

Abstract
Cellular immunity is mediated by the interaction of an αβ T cell receptor (TCR) with a peptide presented within the context of a major histocompatibility complex (MHC) molecule. Alloreactive T cells have αβ TCRs that can recognize both self- and foreign peptide–MHC (pMHC) complexes, implying that the TCR has significant complementarity with different pMHC. To characterize the molecular basis for alloreactive TCR recognition of pMHC, we have produced a soluble, recombinant form of an alloreactive αβ T cell receptor in Drosophila melanogaster cells. This recombinant TCR, 2C, is expressed as a correctly paired αβ heterodimer, with the chains covalently connected via a disulfide bond in the C-terminal region. The native conformation of the 2C TCR was probed by surface plasmon resonance (SPR) analysis by using conformation-specific monoclonal antibodies, as well as syngeneic and allogeneic pMHC ligands. The 2C interaction with H-2Kb-dEV8, H-2Kbm3-dEV8, H-2Kb-SIYR, and H-2Ld-p2Ca spans a range of affinities from Kd = 10−4 to 10−6M for the syngeneic (H-2Kb) and allogeneic (H-2Kbm3, H-2Ld) ligands. In general, the syngeneic ligands bind with weaker affinities than the allogeneic ligands, consistent with current threshold models of thymic selection and T cell activation. Crystallization of the 2C TCR required proteolytic trimming of the C-terminal residues of the α and β chains. X-ray quality crystals of complexes of 2C with H-2Kb-dEV8, H-2Kbm3-dEV8 and H-2Kb-SIYR have been grown.
Antigen-presenting cells present intracellularly processed peptides bound to major histocompatibility complex (MHC) class I molecules (pMHC) to αβ T cell receptors (TCRs) on CD8+ cytotoxic T lymphocytes (CTLs) (1). Engagement of the class I-specific αβ TCR and its coreceptors leads to activation of the CTL and lysis of target cells. CTLs are selected during thymic development through a process of negative and positive selection (2). A lower threshold of affinity between TCR and self-pMHC appears to lead to positive selection, whereas a higher threshold results in negative selection by cell death (3). Peripheral CD8+ CTL responses result when foreign peptides originating, for example, from a viral or tumor antigen are presented by self-MHC, but usually there is tolerance to self-peptide MHC complexes within the same species. CTL responses to self-peptides can occur, however, during autoimmune reactions. A large number of peripheral T cells can also recognize foreign pMHC complexes in a phenomenon known as alloreactivity (4). Because CTLs cannot encounter allo-MHC during maturation, alloreactivity must then result from cross-reactivity of the TCR with both self- and foreign pMHC (5, 6). One possibility is that cross-reactivity results from shared structural features between the self- and allo-MHC. However, alloreactive TCRs also recognize conformationally distinct MHC (7), amino acid mutations in the α-helices comprising the MHC groove (8), and different bound peptides (9). An important question, then, is whether cross-reactivity is a result of highly efficient molecular mimicry by the pMHC alloantigen or whether αβ TCRs are able to accommodate a wider range of ligands than one would expect in comparison with the restricted fidelity of high-affinity antibodies.
We have focused our efforts on the 2C TCR system and its ligands to characterize biochemically and structurally the molecular basis of TCR alloreactivity. The TCR 2C is the clonotypic receptor of a CTL clone educated on H-2Kb molecules, but with reactivity toward the class I MHC alloantigen H-2Ld complexed with the self-peptide p2Ca (or QL9), derived from a mitochondrial protein (10–13). Recently, self- and artificial peptide ligands for H-2Kb and H-2Kbm3 have been identified that can be recognized specifically as pMHC complexes by 2C TCR (14, 15). Thus, for 2C, distinct, defined pMHC complexes are now available that make it possible to address questions about the structural basis of alloreactivity (16). Our approach has been to engineer and express recombinant 2C TCR, to characterize the affinities of 2C for its various recombinant self- and foreign ligands, and to determine not only the individual crystal structures of the αβ TCR and the various reactive pMHC complexes, but complexes of the TCR with its pMHC ligands.
Here, we report high-level expression of a disulfide-linked 2C TCR αβ heterodimer glycoprotein from Drosophila melanogaster cells and the characterization of its interactions with self- and foreign ligands by surface plasmon resonance (SPR) and crystallization. This recombinant material is biologically active and displays similar relative affinities to those reported previously for its MHC ligand H-2Ld bound to the self-peptide p2Ca (LSPFPFDL) peptide (17). SPR affinity measurements of 2C have been made with recently discovered peptides, the self dEV-8 (EQYKFYSV) (14) and the synthetic SIYR (SIYRYYGL) (15), in association with H-2Kb and H-2Kbm3. The affinity of 2C for the self-MHC H-2Kb in complex with dEV8 and SIYR is almost an order of magnitude weaker than for H-2Ld bound to p2Ca. An ultrapurification scheme for 2C has been combined with proteolytic digestion of the free C termini of the α and β chains to achieve growth of high-quality, three-dimensional TCR crystals that diffract to at least 2.5Å. Crystallization of 2C in complex with the lower-affinity pMHC ligands H-2Kb-dEV8, H-2Kbm3-dEV8, and H-2Kb-SIYR also has been achieved to provide valuable insights into the structural basis of TCR/MHC recognition and alloreactivity.
