many emerging or resurgent infectious diseases are vector transmitted (Gubler 1998; Gratz 1999; Daszak et al. 2000; Dobson & Foufopoulos 2000; Taylor et al. 2001).
Although there has been an increased emphasis on studying host–pathogen dynamics within a community context, research has largely focused on a limited set of interactions. Community epidemiology studies have tended to focus on interactions between hosts sharing a common pathogen (Holt & Pickering 1985; Begon et al. 1992; Woolhouse et al. 2001; Holt et al. 2003), or between pathogens that infect the same hosts (Holmes & Price 1986; Esch et al. 1990; Kuris & Lafferty 1994). Previous theoretical studies have explored the effect of predation on host–pathogen dynamics when a predator and pathogen compete for the host as a resource (Hochberg et al. 1990; Packer et al. 2003; Ostfeld & Holt 2004; Hall et al. 2005; Holt & Roy 2007). Predation intensity on host populations can alter host–pathogen dynamics (Hudson et al. 1992; Packer et al. 2003; Dwyer et al. 2004) and even affect pathogen persistence in the host population (Grenfell et al. 1995; Duffy et al. 2005; Hall et al. 2005). In general, predation often has major impacts on community structure via direct suppression of prey populations and indirect effects such as trophic cascades (Hairston et al. 1960; Paine 1966; Price et al. 1980; Sih et al. 1985; Schmitz et al. 2000). Because vector-borne pathogens are dependent on a vector species for transmission, pathogen persistence may be affected by predator–vector dynamics in addition to predator–host interactions. For example, a variety of predators consume the larvae of different disease-transmitting mosquito species (Kumar & Hwang 2006; Floore 2007 for recent reviews), and these predators are capable of regulating mosquito populations (Chase & Knight 2003; Stav et al. 2005; Juliano 2007, 2009; Seng et al. 2008). To date, however, there has not been a theoretical exploration of the impact of predator–vector interactions on the transmission or persistence of vector-borne pathogens.
The potential for predation to prevent pathogen invasion or reduce disease prevalence in a host population also has implications for the biological control of vector populations. Predators have been introduced, or proposed, as biological control agents of vectors for various diseases such as malaria, dengue fever and Lyme disease (Jenkins 1964; Legner 1995; Stauffer et al. 1997; Samish & Rehacek 1999; Scholte et al. 2005; Kumar & Hwang 2006; Ostfeld et al. 2006; Walker & Lynch 2007). Several recent studies suggest that predator introductions led to a decline in local cases of dengue fever in Vietnam and Thailand (Kay & Nam 2005; Kittayapong et al. 2008), and malaria in India (Ghosh et al. 2005; Ghosh & Dash 2007). However, many control efforts have been unsuccessful or have had unintended consequences, such as the displacement of native fish species by mosquito fish (Gambusia affinis) introduced to control malaria (WHO 1982; Pyke 2008). The goal of biological control is not necessarily to eliminate the vector (Murdoch et al. 1985), but to reduce pathogen prevalence and the risk of disease outbreaks in the host population. Natural enemies and introduced predators could accomplish this goal by directly reducing the density of the vector population, or by lowering the infectious proportion of the vector population. The identification of a target vector population threshold density, below which the pathogen cannot persist, is an important step in determining whether predation could sufficiently lower the vector population density. Biological control efforts are often used in concert with other vector control efforts (WHO 2004), such as insecticide spraying; therefore, it is also important to investigate how predator–vector dynamics may influence or be influenced by additional vector mortality factors.
Although recent theoretical work has incorporated vector interactions with pathogen and host species, current models of disease dynamics have not yet considered the potential role of predators in regulating vector and pathogen populations. Here we developed a mathematical model that integrates predator–prey and host–pathogen theories to examine the indirect effect of predators, via a vector, on pathogen dynamics. Using the model, we examined how predation on a vector population may affect disease prevalence as measured by the proportion of infected individuals in a host population. In particular, we investigated the relationship between predation strength and pathogen prevalence. We also determined whether predation can prevent pathogen persistence and examined different scenarios to determine under what conditions predation is most likely to effectively eradicate a pathogen. Lastly, we determined whether the form of disease transmission affects the model results.
2. The model
We designed our model to examine the effects of a vector's predator on disease dynamics, by incorporating both vector population dynamics and a predator. The main version of our model assumes that the host population is at a constant equilibrium and that pathogen transmission is density dependent (e.g. Anderson & May 1991; Hethcote 2000). We also examined model formulations incorporating host population demographics (§4), frequency-dependent transmission (§2.2), host immunity, vector latency, a saturating predator functional response and selective predation (appendix C). Incorporating host population demographics can be particularly important because disease-induced mortality and reductions in host fecundity often have a large effect on host–pathogen dynamics. We begin without host dynamics to set a baseline for us to examine the potentially interactive effects of predation and the vector population's growth rate. Predator–vector dynamics are represented by a set of Lotka–Volterra predator–prey equations (Gurney & Nisbet 1998). For simplicity, the predator is modelled as an obligate dietary specialist, dependent on the vector population for survival. However, this model also adequately represents a generalist predator capable of regulating the vector population.
We initially assumed a constant host population size based on the Ross–MacDonald susceptible–infected (SI) model for malaria (Ross 1910; MacDonald 1957; Anderson & May 1991). We modified the Ross–MacDonald model by adding a predator (P) to the system and making the vector population dynamic:
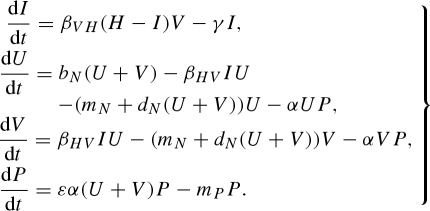 |
2.1 |
In these equations, I represents infected hosts. Because the total host population size is constant, we only need to keep track of infecteds, and represent susceptible hosts as S = H − I. βVH is the transmission coefficient for hosts acquiring infection from vectors, and γ represents the removal rate of individuals from the infected class as a result of death or recovery to the susceptible class. Disease transmission from the vector to the host is dependent on the densities of susceptible hosts (H − I) and infectious vectors (V).
Unlike hosts, vectors have a dynamic population size. U and V represent uninfected and infectious vectors, respectively. All vectors are born uninfected into class U with a per-capita birth rate of bN. Infectiousness does not affect vector birth rate or mortality. Vectors only acquire infection from hosts, with the transmission coefficient βHV. Host-to-vector disease transmission is also density dependent, determined by the densities of infected hosts (I) and uninfected vectors (U). Vectors in both classes experience density-independent (mN) and density-dependent (dN) mortality in addition to death from predation. Vector predators (P) have a conversion efficiency of ε, an attack rate of α and a density-independent mortality rate of mP. Thus, the vectors experience logistic growth, while their predators have a linear, type I functional response.
2.1. Equilibrium and invasion analysis
The main model with a constant host population has five biologically relevant equilibria (negative equilibrium values are excluded): (i) populations other than the host (H) are absent; (ii) the host and vector (N) are present, but the pathogen and predator are absent; (iii) vectors (N) and predators (P) are present in the absence of the pathogen; (iv) the pathogen is present in the host and vector populations, but the predator is absent; and (v) all populations including the pathogen coexist at non-zero densities (table 1). Because host density remains constant, the host is present in each of these equilibria as long as the parameter H > 0.
Table 1.
Equilibrium equations for the vector–predator SI model with a constant host population. (Equilibria for the constant host population model described by equation (2.1). Because host density remains constant, the host population is present in each of these equilibria as long as the parameter H > 0. Equilibrium (i) is host (H) population only; (ii) only uninfected hosts and vectors (U) are present; (iii) the vector and predator equilibria in the absence of the pathogen (U* and P*) match those for a Lotka–Volterra predator–prey model with prey self-regulation; (iv) the pathogen is present in the host and vector populations but the predator is absent; and (v) all populations including the pathogen coexist at non-zero densities. Note: for equilibria (iii) and (v), we denote the population size of the vectors without the predators as KV (equivalent to U* in (ii).)
| equilibrium | I* | U* | V* | P* |
|---|---|---|---|---|
| (i) | 0 | 0 | 0 | 0 |
| (ii) | 0 | 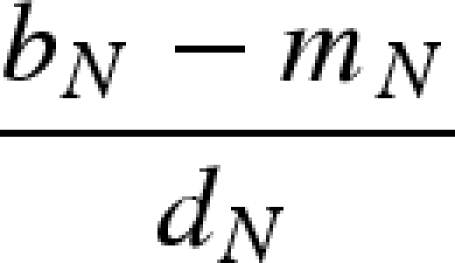 |
0 | 0 |
| (iii) | 0 | 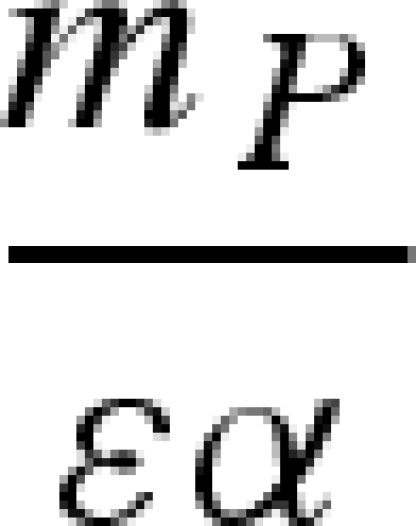 |
0 | a |
| (iv) | 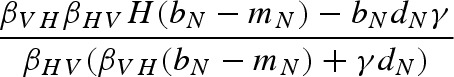 |
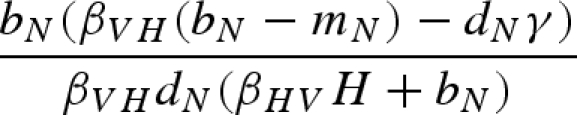 |
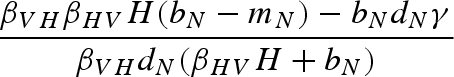 |
0 |
| (v) | 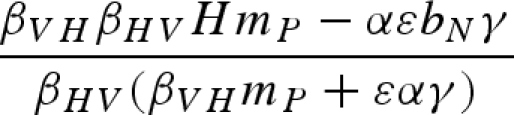 |
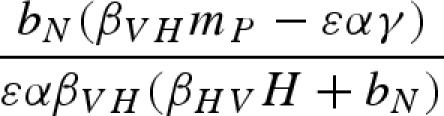 |
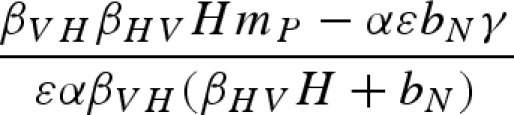 |
a |
a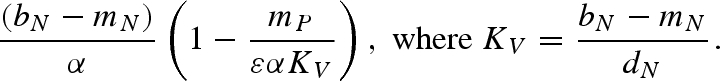
Equilibrium (i) is stable and the host population (H) is disease-free if bN ≤ mN. If bN > mN, then equilibrium (i) is unstable and, at a minimum, the vector population can invade the system. Equilibrium (ii) represents the disease-free, predator-free system where the host population, H, is disease-free (i.e. 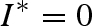 ) and the vector population, N, is at equilibrium
) and the vector population, N, is at equilibrium 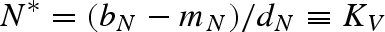 , which is the vector's carrying capacity. When predation is strong enough to regulate vector dynamics—as determined by the inequality mP/εα < KV—the predator can invade and shift the system to equilibrium (iii). When the pathogen is absent, dynamics between the vector and predator are described by the traditional Lotka–Volterra predator–prey model with a self-limiting prey population (Gurney & Nisbet 1998).
, which is the vector's carrying capacity. When predation is strong enough to regulate vector dynamics—as determined by the inequality mP/εα < KV—the predator can invade and shift the system to equilibrium (iii). When the pathogen is absent, dynamics between the vector and predator are described by the traditional Lotka–Volterra predator–prey model with a self-limiting prey population (Gurney & Nisbet 1998).
For analysing the effect of predation on the invasion or persistence of a pathogen, it is useful to consider the pathogen's basic reproduction number, R0—the number of individuals infected by the initial infectious case in an entirely susceptible population (Diekmann et al. 1990). If R0 > 1, each infectious host will infect more than one susceptible individual, and the pathogen can invade and persist upon introduction into a susceptible population. If R0 < 1, infected individuals do not fully replace themselves in the population, which leads to the elimination of the pathogen from the host population. Therefore, R0 = 1 serves as a threshold parameter for the invasion of the pathogen into a susceptible population. With the next-generation method (Diekmann et al. 1990; van den Driessche & Watmough 2002), and the total vector population defined as N = U + V for convenience, the pathogen's basic reproduction number from equation (2.1) is
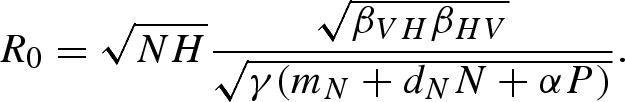 |
2.2 |
This equation for R0 is sensitive to our initial assumptions; R0 will be altered when host demographics are included or disease transmission is frequency-dependent. When the predator is absent, as in equilibrium (ii), 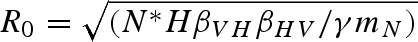 . From this equation, we can see that there is a host density threshold
. From this equation, we can see that there is a host density threshold 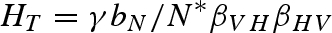 , such that H > HT is required for pathogen invasion and persistence. If the predator is present, the host density threshold for a successful pathogen invasion is HT = εαγbN / mPβVHβHV.
, such that H > HT is required for pathogen invasion and persistence. If the predator is present, the host density threshold for a successful pathogen invasion is HT = εαγbN / mPβVHβHV.
In addition to a host density threshold, there is also a minimum vector density required for pathogen persistence:
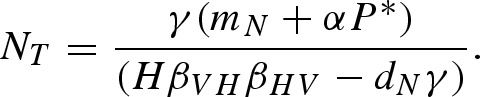 |
2.3 |
The vector density threshold depends on the equilibrium predator density (P*). In the absence of a predator, the minimum vector density for pathogen persistence simplifies to NT = γbN / HβVHβHV.
2.2. Frequency-dependent transmission model
The initial model assumed density-dependent transmission, in which the number of contacts between the vector and host is proportional to host density. In contrast, when disease transmission is frequency-dependent, the number of contacts depends on the proportion of infected host individuals rather than host density. It has been argued that the transmission of many vector-borne pathogens is better described by the frequency, rather than the density, of infected individuals in the host population (Getz & Pickering 1983; Thrall et al. 1993; Antonovics et al. 1995; Rudolf & Antonovics 2005).
Disease transmission is likely to be frequency-dependent when the vector only feeds on one or a few hosts during its lifetime, a feeding strategy employed by many mosquitoes, ticks and other arthropods that transmit disease (Antonovics et al. 1995). Many models with frequency-dependent transmission predict host–pathogen dynamics that differ from the results of density-dependent transmission models (Getz & Pickering 1983; Thrall et al. 1993; Wonham et al. 2006). Therefore, we modify our model by making disease transmission frequency-dependent; formulation details for the frequency-dependent transmission model are described in appendix B.
3. Results: constant host population
Adding a predator reduces the region of pathogen persistence at equilibrium (figure 1). Because an increase in the predator's attack rate, α, or conversion efficiency, ε, leads to a decrease in R0, an increase in predation strength leads to a decrease in the equilibrium proportion of infected hosts (figure 2). In addition to reducing equilibrium infection levels, predation can also delay the onset of an epidemic (figure 3). By decreasing the vector's lifespan, predation decreases the average number of new hosts infected during the lifespan of each infectious vector, thereby slowing the spread of disease. Even the relatively moderate predation rates used in figure 3 decrease the equilibrium pathogen prevalence by 30 per cent and increase the time to equilibrium from 50 to 150 days.
Figure 1.

Regions of stability in parameter space for each of the system equilibria as a function of vector birth rate (bN) and predator attack rate (α). (a) When there is no predator in the system, the pathogen reproduction number, R0, and pathogen persistence are a function of vector birth rate. (b) When a predator is added, the pathogen reproduction number and pathogen persistence depend on bN and α. The solid line represents the R0 = 1 isocline and the dashed line represents the P* = 0 isocline. The other model parameter values are H = 1, βVH = 0.15, βHV = 0.15, γ = 0.05, mN = 0.1, dN = 0.05, ε = 0.25 and mP = 0.1.
Figure 2.

Pathogen prevalence in the host and vector populations as a function of the predator attack rate (α). Pathogen prevalence is measured as the proportion of infected hosts (I*/H) and vectors (V*/N*) in their respective populations at equilibrium. The other model parameter values are H = 1, βVH = 0.15, βHV = 0.15, γ = 0.05, bN = 0.35, mN = 0.1, dN = 0.05, ε = 0.25, mP = 0.1. (Filled squares, host population; filled triangles, vector population.)
Figure 3.

Model of an epidemic outbreak in the presence or absence of a predator. At t = 0, the host population is entirely susceptible and 1 per cent of the vector population is infectious. Parameter values are H = 1, βVH = 0.15, βHV = 0.15, γ = 0.05, bN = 0.35, dN = 0.05, mN = 0.1, α = 0.2, ε = 0.25 and mP = 0.1. R0 = 2.54 when predator is absent and R0 = 1.60 when predator is present at its equilibrium density. (Solid line, predator present; dashed line, predator absent.)
In the absence of predation, an increase in the vector birth rate leads to an increase in the proportion of infected hosts (figure 4a). However, when the predator is introduced into the system, an increase in the vector birth rate leads to a decline in the prevalence of disease in the host population (figure 4a) because an increase in the vector birth rate leads to an increase in the predator population. Predation increases turnover in the vector population, and the average individual vector is infectious for a shorter period because it is alive for a shorter period. In addition, higher vector fecundity leads to more non-infectious vectors, thereby diluting the infectious potential of the vector population. Non-intuitively, in a tri-trophic system, increased vector fecundity leads to reduced host infection. When the predator is present, the proportion of infected hosts at equilibrium is sensitive to the vector birth rate, bN, but not to the vector mortality rate, mN, or density-dependent mortality term, dN (figure 4b). Increases in the vector mortality rate lead to a gradual reduction in pathogen prevalence in the absence of the predator. However, when a predator is present, moderate increases in the non-predation vector mortality rate do not affect pathogen prevalence because the predator regulates the vector population. At equilibrium, vector density is determined solely by the predator population's parameter values (table 1). Therefore, increasing mN reduces predator density, but does not change vector density or pathogen prevalence until the additional vector mortality is high enough that the predator cannot persist by feeding on the vector.
Figure 4.

(a) Pathogen prevalence, represented as the proportion of the host and vector population that is infected, as a function of the vector birth rate (bN) either in the absence of predation (open symbols) or with the predator present at its equilibrium density (filled symbols). When bN < mN (mN = 0.1), the vector population is absent at equilibrium, and by necessity the pathogen cannot persist. (b) Pathogen prevalence in the host and vector populations as a function of the non-predation vector mortality rate (mN). In the absence of the predator, an increase in the vector mortality rate leads to a decrease in pathogen prevalence, while pathogen prevalence does not respond to an increase in vector mortality when a predator is present. The other model parameter values are H = 1, bV = 0.5 (b only), βVH = 0.15, βHV = 0.15, γ = 0.05, dN = 0.05, α = 0.2, ε = 0.25 and mP = 0.1. (Open squares, predator absent—host population; open inverse triangles, predator absent—vector population; filled squares, predator present—host population; filled inverse triangles, predator present—vector population.)