MATERIALS AND METHODS
Construction of the 2C TCR Expression Vector.
The cDNAs were modified by PCR by using overhang primers retaining modified sequences and cloning sites. The α and β chain cDNA expression vectors were individually constructed and then cotransfected into Drosophila melanogaster cells. Soluble, secreted forms of both α and β chains were constructed by fusing the short intracellular coding sequences directly to the extracellular coding sequences at the point where the putative transmembrane domains begin, thereby splicing out the transmembrane sequence (Fig. 1A). For the 2C TCR α chain (Vα3-Jα58Cα), the cDNA was interrupted after codon 221 (Asn) and extended by the sequence corresponding to residues 244–248 of its cytoplasmic tail (RLWSS). The 2C TCR β chain (Vβ8.2-Dβ2Jβ2.4Cβ2) was interrupted at codon 246 (Gln) and extended by the sequence corresponding to residues 278–283 of the β chain cytoplasmic tail (VKKKNS). Hexa-histidine sequences were added to the C termini of both chains. The modified cDNAs were subcloned between the EcoRI and SalI sites of the polylinker of pRMHa3, a metallothionein promoter-driven vector. The α and β chain constructs were cotransfected into SC2 fly cells by calcium phosphate precipitation together with pUChsneo, a neomycin-resistance gene. Stable cell lines were derived by G418 selection and kept under selection in Schneider’s medium (GIBCO/BRL) containing 500 μg of G418 per ml.
Figure 1.
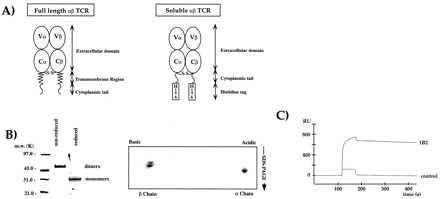
Expression of the 2C TCR as a covalently linked αβ heterodimer from Drosophila melanogaster cells. (A) Schematic diagram of the 2C expression construct design (see Materials and Methods). (B) The expressed 2C is a covalent heterodimer. On the left, SDS/PAGE analysis of Ni2+-agarose purified 2C. On the right, 2D nonequilibrium, pH-gradient electrophoresis. Both gels are Coomassie-stained. (C) The recombinant TCR 2C binds with high affinity to the Fab of the anti-idiotypic mAb 1B2 (21) using SPR on a BIAcore 2000 machine.
The construction of the class I MHC H-2Kb and H-2Ld expression vectors for Drosophila melanogaster cells have been reported previously (18, 19). The H-2Kbm3 heavy chain was constructed by site-directed mutagenesis of H-2Kb (D77S and K89A) and expressed as described for H-2Kb.
Protein Expression and Purification.
Stable transfectants were expanded in serum-free medium (Insect Xpress, BioWhittaker). Protein was induced over a period of 72 hr by the addition of copper sulfate to a final concentration of 0.7 mM. Supernatants were collected, concentrated, dialyzed by ultrafiltration (Filtron Technology, Northborough, MA), and filtered. The recombinant, histidine-tagged molecules were purified by Ni-agarose (Ni2+-NTA-agarose; Qiagen) affinity chromatography. The Ni-agarose column was washed with 20 mM Imidazole/PBS, and then the protein was step-eluted with 200 mM Imidazole/PBS. The fractions containing TCR were pooled and concentrated by centrifugation (Ultra-free 15; Millipore) to 5 ml. The pooled TCR fractions were then loaded onto a 26/100 cm Sephacryl S-100 gel filtration column (Pharmacia) run in PBS at a flow rate of 0.4 ml/min and monitored at OD280 at 4°C. Heterodimer-containing fractions from the gel filtration column were analyzed by nonreducing 10–20% gradient SDS/PAGE (Bio-Rad). Two-dimensional (2D) gel electrophoresis was performed as described (20) with 3.5–10.0 nonequilibrium, pH-gradient electrophoresis in the first dimension and a 12% SDS/PAGE in the second dimension.
The size-purified, recombinant αβ TCR was digested at this point with a mixture of carboxypeptidases A and B (Boehringer Mannheim) at an enzyme:substrate ratio of 1:100 (wt/wt). The entire reaction mixture was then brought to a concentration of 1.2 M ammonium sulfate (AS), 50 mM sodium phosphate, pH 7.4, by addition of solid AS. The TCR was then loaded onto a HIC column (Phenyl Superose 10/10; Pharmacia) at 1.2 M AS, and an elution gradient was developed from 1.2 M AS to 0.0 M AS over a volume of 120 ml. The peak fractions that contained the fully digested αβ TCR were pooled and concentrated to approximately 50 mg/ml in 10 mM Na⋅Hepes, 25 mM NaCl, pH 7.5, by Centricon-30 (Amicon).
The native conformation of the purified TCR was verified by sequential antibody binding by using SPR with anti-idiotype 1B2 (21), anti-Vβ F23.1 (22), anti-Cα H28–710 (23), and anti-Cβ H57–597 (PharMingen) antibodies. Antibodies were injected at 2.0, 1.0, 0.5, 0.25, 0.125, and 0.0625 μM using a flow of 20 μl/min.
The expression, purification, and crystallization of murine MHC H-2Kb molecules from Drosophila melanogaster cells has been reported previously (24–26). The specific MHC–peptide complexes were produced by incubation of the Drosophila-expressed MHC molecules with a slight molar excess of peptide, followed by mono-Q (Pharmacia) chromatography. Peptide occupancy of the pMHC complexes was verified prior to all experiments by native gel analysis and isoelectric focusing.