Introducing a predator to the system will lower the proportion of infected hosts, except in a narrow parameter range where the vector's birth rate is barely higher than its mortality rate. Equation (2.3) for the minimum vector population threshold, NT, suggests that an increase in bN increases the minimum vector density required for pathogen persistence. There is also an inverse relationship between vector productivity (as measured by bN) and the strength of predation, α. At low vector productivity levels, pathogen persistence is possible except at very high levels of predation. As vector productivity increases, the predation strength needed to exclude the pathogen from the system decreases (figure 1b). Increasing the predator's conversion efficiency, ε, also reduces disease prevalence.
The presence of the predator also increases the minimum host density, HT, required for disease persistence when disease transmission is density-dependent. As the strength of predation increases, HT also increases. The other parameters that affect pathogen persistence and prevalence are identifiable by examination of equation (2.2) for R0. An increase in transmission rates βVH or βHV leads to increased pathogen prevalence. A decrease in the density of the host population causes a decrease in the proportion of infected hosts, and below a critical threshold density, HT, the pathogen cannot persist in the host population. There is also a negative relationship between the rate of host recovery or turnover, γ, and pathogen persistence. At high recovery rates, the pathogen cannot invade, and persist in, the host population.
3.1. Frequency-dependent model results
When disease transmission is frequency-dependent, increasing host density leads to a decrease in disease prevalence. R0 is no longer positively related to host density but instead scales with the inverse of host density (see appendix B). Likewise, the predation strength required to exclude the pathogen is also lower at higher host densities. Except at low host densities, the region of parameter space permitting pathogen persistence will be greater under density-dependent transmission than under frequency-dependent transmission. However, the mode of transmission does not affect the relationship between vector productivity, predation strength and disease prevalence. An increase in predation strength or the vector birth rate, with predators present, still leads to a decrease in the proportion of infected hosts.
4. Dynamic host population model
The assumption of a constant host population is useful for simplifying the dynamics of a relatively complex system and may be justified in the case of a disease with a short infectious period and limited effects on host mortality or fecundity. However, when a pathogen has an effect on host fecundity or mortality, or a long infectious period, the presence of a pathogen can have a large impact on host population dynamics. Including host population dynamics is also appropriate if host and vector population dynamics occur on a similar time scale. Here, the initial model of density-dependent transmission in a constant host population (equation (2.1)) is altered to include host demographics. The host has a per-capita mortality rate, mH, and a per-capita birth rate, bH, with a density-dependent control, ϕ. The modified model also explores the effect of the pathogen on host fecundity, ρ, and an additional disease-induced mortality rate, δ:
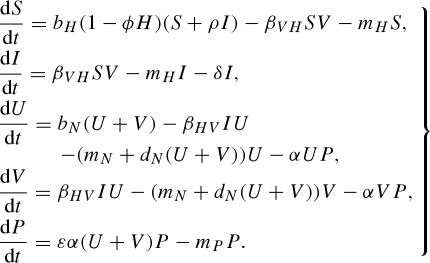 |
4.1 |
After including host demographics, the pathogen reproduction number is
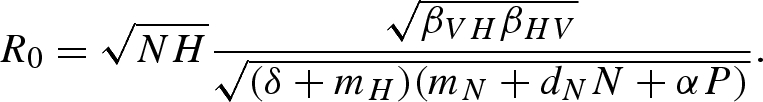 |
4.2 |
The only difference between this equation and equation (2.2) is that, instead of 1/γ, the average infectious period of an infected host is now 1/(δ + mH).
The equilibrium equations for the total host density (H*) when the pathogen is present are too complex to display succinctly. However, the equilibrium values for the other model equations can be solved as a function of H* (see appendix A). The equilibrium solutions to the dynamic host population model as a function of H* are very similar to those for the constant host population model, where host density was represented as a parameter, H. In fact, when there is no disease-induced mortality (δ = 0) and no reduction in the fecundity of infected individuals (ρ = 1), the equilibrium solutions of the two models are identical when H* = H.
4.1. Results: dynamic host model
Disease-induced changes in host mortality and fecundity decrease the proportion of infected hosts at equilibrium, but they do not modify the qualitative relationship between predation intensity and disease prevalence in the host population compared with the initial model. If infected hosts are subject to additional mortality, the proportion of infected hosts at equilibrium decreases (figure 5a). In addition, an increase in the disease-induced mortality rate leads to a decrease in the predation intensity required to prevent pathogen persistence. Reducing the fecundity of infected hosts also leads to a reduction in pathogen prevalence, but it does not affect pathogen persistence (figure 5b). Even if infected hosts are sterile (ρ = 0), the threshold for pathogen persistence does not change.
Figure 5.

Disease prevalence as a function of vector productivity with varying rates of (a) disease-induced mortality (solid line, δ = 0; dashed line, δ = 0.01; dotted line, δ = 0.05; dot and dashed line, δ = 0.10) and (b) reductions in host fecundity (solid line, ρ = 1.0; dashed line, ρ = 0.5; dotted line, ρ = 0.25; dot and dashed line, ρ = 0). The other model parameter values are bH = 0.20, mH = 0.05, ϕ = 0.2, δ = 0, βVH = 0.15, βHV = 0.15, mN = 0.1, dN = 0.05, α = 0.2, ε = 0.25, mP = 0.1, ρ = 1 (a only) and δ = 0 (b only).
When host demographics are included in the model, the pathogen can also affect the equilibrium host density. When the pathogen does not affect host mortality or fecundity, the equilibrium host density is at its carrying capacity, KH = (bH − mH)/(bHϕ), whether or not the pathogen is present. If infection increases the host mortality rate (δ > 0) or reduces fecundity (ρ < 1), the host population density will remain below the carrying capacity when the pathogen is present at equilibrium. Increasing the negative effect of infection on host fecundity reduces the host population density, but the relationship between the disease-induced mortality rate and host density is nonlinear. Initial increases in the disease-induced mortality rate reduce the equilibrium host population density; further increases minimally increase host population density, although equilibrium host density remains below the carrying capacity unless the pathogen is extirpated. Equilibrium host population density is higher in the presence of the predator for a given set of parameter values. In addition, increasing the predator attack rate or predator conversion efficiency increases the equilibrium host density (a standard trophic cascade) and decreases pathogen prevalence in the host population as we saw when host population size was constant.
5. Discussion
Numerous ecological studies have shown that, in addition to directly affecting their prey, predators often indirectly affect other species in a community (Holt 1977; Sih et al. 1985; Wootton 1994; Shurin et al. 2002; Caceres et al. in press). Our analysis indicates that predators may have important indirect effects on the prevalence or persistence of a vector-borne pathogen in a host population by controlling the vector population. If predation intensity is strong enough, the predator can prevent the establishment of a pathogen in a susceptible population. In addition, introducting a predator into a system where an endemic pathogen is at equilibrium with the host and vector populations can eliminate the pathogen (as long as R0 < 1 in the presence of the predator). These predictions are robust to assumptions about host population dynamics and disease transmission as well as the addition of acquired immunity in the host population, a vector latency period, a saturating response of predation to vector density and selective predation on infectious or non-infectious vectors (appendix C).
A non-intuitive prediction of this model is that predation on the vector population reverses the relationship between vector productivity and pathogen prevalence. Pathogen prevalence increases with vector productivity (defined here as the vector's birth rate, bN) in the absence of predation, whereas in the presence of the predator, pathogen prevalence declines with increasing vector productivity. The effect of predation on pathogen prevalence or persistence is not dependent on whether disease transmission is modelled as density- or frequency-dependent, and provides an indication of situations where natural predation or biological control may successfully control vector-borne diseases. Interestingly, the results of several studies are consistent with this prediction. In particular, vector control methods are essential for managing dengue fever and dengue hemorrhagic fever—a common mosquito-transmitted viral disease in humans—because of limited treatment options and no approved vaccine (Kroeger & Nathan 2007). Predaceous copepods in the genera Mesocyclops and Macrocyclops have been used successfully as biological control agents to control Aedes spp. mosquitoes that transmit dengue (Marten et al. 1994; Kay et al. 2002; Kay & Nam 2005; Nam et al. 2005; Kittayapong et al. 2008). In Vietnam, biological control efforts targeted larval breeding sites where the productivity of Aedes aegypti was especially high (Kay et al. 2002). These control efforts helped eradicate the vector from 32 of the 37 communities; no new dengue cases were reported in any of the treated communities over a period of several years (Kay & Nam 2005; Nam et al. 2005). The introduction of Mesocyclops in combination with other control measures also led to a significant reduction in dengue cases in Chachoengsao Province, Thailand (Kittayapong et al. 2008), and larvivorous fishes significantly reduced the long-term (>12 months) density of A. aegypti in rural Cambodia (Seng et al. 2008).
Targeting sites of high vector productivity is likely to have the largest effect on vector abundance. Our model suggests that introducing a predator to these sites could also lead to large reductions in pathogen prevalence due to a predator-induced reversal in the relationship between vector productivity and pathogen prevalence. In addition to targeting highly productive larval sites for dengue control, several researchers have suggested that malarial control efforts should target the most productive larval habitats (Gu & Novak 2005; Gu et al. 2008). Although these efforts are aimed at targeting productive habitats for environmental management and source reduction, our model predicts that these productive habitats could be targets for successful biological control efforts, particularly when elimination of the habitat is not feasible. Although controlling malaria by introducing predators has a controversial history (Pyke 2008), several recent reviews suggest that native or introduced predators can reduce the abundance of Anopheles larvae in certain habitats (Walker & Lynch 2007; Chandra et al. 2008). For example, Wu et al. (1991) found that introducing carp to rice paddies in Guangxi, China, significantly reduced larval mosquito density and may have reduced malarial transmission at the village and county levels. Kumar et al. (1998) found that replacing DDT and pyrethum treatments with the introduction of Bacillus thuringiensis and a native larvivorous fish Aplocheilus blocki to the major breeding habitats of Anopheles stephensi in Goa, India, led to a significant reduction in larval A. stephensi abundance. In addition, malarial incidence declined when compared with nearby towns that did not receive the new treatments. These studies, along with others reviewed by Walker & Lynch (2007) and Chandra et al. (2008), suggest that predator control of Anopheles mosquitoes can reduce malarial incidence in certain situations. Although quantitative evaluation of vector productivity is often difficult (Killeen et al. 2005), recent efforts to quantify the productivity of Anopheles (Mutuku et al. 2006) and Aedes (Kay et al. 2002; Chadee 2007) mosquitoes show promise and suggest that using measures of habitat-based vector productivity to guide biological control efforts may prove useful. Our model suggests that targeting these highly productive sites would lead to the largest reduction in pathogen prevalence.