SPR.
All SPR experiments were done on a BIAcore 2000 machine (Biosensor, Piscataway, NJ) at 25°C. Purified molecules were immobilized on a CM5 sensor chip at pH 5.5 in 10 mM sodium acetate buffer by classic amine coupling. Blank surfaces were made by ethanolamine deactivation of the activated surface. Approximately 250–2,000 resonance units (RU) of purified 2C TCR were immobilized to the chip. For the H-2Ld measurements, Ld–peptide complexes were injected at 20, 10, 5, and 2.5 μM at a flow rate of 20 μl/min. For H-2Kb and H-2Kbm3, the pMHC complexes were injected at concentrations of 40, 20, 10, and 5 μM at the same flow rate. Data were analyzed with the biaevaluation 2.1 software program. Model curve fitting was chosen from a plot of ln (Ro/R) vs. time of the raw data. Scatchard plots were derived from the RU/concentration (C) vs. RU plot and linear regression analysis (cricket graph software) of specific binding.
Crystallization of the 2C TCR.
Crystallization trials were carried out using the sitting-drop methodology. A limited 18-condition screen (K.C.G., unpublished data) of the carboxypeptidase-digested 2C (15 mg/ml) yielded intergrown bundles of crystals in 1.8 M AS, 0.1 M sodium citrate, pH 6.0, at 22°C. These bundles could be broken up into single microcrystals and seeded into a mother liquor of 1.8 M AS, 0.1 M sodium acetate, 50 mM sodium citrate, pH 6.5, containing 15 mg/ml 2C. A single crystal of approximate dimensions of 0.2 × 0.2 × 0.1 mm3 was successfully cryo-cooled at −170°C for low-temperature data collection by the addition of glycerol at a rate of 2% every 20 min up to a final concentration of 24%.
Crystallization of the TCR/MHC Complexes.
The purified, carboxypeptidase-treated 2C was mixed in a 1:1 molar ratio (≈10 mg/ml) with either the H-2Kb or H-2Kbm3 ligands complexed to the dEV8 or SIYR peptides (plus addition of 1.5-fold molar excess to account for any peptide dissociation). A limited 18-condition screen yielded crystals in 12% PEG-6000, 0.2 M Tris⋅acetate, 0.5 M NaCl, pH 7.2. These small crystals could be repeatedly macroseeded into 12% PEG-4000, 0.2 M Tris⋅acetate, 0.1 M NaCl, pH 7.2, to dimensions of 0.7 × 0.2 × 0.1 mm for data collection. The addition of small amounts (50 μl) of 5 M NaCl to the well solution served to raise the PEG concentration in the protein-containing drop and increase the diffraction limit of the crystals. Crystals of 2C/H-2Kb-dEV8, 2C/H-2Kbm3-dEV8, and 2C/H-2Kb-SIYR complexes grew under identical conditions and can be cryo-cooled by addition of ethylene glycol a rate of 4% every 15 min to a final concentration of 22%.
X-Ray Data Collection.
For the 2C TCR, x-ray data were collected on an MAR Imaging Plate mounted on a Siemens rotating anode operating at 40 kV and 50 mA with Supper double-focusing long mirrors (Siemens, Iselin, NJ). Cryo-cooling of crystals (−170°C) was carried out with a cryo-nozzle from Oxford Cryosystems. Data were indexed, integrated, scaled, and reduced with denzo and scalepack (27, 28). Crystals of 2C/H-2Kbm3-dEV8 were cryo-cooled to −170°C, and data were collected with a 30-cm MAR Imaging Plate. For the 2C/H-2Kb-SIYR and 2C/H-2Kb-dEV8, data were collected on cryo-cooled crystals at beamline 7–1 of the Stanford Synchrotron Radiation Laboratory (SSRL) with a 30-cm MAR Imaging Plate.
Peptides.
The H-2Kb and H-2Ld peptide ligands (SIYR, dEV8, VSV, and MCMV) were synthesized by t-BOC chemistry on an Applied Biosystems instrument and purified to >95% purity on a C18 reverse-phase column (Rainin Instruments). The MCMV sequence is YPHFMPTNL (29).
RESULTS AND DISCUSSION
Expression of the 2C TCR.
The design of the initial expression constructs for the α and β chains were based on the model that we used for the class II MHC expression (20), in which a six-residue histidine tag (His-tag) was added to the truncated C termini (Fig. 1A). After cotransfection of SC2 Drosophila melanogaster cells with α and β chain vector constructs, expression of TCR is induced by addition of copper sulfate to the culture media. Expression of TCR by using these constructs resulted in low yields of 2C (<100 μg/liter), and so a modified expression construct was designed to try to improve the yields. The modified constructs had the short intracellular segments of both chains added onto the extracellular domains (Fig. 1A). The His-tags were again added onto the new TCR C termini. Transfection of SC2 cells with these constructs resulted in dramatically improved expression levels (>2–5 mg/liter) and enhanced proteolytic stability of the TCR.
Purification of the 2C TCR.
The initial purification of the TCR from the insect cell culture media by Ni-agarose chromatography via the His-tags on both chains produced 85% pure TCR. Nonreducing SDS/PAGE indicated an approximate 58-kDa band that, upon reduction, migrated at approximately 28 kDa, indicating that the interchain disulfide bond had been formed (Fig. 1B). The molecular mass of the heterodimer was determined by mass spectrometry to be 58,829 Da. Heterodimer formation was confirmed by N-terminal protein sequencing, which further showed that the predicted N-terminal signal sequence cleavage sites had been used in each chain. To verify further the presence of both α and β chains in the dimer, 2D gel analysis revealed equal quantities of both acidic and basic components that represented the α and β chains, respectively (Fig. 1B). The correct native conformation of 2C was verified with the conformation-specific anti-idiotypic monoclonal antibody (mAb), 1B2 (21) by using SPR (Fig. 1C). Anti-Cα H28–710 (23) and anti-Cβ H57–597 (PharMingen) mAbs also exhibited high-affinity binding to 2C (Table 1).
Table 1.
Affinities of anti-TCR antibodies for the 2C TCR as measured by surface plasmon resonance
| Antibody name and specificity | Kass, (M−1⋅s−1 × 104) | Kdiss, s−1 × 10−4 | Kd, nM |
|---|---|---|---|
| 380, polyclonal anti-TCRαβ | 9.7 ± 3.0 | 6.1 ± 0.5 | 6.3 ± 1.7 |
| H57-597, anti-Cβ | 6.5 ± 1.7 | 1.5 ± 0.3 | 2.4 ± 1.1 |
| H28-710, anti-Cα | 5.2 ± 2.0 | 1.6 ± 0.7 | 3.1 ± 1.7 |
| 1B2, anti-idiotype | 6.9 ± 1.7 | 2.0 ± 0.4 | 2.9 ± 0.2 |
The high-affinity binding of the clonotypic 1B2 antibody (Kd = 3.0 × 10−9 M) clearly demonstrates no inherent structural or physicochemical limit on the affinity with which TCR combining sites can bind a ligand (30).
After the initial Ni-agarose purification, many different purification methods were attempted to obtain large, single crystals (Fig. 2). Although all of the purification methods resulted in functional 2C, as judged by BIAcore analysis, only one protocol resulted in x-ray diffraction quality crystals of 2C. Small, poorly diffracting crystals were obtained from a number of other protocols (Fig. 2). Thus, we focused on the elimination of isoform heterogeneity, as revealed by isoelectric focusing. Because the TCR 2C contains seven consensus N-linked glycosylation sites, we attempted both enzymatic (PNGase F) and chemical (tunicamycin) deglycosylation (data not shown). Enzymatic deglycosylation did not significantly decrease the molecular mass or heterogeneity of 2C. Addition of tunicamycin (5 μg/ml) to the fly cell growth media before induction did inhibit addition of N-linked glycosylation, as judged by an approximate 10-kDa molecular mass decrease in the apparent 2C MW by SDS/PAGE and mass spectrometry (data not shown). The nonglycosylated 2C, however, did not produce any crystals after a variety of purification methods.
Figure 2.
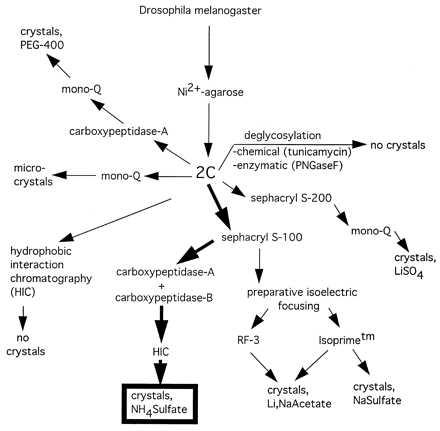
Purification and crystallization scheme for 2C TCR. Crystallization of 2C was achieved after many different purification protocols and covalent modifications were attempted. The final purification and carboxypeptidase digestion schemes that lead to high-resolution crystals are outlined in bold arrows.
The use of preparative isoelectric focusing allowed us to isolate single-charge isoforms of 2C, which again yielded small, unusable crystals (data not shown). Success was finally achieved by an initial gel filtration step to remove large molecular mass aggregates and monomeric α and β chains (Fig. 3A). At this point, carboxypeptidases A and B were added at a substrate:enyme ratio of 100:1 (wt/wt) to see whether the C-terminal tails could be trimmed back. Treatment with the two different carboxypeptidases resulted in an approximate overall 5-kDa molecular mass decrease and a shift in the pI values of both chains to a more acidic range (Fig. 3B). The use of carboxypeptidase A or B alone was insufficient; complete trimming of the C terminus required the simultaneous presence of both enzymes. Following carboxypeptidase digestion, HIC allowed separation of 2C with fully digested and incompletely digested tails (Fig. 3B).
Figure 3.