Classical biological control strategies have emphasized the use of specialist predators to maintain pest populations at low, stable equilibrium levels (Murdoch et al. 1985; Stiling & Cornelissen 2005). Our model also suggests that it is not essential for the predator to extirpate the vector population in order to eradicate the pathogen. The minimum vector population threshold predicted by our model (NT, equation (2.3)) provides a target for vector control. Biological control agents are often successful at reducing vector abundance, at least in the short term (Legner 1995; Kumar & Hwang 2006; Ostfeld et al. 2006; Walker & Lynch 2007; Chandra et al. 2008) although they will not eradicate the pathogen if the equilibrium vector density remains above NT. For example, in three communities examined by Nam et al. (2005) in which predaceous copepods were used to control Aedes mosquitoes, dengue cases dropped to zero in both 2002 and 2003 even though mosquitoes were still present at low densities. These results are consistent with our prediction that biological control can maintain vector density below NT.
Even if predation intensity is not high enough to remove the pathogen from the host population, predation can still decrease disease prevalence in the host population to low levels, potentially delaying the onset of an epidemic. For human diseases, an increase in the time between the introduction of a pathogen and an outbreak would provide additional time for disease control efforts (such as vaccination or quarantine) to be implemented (Anderson & May 1991). In agricultural systems, farmers are often concerned about an economic threshold at which crop losses become great enough to trigger economic losses (Kogan 1998). In this case, the reduction in the rate of disease spread due to predation may be sufficient to allow crop harvest before an epidemic outbreak. For example, vector-transmitted plant pathogens such as the barley and cereal yellow dwarf viruses (BYDV/CYDV) can have detrimental effects on crop yields by causing stunted growth, reduced seed-set or early senescence (Irwin & Thresh 1990; D'Arcy & Burnett 1995); therefore, reducing disease prevalence within the host population could limit crop losses to acceptable levels. Natural predators and biological control agents have been used in agricultural settings to reduce the abundance and slow the population growth of several different aphid species that transmit BYDV/CYDV (Chiverton 1986; Brewer & Elliott 2004), but the corresponding effects on BYDV/CYDV have not yet been investigated. Laboratory studies of aphid predators have found either no impact on BYDV infection rates or a transient reduction in the spread of the virus (Christiansen-Weniger et al. 1998; Smyrnioudis et al. 2001). However, these studies were short term and did not continue long enough for the predator to regulate the aphid population, which is required to observe large reductions in pathogen prevalence in our model.
In the absence of predation, an increase in the vector mortality rate leads to a decline in disease prevalence. However, when a predator with a type I linear functional response is present, an increase in the vector mortality rate from factors other than predation does not change disease prevalence. Because the predator regulates the vector population, an increase in non-predation mortality reduces predator, but not vector, abundance. The overall vector mortality rate stays the same because the increase in the background mortality rate is compensated by a decrease in mortality due to predation. If the predator has a type II saturating functional response, pathogen prevalence may decline with an increase in non-predation mortality if the predator's consumption rate is already saturated (see appendix C for details). However, even with a type II response, once the vector density decreases to a level at which the predator consumption rate is no longer saturated, pathogen prevalence will level off despite further increases in non-predation mortality.
As a result, predator regulation of the vector population may have implications for the use of other vector control methods (such as pesticides) in conjunction with biological control. Biological control is often part of an integrated pest management strategy that includes other vector control methods, such as spraying insecticides, environmental manipulation or the application of larvicides to vector breeding sites (WHO 2004). Ideally, these integrated approaches will have synergistic effects, but our model suggests that predator regulation of the vector population could reduce or prevent the effectiveness of other control methods aimed at increasing vector mortality. Instead, these additional mortality factors may reduce predator abundance, limiting the regulatory effects of predation on the vector population as Chansang et al. (2004) and Snyder & Ives (2001) have observed in mosquitoes and aphids, respectively. Chansang et al. (2004) found that the application of B. thuringiensis alone or in combination with Mesocyclops thermocyclopoides initially reduced A. aegypti densities more than a Mesocyclops-only treatment, but after 16 weeks the Mesocyclops-only treatment had higher Mesocyclops densities and lower A. aegypti densities than the combined treatment. This suggests that B. thuringiensis prevented Mesocyclops from strongly regulating the A. aegypti population. Other sources of density-independent vector mortality besides vector control efforts, such as the presence of other predators, can interfere with pathogen regulation by a specialist predator. Snyder & Ives (2001) found that specialist parasitoids introduced to control the pea aphid (Acyrthosiphon pisum), which transmits several crop viruses, were disrupted by generalist predators that fed on both the aphid and its parasitoid.
Previous work has explored how predation on a host population affects host–pathogen dynamics (Packer et al. 2003; Ostfeld & Holt 2004; Duffy et al. 2005; Hall et al. 2005; Holt & Roy 2007; Duffy & Hall 2008; Caceres et al. in press). Packer et al. (2003) detailed how predation often reduces the incidence of parasitic infections and increases the overall size of the prey population. Our model reveals that predation on a vector can similarly reduce pathogen prevalence in both the host and vector. In addition, when the pathogen regulates the host by increasing mortality or reducing fecundity, predation can weaken the pathogen's negative impact on the host, thereby increasing host abundance. When predation on immune individuals occurs, predation can increase pathogen prevalence in a prey population (Holt & Roy 2007). We found that including acquired immunity in the host does not alter the qualitative effects of predation on pathogen prevalence (see appendix C for details). Because our model does not include recovery or acquired immunity of infectious vectors, predation on the vector population cannot lead to an increase in pathogen prevalence in the host or vector; however, relaxing this assumption by incorporating predation on vectors resistant to infection could increase pathogen prevalence. This could affect disease control efforts; predation on mosquitoes that have been genetically modified to be incapable of transmitting malaria or other pathogens (Alphey et al. 2002; Scott et al. 2002) could limit the effectiveness of their introduction.
In addition to predator effects on the pathogen, predation may indirectly increase host abundance by reducing the negative impacts of infection. Indirect effects of predators on lower trophic levels, i.e. trophic cascades, are common in nature (Pace et al. 1999; Shurin et al. 2002) and are predicted to be stronger in highly productive systems (Oksanen et al. 1981; Polis 1999; but see Borer et al. 2005). In a system with direct predation on the host population, Hall et al. (2005) found that high ecosystem productivity (as measured by the carrying capacity of the host population) could facilitate the invasion of a parasite. However, increasing the strength of predation in a highly productive environment destabilized parasite–host dynamics leading to extinction of both the parasite and its host. In our model, the predator, vector and host populations form a tri-trophic system with both direct trophic interactions and indirect interactions mediated by the pathogen. The indirect effects of predation on pathogen prevalence and host abundance are predicted to be strongest when the vector population experiences high growth rates, as may be expected to occur in highly productive environments. For vectors such as mosquitoes that complete different life stages in different environments, population growth rates are often influenced primarily by productivity in the larval environment rather than by host density (Southwood et al. 1972; Juliano 2007), and therefore we would expect the strength of the indirect effects of predation to be related to larval vector productivity. However, the productivity of other vectors that remain in the same environment throughout their life cycle, such as many fleas, ticks or aphids, may be positively influenced by host density (Dixon 1998).
Our model results suggest that predation on a vector can strongly influence pathogen prevalence and host abundance in both intuitive and non-intuitive ways. The introduction of biological control agents to control vector populations can reduce prevalence or eradicate the pathogen, particularly in productive environments where the vector population experiences a high turnover rate. In the absence of predation, these productive environments would be expected to have the highest infection rates. This predator-induced reversal in disease prevalence indicates that reductions in the abundance of natural predators due to invasive species, habitat modifications or climate change may be partially responsible for the increased frequency of disease outbreaks caused by vector-borne pathogens (Gubler 1998; Gratz 1999; Daszak et al. 2000). The presence of a predator could also decrease the risk of zoonotic diseases such as Lyme disease and West Nile virus spilling over from reservoir hosts to humans (Bernard et al. 2001; Schmidt & Ostfeld 2001; Logiudice et al. 2003). With the emergence and re-emergence of many vector-borne diseases, determining the potential interactions of the host(s) and pathogen with other species in the community is essential for predicting potential disease risks and guiding control efforts. Our model results suggest that empirical research into the role of native and introduced predators across a range of vector productivity and mortality will be essential to determining the influence of predators on vectored disease transmission.
Acknowledgements
We would like to thank E. Seabloom, A. Brandt, P. Zarnetske, C. Benfield, A. Jolles and J. Dauer for comments on this manuscript. S.M.M. was supported by an NSF IGERT graduate fellowship (NSF 0333257) in the Ecosystem Informatics IGERT programme at Oregon State University. Additional support for E.T.B. and P.R.H. was provided by NSF-EID grant 05-25666 to E.T.B. and E. Seabloom.