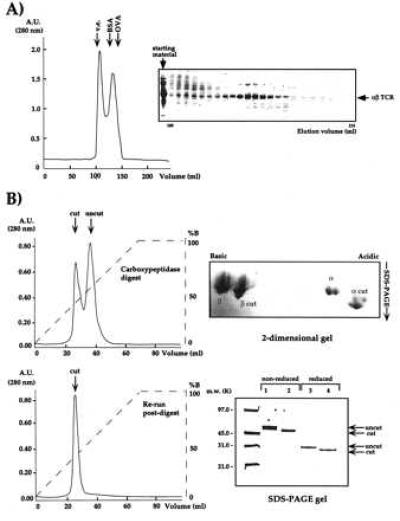
Analysis of the purification and preparation steps of 2C for crystallization. (A) Ni2+-agarose-purified 2C was purified by gel filtration (Sephacryl-S-100) to remove aggregates and monomers. (B) Gel filtration-purified 2C was treated with carboxypeptidases A and B and purified by HIC. The progress of the reaction is monitored by HIC. Partially (Upper Left) and fully digested (Lower Left) 2C clearly separate by HIC and 2D nonequilibrium, pH-gradient electrophoresis (Upper Right). The fully digested 2C is purified by HIC and migrates on reducing and nonreducing SDS/PAGE at a position approximately 5 kDa smaller than the digested band (Lower Right).
The fully digested material yielded large, diffraction-quality crystals (Fig. 4A). To verify the presence of covalent αβ heterodimer in the crystals, a single crystal of 2C was analyzed by nonreducing SDS/PAGE (Fig. 4B) and 2D gels (not shown). The crystal was composed entirely of covalent αβ heterodimer. Isoelectric focusing analysis of the dissolved crystal indicated that the material in the crystal was composed of the predominant charge isoforms of the 2C starting material (Fig. 4C). A complete x-ray data set was collected to 2.5Å from these crystals (31). Inspection of the crystal lattice packing of 2C indicates that the C termini of both chains are in contact with symmetry-related molecules (Fig. 4D) and probably explains the beneficial effect of trimming away the C-terminal tails for crystal growth.
Figure 4.
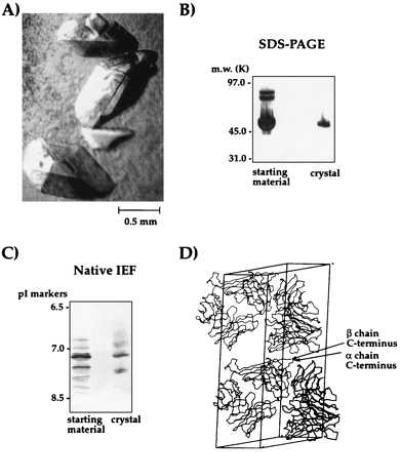
Analysis of crystals of the 2C TCR. (A) Large crystals of 2C (B) SDS/PAGE analysis (silver-stained) of a washed 2C crystal reveals crystal is composed entirely of αβ covalent heterodimer. (C) IEF of a washed 2C crystal indicated that the predominant charge isoforms from the starting material are present in the crystallized protein. (D) Inspection of the crystal lattice packing of the 2C TCR crystals indicates that the α and β chain C termini of 2C are in contact with symmetry-related molecules.
Characterization of 2C Interaction with Its pMHC Ligands.
We analyzed 2C after each purification step to assess whether the purification methods affect the binding properties of the TCR. The kinetics and affinities of 2C binding to its pMHC ligands appears to be unchanged by deglycosylation, carboxypeptidase digestion, or purification method (Table 2).
Table 2.
Affinities of the 2C TCR for its pMHC ligands
| pMHC ligand | 2C TCR | Kass, (M−1⋅s−1 × 103) | Kdiss, s−1 × 10−2 | Kd, μM |
|---|---|---|---|---|
| Ld-p2Ca | C-AB treated | 8.30 ± 1.45 | 2.7 ± 0.4 | 3.3 ± 0.4 |
| Ld-p2Ca | Deglycosylated | 9.40 ± 2.10 | 1.5 ± 0.2 | 1.6 ± 0.1 |
| Ld-QL9 | C-AB treated | 6.35 ± 0.60 | 2.5 ± 0.4 | 3.9 ± 0.2 |
| Ld-QL9 | Deglycosylated | 6.12 ± 0.65 | 1.2 ± 0.1 | 2.0 ± 0.2 |
| Ld-MCMV | C-AB treated | NM | NM | NM |
| Ld-MCMV | Deglycosylated | NM | NM | NM |
| Kb-dEV8 | C-AB treated | 2.20 ± 0.45 | 18.5 ± 2.0 | 84.1 ± 12.0 |
| Kb-SIYR | C-AB treated | 2.35 ± 0.85 | 7.5 ± 2.1 | 31.9 ± 6.2 |
| Kb-VSV | C-AB treated | NM | NM | NM |
| Kbm3-dEV8 | C-AB treated | 0.85 ± 0.15 | 4.8 ± 0.4 | 56.5 ± 8.1 |
| Kbm3-SIYR | C-AB treated | 2.65 ± 0.15 | 7.8 ± 0.9 | 29.4 ± 3.2 |
| Kbm3-VSV | C-AB treated | NM | NM | NM |
For the 2C TCR, C-AB indicates that the 2C has been carboxypeptidase A and B treated to trim the C-terminal tails, and deglycosylated indicates that the 2C TCR was expressed in the presence of tunicamycin and therefore was free of N-linked glycosylation (see Methods). Measurements were made by surface plasmon resonance on a BIAcore 2000 machine as detailed in Materials and Methods. NM, not measurable.