Appendix A. dynamic host population model equilibria
The model incorporating host population demographics represented by equation (4.1) has eight biologically relevant equilibria. Equilibria (i)–(vi) are all disease-free equilibria with different combinations of the host, vector and predator populations present at equilibrium. Because the vector population does not depend on the host as a resource, the vector and predator populations can persist in the absence of the host. Equilibrium (vii) represents an endemic disease equilibrium in the host and vector populations in the absence of the predator; while the host, vector, predator and pathogen populations are all present in equilibrium (viii). The equilibrium equations for the total host density (H*) when the pathogen is present are too complex to display succinctly. However, the equilibrium values for the other populations can be solved as a function of H*. Note that 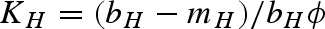 represents the carrying capacity of the host population in the absence of infection.
represents the carrying capacity of the host population in the absence of infection.
Equilibrium (i):
Equilibrium (ii):
Equilibrium (iii):
Equilibrium (iv):
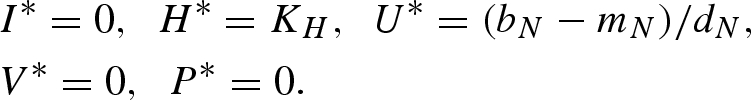 |
Equilibrium (v):
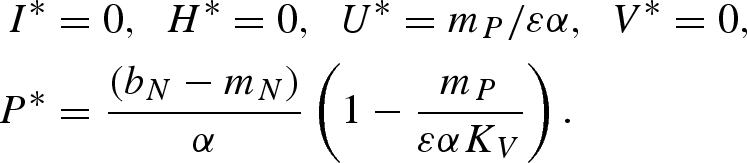 |
Equilibrium (vi):
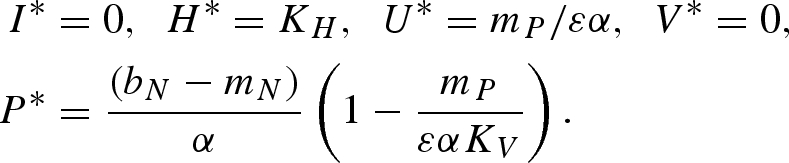 |
Equilibrium (vii):
 |
Equilibrium (viii):
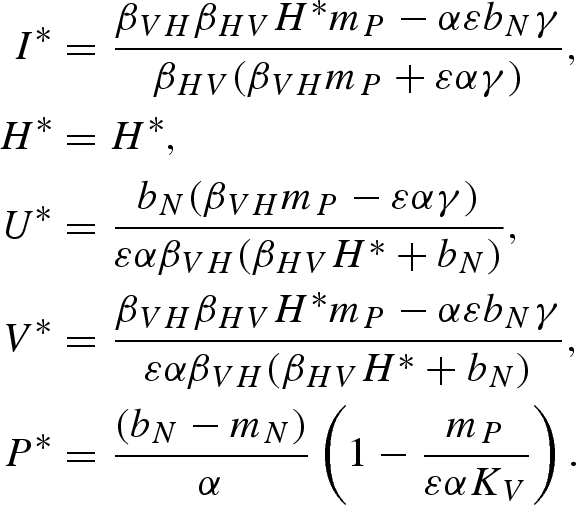 |
Appendix B. frequency-dependent model formulation
The constant host population model with frequency-dependent transmission is
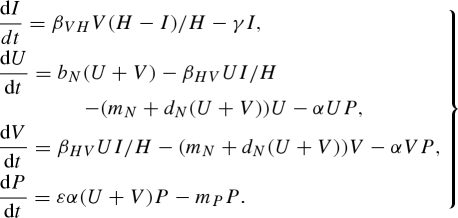 |
B1 |
The vector and predator population dynamics are identical to the density-dependent model described in equation (2.1). The only alteration to the system equations is the alteration of the disease transmission terms from βVHSV and βHVIU, to βVHSV/H and βHVIU/H. Because disease transmission depends on the frequency of infection in the host population as opposed to the density of the host population, the pathogen reproduction rate, R0, differs between the two models. The equation for R0 with frequency-dependent disease transmission is
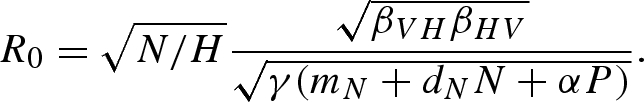 |
B2 |
The equilibrium values for the total vector population (N) and the predator population (P) are the same whether disease transmission is density- or frequency- dependent. The equilibrium densities of infected hosts (I) and infectious vectors (V) differ from the equilibrium densities in the density-dependent model as follows.
Equilibrium (i):
Equilibrium (ii):
Equilibrium (iii):
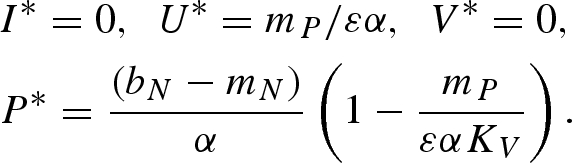 |
Equilibrium (iv):
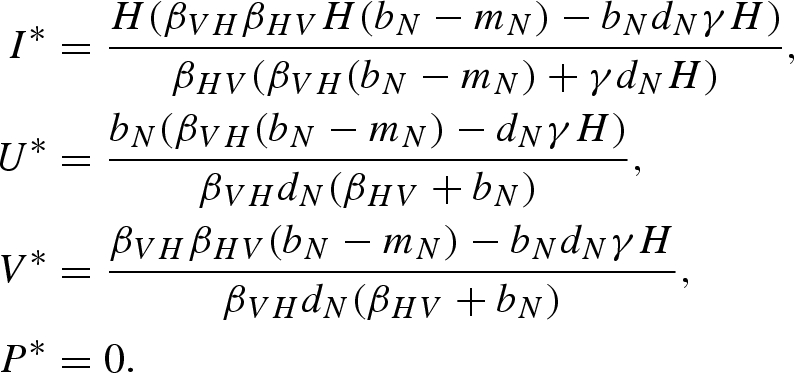 |
Equilibrium (v):
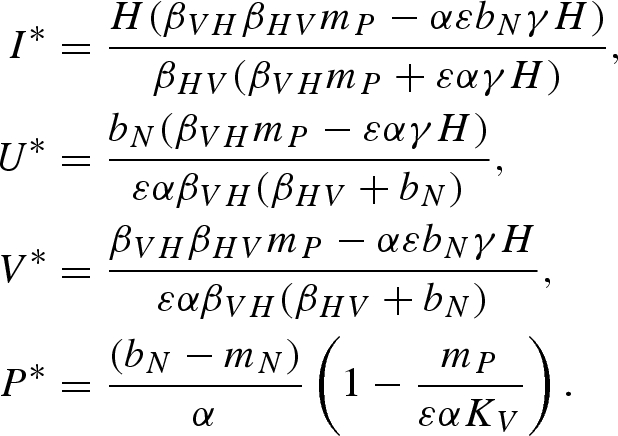 |
Appendix C. additional model extensions
C.1. Acquired host immunity
The addition of a recovered class (R) with acquired immunity in the host population does not change the qualitative effects of predation on pathogen prevalence or persistence. The constant host population model with host immunity is
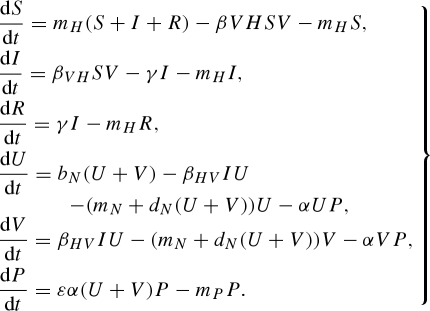 |
C1 |
The equations representing the predator (P) and vector (U and V) populations are indentical to the basic model (equation (2.1)). Infected host individuals recover at a rate γ, and then remain immune to reinfection for life. The modified pathogen reproduction number with host immunity is
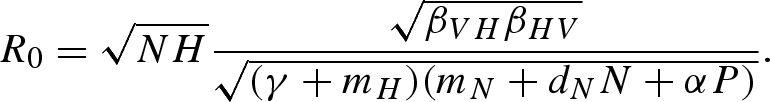 |
C2 |
The rate at which infected hosts recover and become immune (γ) appears in the denominator of R0, indicating that acquired host immunity lowers the pathogen reproduction number, and lowers the threshold at which predation can eliminate the pathogen from the system (figure 6). Figure 6 shows that predation decreases pathogen prevalence in the host population whether or not hosts are immune to infection, and an increase in the recovery rate decreases pathogen prevalence.
Figure 6.

Disease prevalence in the host population as a function of predator attack rate (α) for three different host recovery rates (γ) with lifelong immunity for recovered individuals. The other model parameter values are H = 1, βVH = 0.2, βHV = 0.2, γ = 0.05, bN = 0.35, mN = 0.1, dN = 0.05, ε = 0.25 and mP = 0.1. (Solid line, γ = 0; dashed line, γ = 0.01; dotted line, γ = 0.05.)
C.2. Vector latency period
For many diseases, vectors experience a latent period between their initial exposure to a pathogen and when they become infectious (Anderson & May 1991). For some diseases, such as malaria, the length of this latent period relative to the lifespan of the vector is an important factor in determining pathogen persistence (MacDonald 1957). In the malarial model originally developed by Ross (1910), extending the latent period decreases the basic pathogen reproduction number, R0, and reduces equilibrium prevalence. Similarly, our basic model can be extended by adding an exposed vector class, E, to equation (2.1):
| C3 |
When the pathogen is transmitted to an uninfected vector, the vector enters the exposed class and then becomes infectious at a rate η. Because vectors in the exposed class may be killed by the predator before they become infectious, adding a vector latency period raises the minimum vector threshold density, NT, for pathogen persistence, and further lowers pathogen prevalence at equilibrium. The adjusted pathogen reproduction number with vector latency is
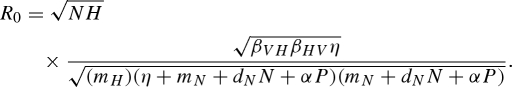 |
C4 |
Increasing the length of this latent period (reducing η) decreases R0 and increases NT, increasing the chance that predation will be sufficient to eradicate the pathogen by driving R0 < 1.