The availability of correctly folded recombinant 2C enabled us to measure the binding affinities and kinetic rate constants for the interaction of 2C with a variety of its known pMHC ligands. The 2C CTL was raised by injecting H-2b mice with cells of the H-2d haplotype (10). The negatively selecting element was determined to be H-2Ld, and 2C was positively selected on H-2Kb (11). Later, it was determined that 2C was also reactive to a mutant of Kb that has two amino acid changes in the α1 helix (positions 77 and 89) termed Kbm3 (12, 32). The alloreactivity of H-2Ld is associated with a single octameric peptide, p2Ca (or the nonameric form, QL9), derived from the mitochondrial protein α-ketoglutarate dehydrogenase (13, 36). Recently, attempts have been made to find a positively selecting peptide bound to H-2Kb. Elution experiments have identified a peptide, dEV8 (EQYKFYSV), which renders H-2Kb and H-2Kbm3 reactive with 2C and is also derived from a mitochondrial enzyme (14). Whether the dEV8 peptide is one of the positively selecting peptide elements is not yet known. Another peptide, SIYR (SIYRYYGL), which renders 2C reactive with H-2Kb, has been discovered by using a synthetic peptide library and is rather similar in aspects of its sequence to dEV8 (15). The bioactivity of the SIYR peptide in the context of Kb is extremely potent (∼pM) compared with p2Ca in the context of Ld (∼nM), as measured by concentration required for half-maximal lysis in a CTL assay (15) (direct comparisons have not been made for dEV8). These results lead to the question of whether the bioactivity difference is paralleled by corresponding affinity differences of the two pMHC complexes for the 2C TCR. We now can compare this binding with allogeneic ligands (Ld-p2Ca, Ld-QL9, Kbm3-dEV8, Kbm3-SIYR) to address the question of how TCR/pMHC affinity relates to cross-reactivity.
Identical affinity (Kd = Kdiss/Kass) values were obtained for both the carboxypeptidase- and noncarboxypeptidase-treated 2C, and so the values reported here (Table 2) are for the carboxypeptidase-treated form because this is the form that crystallized (31). The affinities of 2C for H-2Ld-p2Ca and H-2Ld-QL9 are in the range of Kd = 2–3 μM, which are in the same range but differ slightly from previously reported values (19, 29, 33). The discrepancy in the measured affinities probably reflects the fact that, in one case, the measurements were carried out on live CD8+ cells (33), which has been shown to enhance the TCR/pMHC interaction, and in another, the recombinant H-2Ld peptide complexes were prepared differently (29). The negative control of H-2Ld bound to the MCMV peptide (29) showed no binding to 2C. The affinities of the H-2Kb-peptide ligands are, in general, an order of magnitude weaker than the H-2Ld-peptide ligands. H-2Kb-dEV8 has an affinity for 2C of Kd = 85 μM, and the H-2Kbm3-dEV8 has a slightly higher affinity of 56 μM. H-2Kb-SIYR and H-2Kbm3-SIYR both show affinities of approximately 30 μM, which are higher (i.e., tighter binding) by factors of approximately 2 and 3, respectively, than the Kb and Kbm3–dEV8 complexes. The difference in the affinity of 2C for H-2Kb-dEV8 and -SIYR is mainly due to a slower off-rate for the SIYR complex. It is interesting that the affinity of 2C for Kb-SIYR is lower than that for Ld-p2Ca, in spite of the fact that the bioactivity of Kb-SIYR is ∼pM, compared with ∼nM for Ld-p2Ca. The explanation of this discrepancy between bioactivity and affinity is clearly dependent on the mechanism of signal transduction by the TCR signaling complex and whether any aggregation and/or conformational changes are specific to the activating ligands.
In the 2C system, the H-2Ld-p2Ca, H-2Kbm3-dEV8, and H-2Kbm3-SIYR can be considered as allogeneic ligands. Cells presenting these peptide complexes are lysed by the 2C CTL. H-2Kb-dEV8 can be considered as a syngeneic ligand, and H-2Kb-SIYR is a self-MHC presenting an artificial peptide. Nineteen residues differ in the heavy chain between Kb and Ld, and the peptides normally presented by each MHC module are very different. However, Kb and Kbm3 have only two amino acid differences (32, 36) in the heavy chain at positions 77 (D77S) and 89 (K89A), only one of which (position 77) points into the binding site from the long α1 helix (26). The Ld–peptide pMHC complexes have significantly higher affinity for 2C than both the Kb and Kbm3–peptide complexes. Within the Kb system, the allogeneic Kbm3-dEV8 has a slightly higher affinity for 2C than the syngeneic Kb-dEV8. However, the Kb- and Kbm3-SIYR ligands have identical affinities. So, the conclusion from our measurements is that for Ld and Kb, the difference in affinity between this particular set of syngeneic and allogeneic ligands is quite dramatic. However, within the Kb system, the affinity differences are small.