C.3. Predator functional response
We initially assumed that the predator has a Holling type I linear functional response to vector abundance—as vector density increases, per-capita predation scales linearly at the rate αN. However, this assumption becomes biologically unrealistic at high vector densities as the individual predator attack rate becomes limited by the time required to handle and digest each prey item. This saturating predator functional response can be represented by a Holling type II functional response (Holling 1959):
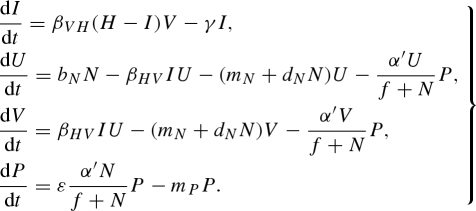 |
C5 |
The adjusted pathogen reproduction number is
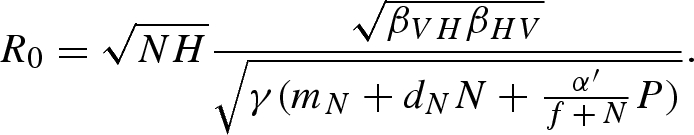 |
C6 |
Introducing a type II functional response does not change the qualitative effect of predation on pathogen prevalence and persistence. The presence of a predator still lowers pathogen prevalence, and increasing the strength of predation leads to a decline in pathogen prevalence. With a type I functional response, the proportion of infected hosts always reaches a stable equilibrium. In contrast, increasing predation strength with a type II functional response leads to cycles in pathogen prevalence in the host (figure 7). These cycles occur because predator and vector densities oscillate, leading to oscillation in the force of infection experienced by the host population. Despite these oscillations, predation can still eliminate the pathogen from the system while vector densities are greater than 0.
Figure 7.

Model of an epidemic outbreak for different values of α′ for a predator with a type II functional response. At t = 0, the host population is entirely susceptible and 1 per cent of the vector population is infectious. Parameter values are H = 1, βVH = 0.15, βHV = 0.15, γ = 0.05, bN = 0.35, dN = 0.05, mN = 0.1, ε = 0.25, mP = 0.1 and f = 1. (Solid line, α′ = 0; dotted line, α′ = 0.4; dot and dashed line, α′ = 0.5; dashed line, α′ = 0.9.)
When the predator has a type I functional response, increasing the vector birth rate leads to a decline in pathogen prevalence. With a type II functional response, increasing the vector birth rate initially leads to a decline in prevalence. However, at higher vector birth rates, the predator's per-capita consumption rate becomes saturated and further increases in the birth rate do not reduce the mean pathogen prevalence (figure 8a). The relationship between the vector's non-predation mortality rate (mN) and pathogen prevalence also depends on the predator's functional response. With a type I response, increasing mN does not affect pathogen prevalence in the host population as long as the predator is present (figure 4b). If the predator has a type II response, increasing mN leads to a decrease in pathogen prevalence when mN is low enough that the predator's per-capita consumption rate is saturated (figure 8b). Once mN is high enough that the predator's per-capita consumption rate is no longer saturated, further increases in mN do not affect pathogen prevalence.
Figure 8.

(a) Pathogen prevalence, represented as the proportion of the host population that is infected, as a function of the vector birth rate (bN) when the predator has a type II functional response. (b) Pathogen prevalence in the host population as a function of the non-predation vector mortality rate (mN). Solid lines represent mean prevalence and dashed lines represent the minimum and maximum prevalence values when prevalence is cyclical. Parameter values are H = 1, βVH = 0.15, βHV = 0.15, γ = 0.05, bN = 0.7 (b only), dN = 0.05, mN = 0.1 (a only), α′ = 0.5, ε = 0.25 mP = 0.1 and f = 1.
C.4. Predator selectivity
The capacity of predators to selectively prey on infectious or non-infectious vectors could also alter the quantitative effect of predation on pathogen prevalence. For example, East African jumping spiders (Evarcha culicivora) preferentially prey on female Anopheles mosquitoes carrying blood meals, and therefore are more likely to be carrying the malarial parasite (Nelson & Jackson 2006). Our model predicts that if the predator preferentially preys on susceptible vectors, the negative effect of predation on pathogen prevalence is weakened, but predation never increases prevalence. Preferential predation on exposed or infectious vectors enhances the negative effects of predation on prevalence and lowers the threshold for disease eradication. Predation consistently reduces pathogen prevalence in the host and vector populations under different assumptions about host demographics or immunity, latency and predator selectivity.
Footnotes
Present address: Wildlife Trust, 17th floor, 460 West 34th Street, New York, NY 10001, USA.
References
- Alphey L., et al. 2002. Malaria control with genetically manipulated insect vectors. Science 298, 119–121. ( 10.1126/science.1078278) [DOI] [PubMed] [Google Scholar]
- Anderson R. M., May R. M. C. 1991. Infectious diseases of humans: dynamics and control. Oxford, UK: Oxford University Press. [Google Scholar]
- Antonovics J., Iwasa Y., Hassell M. P. 1995. A generalized model of parasitoid, venereal, and vector-based transmission processes. Am. Nat. 145, 661–675. ( 10.1086/285761) [DOI] [Google Scholar]
- Begon M., Bowers R. G., Kadianakis N., Hodgkinson D. E. 1992. Disease and community structure: the importance of host self-regulation in a host–host–pathogen model. Am. Nat. 139, 1131–1150. ( 10.1086/285379) [DOI] [Google Scholar]
- Bernard K. A., Maffei J. G., Jones S. A., Kauffman E. B., Ebel G. D., Dupuis A. P. 2001. West Nile virus infection in birds and mosquitoes, New York State, 2000. Emerg. Infect. Dis. 7, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borer E. T., Seabloom E. W., Shurin J. B., Anderson K. E., Blanchette C. A., Broitman B., Cooper S. D., Halpern B. S. 2005. What determines the strength of a trophic cascade? Ecology 86, 528–537. ( 10.1890/03-0816) [DOI] [Google Scholar]
- Brewer M. J., Elliott N. C. 2004. Biological control of cereal aphids in North America and mediating effects of host plant and habitat manipulations. Annu. Rev. Entomol. 49, 219–242. ( 10.1146/annurev.ento.49.061802.123149) [DOI] [PubMed] [Google Scholar]
- Caceres C. E., Knight C. J., Hall S. R. In press Predator spreaders: predation can enhance parasite success in a planktonic host–parasite system. Ecology. [DOI] [PubMed] [Google Scholar]
- Chadee D. 2007. Key premises, a guide to Aedes aegypti (Diptera: Culicidae) surveillance and control. Bull. Entomol. Res. 94, 201–207. [DOI] [PubMed] [Google Scholar]
- Chandra G., Bhattacharjee I., Chatterjee S. N., Ghosh A. 2008. Mosquito control by larvivorous fish. Indian J. Med. Res. 127, p. 13. [PubMed] [Google Scholar]
- Chansang U.-R., Bhumiratana A., Kittayapong P. 2004. Combination of Mesocyclops thermocyclopoides and Bacillus thuringiensis var. israelensis: a better approach for the control of Aedes aegypti larvae in water containers. J. Vector Ecol. 29, 218–226. [PubMed] [Google Scholar]
- Chase J. M., Knight T. M. 2003. Drought-induced mosquito outbreaks in wetlands. Ecol. Lett. 6, 1017–1024. ( 10.1046/j.1461-0248.2003.00533.x) [DOI] [Google Scholar]
- Chiverton P. A. 1986. Predator density manipulation and its effects on populations of Rhopalosiphum padi (Hom.: Aphididae) in spring barley. Ann. Appl. Biol. 109, 49–60. ( 10.1111/j.1744-7348.1986.tb0318.x) [DOI] [Google Scholar]
- Christiansen-Weniger P., Powell G., Hardie J. 1998. Plant virus and parasitoid interactions in a shared insect vector/host. Entomol. Exp. Appl. 86, 205–213. ( 10.1046/j.1570-7458.1998.00282.x) [DOI] [Google Scholar]
- Collinge S. K., Ray C. 2006. Disease ecology: community structure and pathogen dynamics. Oxford, UK: Oxford University Press. [Google Scholar]
- D'Arcy C., Burnett P. A. 1995. Barley yellow dwarf: 40 years of progress. St Paul, MN: APS Press. [Google Scholar]
- Daszak P., Cunningham A. A., Hyatt A. D. 2000. Emerging infectious diseases of wildlife: threats to biodiversity and human health. Science 287, 443–449. ( 10.1126/science.287.5452.443) [DOI] [PubMed] [Google Scholar]
- Diekmann O., Heesterbeek J. A. P., Metz J. A. J. 1990. On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. J. Math. Biol. 28, 365–382. ( 10.1007/BF00178324) [DOI] [PubMed] [Google Scholar]
- Dixon A. F. G. 1998. Aphid ecology. London, UK: Chapman & Hall. [Google Scholar]
- Dobson A., Foufopoulos J. 2000. Emerging infectious pathogens of wildlife. Phil. Trans. R. Soc. Lond. B 356, 1001–1012. ( 10.1098/rstb.2001.0900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M. A., Hall S. R. 2008. Selective predation and rapid evolution can jointly dampen effects of virulent parasites on Daphnia populations. Am. Nat. 171, 499–510. ( 10.1086/528998) [DOI] [PubMed] [Google Scholar]
- Duffy M. A., Hall S. R., Tessier A. J., Huebner M. 2005. Selective predators and their parasitized prey: are epidemics in zooplankton under top-down control? Limnol. Oceanogr. 50, 412–420. [Google Scholar]
- Dwyer G., Dushoff J., Yee S. H. 2004. The combined effects of pathogens and predators on insect outbreaks. Nature 430, 299–300. ( 10.1038/nature02569) [DOI] [PubMed] [Google Scholar]
- Esch G. W., Bush A. O., Aho J. M. 1990. Parasite communities: patterns and processes. New York, NY: Chapman and Hall. [Google Scholar]
- Floore T. G. (ed.) 2007. Biorational control of mosquitoes. AMCA Bulletin 7 Mount Laurel, NJ: American Mosquito Control Association. [Google Scholar]
- Getz W. M., Pickering J. 1983. Epidemic models: thresholds and population regulation. Am. Nat. 121, 892–898. ( 10.1086/284112) [DOI] [Google Scholar]
- Ghosh S., Dash A. 2007. Larvivorous fish against malaria vectors: a new outlook. Trans. R. Soc. Trop. Med. Hyg. 101, 1063–1064. ( 10.1016/j.trstmh.2007.07.008) [DOI] [PubMed] [Google Scholar]
- Ghosh S., Tiwari S., Sathyanarayan T., Sampath T., Sharma V., Nanda N., Joshi H., Adak T., Subbarao S. 2005. Larvivorous fish in wells target the malaria vector sibling species of the Anopheles culicifacies complex in villages in Karnataka, India. Trans. R. Soc. Trop. Med. Hyg. 99, 101–105. ( 10.1016/j.trstmh.2004.03.009) [DOI] [PubMed] [Google Scholar]
- Gratz N. G. 1999. Emerging and resurging vector-borne diseases. Annu. Rev. Entomol. 44, 51–75. ( 10.1146/annurev.ento.44.1.51) [DOI] [PubMed] [Google Scholar]
- Grenfell B. T., Dobson A. P. (eds) 1995. Ecology of infectious diseases in natural populations. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Gu W., Novak R. J. 2005. Habitat-based modeling of impacts of mosquito larval interventions on entomological inoculation rates, incidence, and prevalence of malaria. Am. J. Trop. Med. Hyg. 73, 546–552. [PubMed] [Google Scholar]
- Gu W., Utzinger J., Novak R. J. 2008. Habitat-based larval interventions: a new perspective for malaria control. Am. J. Trop. Med. Hyg. 78, 2–6. [PubMed] [Google Scholar]
- Gubler D. J. 1998. Resurgent vector-borne diseases as a global health problem. Emerg. Infect. Dis. 4, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney W. S. C., Nisbet R. M. 1998. Ecological dynamics. New York, NY: Oxford University Press. [Google Scholar]
- Hairston N. G., Smith F. E., Slobodkin L. B. 1960. Community structure, population control, and competition. Am. Nat. 94, 421–425. ( 10.1086/282146) [DOI] [Google Scholar]
- Hall S. R., Duffy M. A., Caceres C. E. 2005. Selective predation and productivity jointly drive complex behavior in host–parasite systems. Am. Nat. 165, 70–81. ( 10.1086/426601) [DOI] [PubMed] [Google Scholar]
- Hethcote H. W. 2000. The mathematics of infectious diseases. SIAM Rev. 42, 599–653. ( 10.1137/s0036144500371907) [DOI] [Google Scholar]
- Hochberg M. E., Hassell M. P., May R. M. 1990. The dynamics of host–parasitoid–pathogen interactions. Am. Nat. 135, 74–94. ( 10.1086/285033) [DOI] [Google Scholar]
- Holling C. S. 1959. Some characteristics of simple types of predation and parasitism. Can. Entomol. 91, 385–398. [Google Scholar]
- Holmes J. C., Price P. W. 1986. Communities of parasites. In Community biology: patterns and processes (eds Kikkawa D., Anderson D. J.), pp. 187–213. Oxford, UK: Blackwell Publishing. [Google Scholar]
- Holt R. D. 1977. Predation, apparent competition, and the structure of prey communities. Theor. Popul. Biol. 12, 197–229. ( 10.1016/0040-5809(77)900429-9) [DOI] [PubMed] [Google Scholar]
- Holt R. D., Pickering J. 1985. Infectious disease and species coexistence: a model of Lotka–Volterra form. Am. Nat. 126, 196–211. ( 10.1086/284409) [DOI] [Google Scholar]
- Holt R. D., Roy M. 2007. Predation can increase the prevalence of infectious disease. Am. Nat. 169, 690–699. ( 10.1086/513188) [DOI] [PubMed] [Google Scholar]
- Holt R. D., Dobson A. P., Begon M., Bowers R. G., Schauber E. M. 2003. Parasite establishment in host communities. Ecol. Lett. 6, 837–842. ( 10.1046/j.1461-0248-2003.00501.x) [DOI] [Google Scholar]
- Hudson P. J., Dobson A. P., Newborn D. 1992. Do parasites make prey vulnerable to predation? Red grouse and parasites. J. Anim. Ecol. 61, 681–692. ( 10.2307/5623) [DOI] [Google Scholar]
- Hudson P. J., Rizzoli A., Grenfell B. T., Heesterbeek H., Dobson A. P. 2002. The ecology of wildlife diseases, 1st edn. Oxford, UK: Oxford University Press. [Google Scholar]
- Irwin M. E., Thresh J. M. 1990. Epidemiology of barley yellow dwarf: a study in ecological complexity. Annu. Rev. Phytopathol. 28, 393–424. ( 10.1146/annurev.py.28.090190.002141) [DOI] [Google Scholar]
- Jenkins D. W. 1964. Pathogens, parasites and predators of medically important arthropods. Annotated list and bibliography. Bull. World Health Organ. 30(suppl.), 1–150. [PMC free article] [PubMed] [Google Scholar]
- Juliano S. A. 2007. Population dynamics. J. Am. Mosq. Control Assoc. 23, 265–275. ( 10.2987/8756-971x(2007)23[265:PD]2.0.cO;2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano S. A. 2009. Species interactions among larval mosquitoes: context dependence across habitat gradients. Annu. Rev. Entomol. 54, 37–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay B., Nam V. S. 2005. New strategy against Aedes aegypti in Vietnam. Lancet 365, 613–617. ( 10.1016/S0140-6736(05)17913-6) [DOI] [PubMed] [Google Scholar]
- Kay B. H., Nam V. S., Tien T. V., Yen N. T., Phong T. V., Diep V. T., Ninh T. U., Bektas A., Aaskov J. G. 2002. Control of aedes vectors of dengue in three provinces of Vietnam by use of Mesocyclops (Copepoda) and community-based methods validated by entomologic, clinical, and serological surveillance. Am. J. Trop. Med. Hyg. 66, 40–48. [DOI] [PubMed] [Google Scholar]
- Keesing F., Holt R. D., Ostfeld R. S. 2006. Effects of species diversity on disease risk. Ecol. Lett. 9, 485–498. ( 10.1111/j.1461-0248.2006.00885.x) [DOI] [PubMed] [Google Scholar]
- Killeen G. F., Tanner M., Mukabana W. R., Kalongolela M. S., Kannady K., Lindsay S. W., Fillinger U., de Castro M. C. 2005. Habitat targeting for controlling aquatic stages of malaria vectors in Africa. Am. J. Trop. Med. Hyg. 73, 546–552. [PubMed] [Google Scholar]
- Kittayapong P., Yoksan S., Chansang U., Chansang C., Bhumiratana A. 2008. Suppression of dengue transmission by application of integrated vector control strategies at sero-positive GIS-based foci. Am. J. Trop. Med. Hyg. 78, p. 70. [PubMed] [Google Scholar]
- Kogan M. 1998. Integrated pest management: historical perspectives and contemporary developments. Annu. Rev. Entomol. 43, 243–270. ( 10.1146/annurev.ento.43.1.243) [DOI] [PubMed] [Google Scholar]
- Kroeger A., Nathan M. B. 2007. Dengue: setting the global research agenda. Lancet 368, 2193–2195. ( 10.1016/s0140-6736(06)69873-5) [DOI] [PubMed] [Google Scholar]
- Kumar R., Hwang J. S. 2006. Larvicidal efficiency of aquatic predators: a perspective for mosquito biocontrol. Zool. Stud. 45, 447–466. [Google Scholar]
- Kumar A., Sharma V. P., Sumodan P. K., Thavaselvam D. 1998. Field trials of biolarvicide Bacillus thuringiensis var. israelensis strain 164 and the larvivorous fish Aplocheilus blocki against Anopheles stephensi for malaria control in Goa, India. J. Am. Mosq. Control Assoc. 14, 457–462. [PubMed] [Google Scholar]
- Kuris A. M., Lafferty K. D. 1994. Community structure: larval trematodes in snail hosts. Annu. Rev. Ecol. Syst. 25, 189–217. ( 10.1146/annurev.es.25.110194.001201) [DOI] [Google Scholar]
- Legner E. 1995. Biological control of diptera of medical and veterinary importance. J. Vector Ecol. 20, 59–120. [Google Scholar]
- LoGiudice K., Ostfeld R. S., Schmidt K. A., Keesing F. 2003. The ecology of infectious disease: effects of host diversity and community composition on Lyme disease risk. Proc. Natl Acad. Sci. USA 100, 567–571. ( 10.1073/pnas.0233733100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald G. 1957. The epidemiology and control of malaria. Oxford, UK: Oxford University Press. [Google Scholar]
- Marten G. G., Bordes E. S., Nguyen M. 1994. Use of cyclopoid copepods for mosquito control. Hydrobiologia 292–293, 491–496. ( 10.1007/BF00229976) [DOI] [Google Scholar]
- Murdoch W. W., Chesson J., Chesson P. L. 1985. Biological control in theory and practice. Am. Nat. 125, 344–366. ( 10.1086/284347) [DOI] [Google Scholar]