These results can possibly, although imprecisely, be reconciled with threshold models of positive and negative selection. The Ld–peptide pMHC ligands are quite divergent in sequence, and presumably structure, from the positively selecting Kb element and also the negatively selecting H-2Kbm3. H-2Ld undergoes negative selection (complete clonal deletion) in the thymus in the absence of CD8 (11) and thus may be more strongly reactive with the 2C TCR than H-2Kb, and also Kbm3, which requires the assistance of CD8 for negative selection (12). The extensive structural differences between Ld and Kb may result in 2C using a higher-affinity epitope on Ld that is not present on Kb, thus minimizing the potential of cross-reactivity with the positively selecting Kb. The higher affinity, then, may result in more efficient negative selection (35). The structural differences between Kb and Kbm3 are presumably so subtle that it would be difficult to achieve high affinity for Kbm3 and not Kb. When it was originally observed that mice bearing Kbm3 negatively select 2C-bearing T cells in a transgenic mouse (12), there was not a complete clonal deletion and the level of CD8 coreceptor expression was critical. When CD8 was overexpressed, both Kb and Kbm3 were negatively selected, but thymocytes expressing low levels of CD8 were spared clonal deletion (12). These results seem to point to a situation in which small structural differences between Kb and Kbm3 cannot be distinguished in the thymus based purely on affinity differences for 2C but require the assistance of CD8 to make the decision of negative vs. positive selection. An interesting question is whether the affinity of a TCR for its allo-ligand can be correlated with how far the ligand deviates from the positively selecting haplotype in structure. A previous study observed that the affinity of 2C for H-2Ld-p2Ca was significantly higher than the affinity of an unrelated Kb-OVA restricted TCR for its syngeneic Kb-OVA ligand (33). However, this comparison was between two different TCRs. Here, we observe that for one αβ TCR, 2C, the higher-affinity ligand is the allogeneic pMHC of a different haplotype.
Crystallization of 2C with its pMHC Ligands.
An integral part of our characterization of the 2C TCR system was the structure determination of 2C in complex with its pMHC ligands. Therefore, crystallization trials were undertaken with all of the ligands that were used for SPR analysis. The affinity of 2C for H-2Kb-peptide complexes was too weak to purify preformed complexes by gel filtration, so these components (2C and H-2Kb-peptide) were mixed in stoichiometric amounts in the crystallization drops. Crystallization of noncarboxypeptidase-treated 2C in complex with the Kb-peptide ligands resulted in large crystals that did not diffract x-rays. However, success was met with carboxypeptidase-treated 2C cocrystallized with H-2Kb-dEV8, Kbm3-dEV8, and K-2Kb-SIYR ligands. Bundles of microcrystals were initially obtained for carboxypeptidase-treated 2C in complex with H-2Kb-dEV8 from PEG-4000. Macroseeding of isolated microcrystals produced single crystals of each of the three 2C/H-2Kb-peptide complexes suitable for data collection. The most efficient protocol for cocrystallization was simply a 1:1 stoichiometric mixture of TCR:pMHC in the presence of a 1.5-fold molar excess of peptide to account for any peptide dissociation (Fig. 5A). We confirmed the presence of the TCR/pMHC complex in the crystals by harvesting a single, large crystal and analyzing the contents by SDS/PAGE (Fig. 5B). This gel analysis revealed the presence of an approximate 1:1:1 ratio of αβ TCR, H-2Kb heavy chain, and β2m.
Figure 5.

Crystals of the 2C/H-2Kb-dEV8 complex. (A) A large, single crystal of 2C/H-2Kb-dEV8. (B) SDS/PAGE analysis of a washed crystal of 2C/H-2Kb-dEV8 reveals the presence of αβ TCR, Kb heavy chain, and β2m in stoichiometric amounts.
Complete x-ray data sets have been collected to date on the complex crystals 2C/H-2Kb-dEV8 (84% complete to 3.0 Å), 2C/H-2Kb-SIYR (91% complete to 2.8 Å), and 2C/H-2Kbm3-dEV8 (88% complete to 3.2 Å). All of the different complex crystals are isomorphous, with space group P21212, and unit cell dimensions a = 296.7 Å, b = 89.3 Å, and c = 84.5 Å with two TCR/pMHC complexes in the asymmetric unit. The two independent TCR/MHC complexes do not form a dimer. The molecular replacement solution for one of these complexes has already been reported (31).
Acknowledgments
L.T. gratefully acknowledges support from R. W. Johnson Pharmaceutical Research Institute. We thank Randy Stefanko for technical assistance. This work was supported in part by National Institutes of Health Grant CA58896 (I.A.W.) and Training Grant T32-A107244 (K.C.G). This is The Scripps Research Institute manuscript 10964-MB.
ABBREVIATIONS
- MHC
major histocompatibility complex
- pMHC
peptide-MHC
- TCR
T cell receptor
- Kass
association rate
- Kdiss
dissociation rate
- Kd
dissociation constant
- RU
resonance units
- SPR
surface plasmon resonance
- CTL
cytotoxic T lymphocyte
- 2D
two dimensional
- HIC
hydrophobic-interaction chromatography
References
- 1.Zinkernagel R M, Doherty P C. Nature (London) 1974;248:701–702. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 2.Jameson S C, Hogquist K A, Bevan M J. Annu Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- 3.Alam S M, Travers P J, Wung J L, Nasholds W, Redpath S, Jameson S C, Gascoigne N R. Nature (London) 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 4.Heath W R, Hurd M E, Carbone F R, Sherman L A. Nature (London) 1989;341:749–752. doi: 10.1038/341749a0. [DOI] [PubMed] [Google Scholar]
- 5.Sherman L A, Chattopadhyay S. Annu Rev Immunol. 1993;11:385–402. doi: 10.1146/annurev.iy.11.040193.002125. [DOI] [PubMed] [Google Scholar]
- 6.Eisen H N, Sykulev Y, Tsomides T J. Adv Prot Chem. 1996;49:1–56. doi: 10.1016/s0065-3233(08)60487-8. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay S, Theobald M, Biggs J, Sherman L A. J Exp Med. 1994;179:213–219. doi: 10.1084/jem.179.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pullen J K, Tallquist M D, Melvold R W, Pease L R. J Immunol. 1994;152:3445–3452. [PubMed] [Google Scholar]
- 9.Kuzushima K, Sun R, van Bleek G M, Vegh Z, Nathenson S G. J Immunol. 1995;155:594–601. [PubMed] [Google Scholar]
- 10.Kranz D M, Sherman D H, Sitkovsky M V, Pasternack M S, Eisen H N. Proc Natl Acad Sci USA. 1984;81:573–577. doi: 10.1073/pnas.81.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sha W C, Nelson C A, Newberry R D, Kranz D M, Russell J H, Loh D Y. Nature (London) 1988;336:73–76. doi: 10.1038/336073a0. [DOI] [PubMed] [Google Scholar]
- 12.Sha W C, Nelson C A, Newberry R D, Pullen J K, Pease L R, Russell J H, Loh D Y. Proc Natl Acad Sci USA. 1990;87:6186–6190. doi: 10.1073/pnas.87.16.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Udaka K, Tsomides T J, Eisen H N. Cell. 1992;69:989–998. doi: 10.1016/0092-8674(92)90617-l. [DOI] [PubMed] [Google Scholar]
- 14.Tallquist M D, Pease L R. J Immunol. 1995;155:2419–2426. [PubMed] [Google Scholar]
- 15.Udaka K, Wiesmüller K-H, Kienle S, Jung G, Walden P. J Immunol. 1996;157:670–678. [PubMed] [Google Scholar]
- 16.Tallquist M D, Yun T, Pease L R. J Exp Med. 1996;184:1017–1026. doi: 10.1084/jem.184.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sykulev Y, Brunmark A, Jackson M, Cohen R J, Peterson P A, Eisen H N. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 18.Jackson M R, Song E S, Yang Y, Peterson P A. Proc Natl Acad Sci USA. 1992;89:12117–12121. doi: 10.1073/pnas.89.24.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia K C, Scott C A, Brunmark A, Carbone F, Peterson P, Wilson I A, Teyton L. Nature (London) 1996;384:577–581. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- 20.Scott C A, Garcia K C, Carbone F R, Wilson I A, Teyton L. J Exp Med. 1996;183:2087–2095. doi: 10.1084/jem.183.5.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kranz D M, Tonegawa S, Eisen H N. Proc Natl Acad Sci USA. 1984;81:7922–7926. doi: 10.1073/pnas.81.24.7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staerz U D, Rammensee H G, Benedetto J D, Bevan M J. J Immunol. 1985;134:3994–4000. [PubMed] [Google Scholar]
- 23.Becker M L, Near R, Mudgett-Hunter M, Margolies M N, Kubo R T, Kaye J, Hedrick S M. Cell. 1989;58:911–921. doi: 10.1016/0092-8674(89)90943-4. [DOI] [PubMed] [Google Scholar]
- 24.Matsumura M, Saito Y, Jackson M R, Song E S, Peterson P A. J Biol Chem. 1992;267:23589–23595. [PubMed] [Google Scholar]
- 25.Stura E A, Matsumura M, Fremont D H, Saito Y, Peterson P A, Wilson I A. J Mol Biol. 1992;228:975–982. doi: 10.1016/0022-2836(92)90881-j. [DOI] [PubMed] [Google Scholar]
- 26.Fremont D H, Matsumura M, Stura E A, Peterson P A, Wilson I A. Science. 1992;257:919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend. Sawyer L, Isaacs N, Bailey S, editors. Daresbury, England: Science and Engineering Research Council Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- 28.Minor W. xdisplayf Program. West Lafayette, IN: Purdue University; 1993. [Google Scholar]
- 29.Corr M, Slanetz A, Boyd L, Jelonek M, Khilko S, Al-Ramidi B, Kim Y, Maher S, Bothwell A, Margulies D. Science. 1994;265:946–948. doi: 10.1126/science.8052850. [DOI] [PubMed] [Google Scholar]
- 30.Schlueter C J, Schodin B A, Tetin S Y, Kranz D M. J Mol Biol. 1996;256:859–869. doi: 10.1006/jmbi.1996.0132. [DOI] [PubMed] [Google Scholar]
- 31.Garcia K C, Degano M, Stanfield R L, Brunmark A, Jackson M R, Peterson P A, Teyton L, Wilson I A. Science. 1996;274:209–219. [PubMed] [Google Scholar]
- 32.Pullen J K, Hunt H D, Horton R M, Pease L R. J Immunol. 1989;143:1674–1679. [PubMed] [Google Scholar]
- 33.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horton R M, Loveland B E, Parwani A, Pease L R, Lindahl K F. J Immunol. 1991;147:3180–3184. [PubMed] [Google Scholar]
- 35.Sherman L A, Hesse S V, Irwin M J, La Face D, Peterson P A. Science. 1992;258:815–818. doi: 10.1126/science.1439792. [DOI] [PubMed] [Google Scholar]
- 36.Udaka K, Tsomides T J, Walden P, Fukusen N, Eisen H N. Proc Natl Acad Sci USA. 1993;90:11272–11276. doi: 10.1073/pnas.90.23.11272. [DOI] [PMC free article] [PubMed] [Google Scholar]