- Mutuku F. M., Bayoh M. N., Gimnig J. E., Vulule J. M., Kamau L., Walker E. D., Kabiru E., Hawley W. A. 2006. Pupal habitat productivity of Anopheles gambiae complex mosquitoes in a rural village in western Kenya. Am. J. Trop. Med. Hyg. 74, p. 54. [PubMed] [Google Scholar]
- Nam V. S., Yen N. T., Phong T. V., Ninh T. U. 2005. Elimination of dengue by community programs using Mesocyclops (copepoda) against Aedes aegypti in central Vietnam. Am. J. Trop. Med. Hyg. 72, 67–73. [PubMed] [Google Scholar]
- Nelson X. J., Jackson R. R. 2006. A predator from East Africa that chooses malaria vectors as preferred prey. PLoS ONE 1, e132 ( 10.1371/journal.pone.0000132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen L., Fretwell S. D., Arruda J., Niemela P. 1981. Exploitation ecosystems in gradients of primary productivity. Am. Nat. 118, 240–261. ( 10.1086/283817) [DOI] [Google Scholar]
- Ostfeld R. S., Keesing F. 2000. Biodiversity and disease risk: the case of Lyme disease. Conser. Biol. 14, 722–728. ( 10.1046/j.1523-1739.2009.99014.x) [DOI] [Google Scholar]
- Ostfeld R. S., Holt R. D. 2004. Are predators good for your health? Evaluating evidence for top-down regulation of zoonotic disease reservoirs. Front. Ecol. Environ. 2, 13–20. [Google Scholar]
- Ostfeld R., Price A., Hornbostel V., Benjamin M., Keesing F. 2006. Controlling ticks and tick-borne zoonoses with biological and chemical agents. BioScience 56, 383–394. ( 10.1641/0006-3568(2006)056[0383.CTATZW]2.0.CO;) [DOI] [Google Scholar]
- Pace M. L., Cole J. J., Carpenter S. R., Kitchell J. F. 1999. Trophic cascades revealed in diverse ecosystems. Trends Ecol. Evol. 14, 483–488. ( 10.1016/s0169-5347(99)01723-1) [DOI] [PubMed] [Google Scholar]
- Packer C., Holt R. D., Hudson P. J., Lafferty K. D., Dobson A. P. 2003. Keeping the herds healthy and alert: implications of predator control for infectious disease. Ecol. Lett. 6, 797–802. ( 10.1046/j.1461-0248.2003.00500.x) [DOI] [Google Scholar]
- Paine R. T. 1966. Food web complexity and species diversity. Am. Nat. 100, 65–75. ( 10.1086/282400) [DOI] [Google Scholar]
- Polis G. A. 1999. Why are parts of the world green? Multiple factors control productivity and the distribution of biomass. Oikos 86, 3–15. ( 10.2307/3546565) [DOI] [Google Scholar]
- Price P. W., Bouton C. E., Gross P., McPheron B. A., Thompson J. N., Weis A. E. 1980. Interactions among three trophic levels: influence of plants on interactions between insect herbivores and natural enemies. Annu. Rev. Ecol. Syst. 11, 41–65. ( 10.1146/annurev.cs.11.110180.000353) [DOI] [Google Scholar]
- Pyke G. H. 2008. Plague minnow or mosquito fish? A review of the biology and impacts of introduced gambusia species. Annu. Rev. Ecol. Evol. Syst. 39, 171–191. ( 10.1146/annurev.ecolsys.39.110707.173451) [DOI] [Google Scholar]
- Ross R. 1910. The prevention of malaria. London, UK: John Murray. [Google Scholar]
- Rudolf V. H. W., Antonovics J. 2005. Species coexistence and pathogens with frequency-dependent transmission. Am. Nat. 166, 112–118. ( 10.1086/430674) [DOI] [PubMed] [Google Scholar]
- Samish M., Rehacek J. 1999. Pathogens and predators of ticks and their potential in biological control. Annu. Rev. Entomol. 44, 159–182. ( 10.1146/annurev.ento.44.1.159) [DOI] [PubMed] [Google Scholar]
- Schmidt K. A., Ostfeld R. S. 2001. Biodiversity and the dilution effect in disease ecology. Ecology 82, 609–619. [Google Scholar]
- Schmitz O. J., Hambaeck P. A., Beckerman A. P. 2000. Trophic cascades in terrestrial systems: a review of the effects of carnivore removals on plants. Am. Nat. 155, 141–153. ( 10.1086/303311) [DOI] [PubMed] [Google Scholar]
- Scholte E. J., Ng'habi K., Kihonda J., Takken W., Paaijmans K., Abdulla S., Killeen G. F., Knols B. G. J. 2005. An entomopathogenic fungus for control of adult African malaria mosquitoes. Science 308, 1641–1642. ( 10.1126/science.1108639) [DOI] [PubMed] [Google Scholar]
- Scott T. W., Takken W., Knols B. G. J., Boete C. 2002. The ecology of genetically modified mosquitoes. Science 298, 117–119. ( 10.1126/science.298.5591.117) [DOI] [PubMed] [Google Scholar]
- Seng C. M., Setha T., Nealon J., Socheat D., Chantha N., Nathan M. B. 2008. Community-based use of the larvivorous fish Poecilia reticulata to control the dengue vector Aedes aegypti in domestic water storage containers in rural Cambodia. J. Vector Ecol. 33, 139–144. ( 10.3376/1081-1710(2008)33[139:CUOTLF]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- Shurin J. B., Borer E. T., Seabloom E. W., Anderson K., Blanchette C. A., Broitman B., Cooper S. D., Halpern B. S. 2002. A cross-ecosystem comparison of the strength of trophic cascades. Ecol. Lett. 5, 785–791. ( 10.1046/j.1461-0248.2002.00381.x) [DOI] [Google Scholar]
- Sih A., Crowley P., McPeek M., Petranka J., Strohmeier K. 1985. Predation, competition, and prey communities: a review of field experiments. Annu. Rev. Ecol. Syst. 16, 269–311. ( 10.1146/annurev.es.16.110185.001413) [DOI] [Google Scholar]
- Smyrnioudis I. N., Harrington R., Clark S. J., Katis N. 2001. The effect of natural enemies on the spread of barley yellow dwarf virus (BYDV) by Rhopalosiphum padi (Hemiptera: Aphididae). Bull. Entomol. Res. 91, 301–306. [DOI] [PubMed] [Google Scholar]
- Snyder W. E., Ives A. R. 2001. Generalist predators disrupt biological control by a specialist parasitoid. Ecology 82, 705–716. [Google Scholar]
- Southwood T. R., Murdie G., Yasuno M., Tonn R. J., Reader P. M. 1972. Studies on the life budget of Aedes aegypti in Wat Samphaya, Bangkok, Thailand. Bull. World Health Organ. 46, 211–226. [PMC free article] [PubMed] [Google Scholar]
- Stauffer J. R. J., Arnegard M. E., Cetron M., Sullivan J. J., Chitsulo L. A., Turner G. F., Chiotha S., McKaye K. R. 1997. Controlling vectors and hosts of parasitic diseases using fishes. BioScience 47, 41–49. ( 10.2307/1313005) [DOI] [Google Scholar]
- Stav G., Blaustein L., Margalit Y. 2005. Individual and interactive effects of a predator and controphic species on mosquito populations. Ecol. Appl. 15, 587–598. ( 10.1890/03-5191) [DOI] [Google Scholar]
- Stiling P., Cornelissen T. 2005. What makes a successful biocontrol agent? A metaanalysis of biological control agent performance. Biol. Control 34, 236–246. ( 10.1016/j.biocontrol.2005.02.017) [DOI] [Google Scholar]
- Taylor L. H., Latham S. M., Woolhouse M. E. J. 2001. Risk factors for human disease emergence. Phil. Trans. R. Soc. Lond. B 356, 983–989. ( 10.1098/rstb.2001.0888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrall P. H., Antonovics J., Hall D. W. 1993. Host and pathogen coexistence in sexually transmitted and vector-borne diseases characterized by frequency-dependent disease transmission. Am. Nat. 142, 543–552. ( 10.1086/285554) [DOI] [Google Scholar]
- van den Driessche P., Watmough J. 2002. Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission. Math. Biosci. 180, 29–48. ( 10.1016/s0025-5564(02)00108-6) [DOI] [PubMed] [Google Scholar]
- Walker K., Lynch M. 2007. Contributions of Anopheles larval control to malaria suppression in tropical Africa: review of achievements and potential. Med. Vet. Entomol. 21, 2–21. ( 10.1111/j.1365-2915.2007.00674.x) [DOI] [PubMed] [Google Scholar]
- WHO 1982. Biological control of vectors of disease: sixth report of the WHO expert committee on vector biology and control. WHO Technical Report Series 679, pp. 5–23. [PubMed] [Google Scholar]
- WHO 2004. Global strategic framework for integrated vector management. Geneva, Switzerland: World Health Organization. [Google Scholar]
- Wonham M. J., Lewis M. A., Renclawowicz J., van den Driessche P. 2006. Transmission assumptions generate conflicting predictions in host–vector disease models: a case study in West Nile virus. Ecol. Lett. 9, 706–725. ( 10.1111/j.1461.0248.2006.00912.x) [DOI] [PubMed] [Google Scholar]
- Woolhouse M. E. J., Taylor L. H., Haydon D. T. 2001. Population biology of multihost pathogens. Science 292, 1109–1112. ( 10.1126/science.1059026) [DOI] [PubMed] [Google Scholar]
- Wootton J. T. 1994. The nature and consequences of indirect effects in ecological communities. Annu. Rev. Ecol. Syst. 25, 443–466. ( 10.1146/annurev.es.25.110194.002303) [DOI] [Google Scholar]
- Wu N., Guo-hou L., Duan-fu L., Yu-lin L., Ge-mei Z. 1991. The advantages of mosquito biocontrol by stocking edible fish in rice paddies. Southeast Asian J. Trop. Med. Public Health 22, 436–442. [PubMed] [Google Scholar]
- Zavaleta J. O., Rossignol P. A. 2004. Community-level analysis of risk of vector-borne disease. Trans. R. Soc. Trop. Med. Hyg. 98, 610–618. ( 10.1016/j.trstmh.2003.12.014) [DOI] [PubMed] [Google Scholar]