

Abstract
Elderly individuals display increased susceptibility to chronic inflammatory diseases and microbial infections, such as periodontitis and oral aspiration pneumonia. The resurgent interest in innate immunity in the 2000s has been accompanied by parallel studies to understand the impact of aging on the function of the innate immune system, which not only provides first-line defense but is essential for the development of adaptive immunity. This review summarizes and discusses our current understanding of age-associated molecular alterations in neutrophils and macrophages, key inflammatory phagocytes implicated in both protective and destructive host responses. The analysis of recent literature suggests that, in advanced age, phagocytes undergo significant changes in signal transduction pathways that may affect their ability to perform antimicrobial functions or regulate the inflammatory response. These abnormalities are expected to contribute to the pathology of oral infection-driven inflammatory diseases in the elderly. Moreover, the elucidation of age-associated defects in the innate immune system will facilitate the development of intervention therapeutic strategies to promote or restore innate immune function and improve the quality of health in old age.
Aging and oral immunity: Introduction
Immunosenescence refers to the declined function of the immune system with advanced age resulting in increased susceptibility of elderly individuals to microbial infections. Age-dependent alterations affect both the adaptive and innate arms of immunity, although the impact of aging on the latter is less well characterized (Gomez et al., 2008; Miller 1996). This is probably because the study of innate immunity was, until relatively recently, neglected as being non-specific and unsophisticated, simply serving to “buy time” until the activation of adaptive immunity that comprises the system of B and T lymphocytes (Fearon 1997). However, it is now firmly established that germ-line encoded innate immune receptors, such as the Toll-like receptors (TLR), can distinguish and differentially respond to distinct microbial structures (Medzhitov 2007). Importantly, as a result of a long co-evolution with microbes, the innate system is sophisticated enough to make judgments that instruct the initiation and progression of the adaptive response, whereas the B and T cells by themselves would not be able to decipher the biological context of the encountered antigen (i.e., does it warrant an immune response or not?). Recent evidence suggests that aging exerts a significant influence on innate immunity, although age-related alterations in innate immune function are not necessarily equated with immunodeficiency but rather with dysregulation of the immune response. This is because the underlying processes are dynamic involving both loss and gain of innate immune activity (Franceschi et al., 2000; Gomez et al., 2008). In this regard, the term “inflamm-aging” has been aptly coined to describe the heightened chronic inflammatory status often associated with advanced age in humans (Franceschi et al., 2000), although this progressive proinflammatory state may, at least in part, be attributed to inability to control infections in old age.
Elderly individuals exhibit increased susceptibility to a number of autoimmune, inflammatory, or infectious diseases, including chronic periodontitis (Gomez et al., 2008; Huttner et al., 2009; Miller 1996). Indeed, advanced age is associated with poor periodontal health and increased prevalence and severity of periodontitis (van der Velden 1991). This oral disease affects the majority of the adult population, and an estimated 10–15% develops severe periodontitis, which may lead to increased risk for systemic conditions like atherosclerosis, aspiration pneumonia, diabetes, and adverse pregnancy outcomes (Pihlstrom et al., 2005).
Aging per se leads to physiological loss of periodontal attachment and alveolar bone but these changes are quite modest and have little clinical significance, unless in the presence of concomitant periodontal inflammation, which is actually seen in a significant portion of the elderly population (Huttner et al., 2009; Kornman 2006). This is consistent with the inflammatory etiology of periodontal disease, where tissue damage is primarily caused by excessive host inflammatory reactions to subgingival gram-negative anaerobic pathogens (Gaffen and Hajishengallis 2008). However, it could be argued that the increased severity of periodontitis in old age could be simply the cumulative effect of prolonged exposure to microbial challenge. While this might be a contributory factor, several observations support the significance of age-related alterations in the periodontal immune response. In an experimental gingivitis study involving young (20–25 years of age) and elderly (65 years) individuals, all subjects received professional dental care to establish healthy gingival starting conditions and, subsequently, abstained from oral hygiene. Although young and old subjects displayed comparable dental plaque biofilm accumulation, the latter group developed more severe gingivitis associated with elevated numbers of inflammatory cells (Fransson et al., 1996). Moreover, resident periodontal cells (fibroblasts) from old humans or mice exhibit heightened inflammatory cytokine responses to microbial stimuli relative to their young counterparts (reviewed by Huttner et al., 2009), further indicating that aging influences the periodontal innate immune response.
This review will focus on the impact of aging on inflammatory phagocytes that can be recruited to periodontal tissues in response to subgingival microbial challenge. In this regard, a great emphasis has been placed on the study of age-related alterations in the function of macrophages and neutrophils due to their central role in innate immunity (Boehmer et al., 2005; Fulop et al., 2004; Gomez et al., 2008; Liang et al., 2009a; Plowden et al., 2004). The underlying molecular causes of age-dependent functional changes in phagocytes have been sought in differential receptor expression and/or signal transduction. In addition to periodontitis, the impact of aging on phagocytic cell function will also be viewed from the viewpoint of respiratory infections that can arise as a result of oral aspiration of the dental plaque biofilm (aspiration pneumonia) (Paju and Scannapieco 2007).
Age-related molecular alterations in neutrophils
Neutrophils are normally found in circulating blood, where they constitute the predominant phagocytic cell type, but readily migrate to sites of infection such as the gingival crevice following chemotactic signals by bacterial products, complement components, or chemokines. As professional phagocytes that mediate first-line defense, the neutrophils are equipped with pattern-recognition and complement receptors which detect and respond to conserved microbial structures or host-derived proinflammatory molecules. Their characteristic ability to generate and release toxic reactive oxygen species and granular enzymes renders them ideal cells for pathogen killing but also important effectors of host tissue damage in a variety of diseases, including periodontitis (Van Dyke and Serhan 2003). Perhaps due to the relative ease of isolating sufficient numbers of neutrophils from human peripheral blood (unlike in mice), most age-related studies on neutrophil function were performed in the human system. It should be noted, however, that there are several limitations associated with the use of phagocytes from elderly subjects. For example, various clinical conditions and the frequent use of drugs by elderly individuals are likely to be confounding factors in interpreting intrinsic effects of aging on innate immune cells, and thus more valid results are obtained if the cells are isolated from “healthy elderly subjects”.
Although the total number of circulating neutrophils remains constant with advancing age, their capacity to chemotactically migrate in vitro in response to granulocyte-macrophage colony-stimulating factor (GM-CSF) or the N-formyl-Met-Leu-Phe (FMLP) peptide is significantly reduced in old age, even when the neutrophils are obtained from healthy elderly individuals (Butcher et al., 2000). Similar age-dependent functional defects are seen in phagocytosis and the production of reactive oxygen species, although the latter defect seems to be stimulus-specific (Gomez et al., 2008). Another factor that may contribute to reduced capacity of “old” neutrophils to effectively fight infections is related to alterations in apoptosis delay under priming conditions. Specifically, although there are no significant differences in the spontaneous apoptosis of neutrophils obtained from young and old individuals, the ability of priming agents, such as GM-CSF, complement fragment C5a, or bacterial lipopolysaccharide (LPS) to induce anti-apoptotic signals is reduced in neutrophils isolated from elderly individuals (Butcher et al., 2000). Therefore, not only do “old” neutrophils display decreased mobilization to sites of infection and impaired antimicrobial functions but also die off more rapidly. It could thus be envisioned that these age-associated alterations (Table 1) could result in a continuous influx of impotent neutrophils that fail to contain an infection and, quite probably, contribute to host tissue damage from eventual release of toxic substances. These products, which include reactive oxygen species, may not be produced at high enough quantities on a per-cell basis in old age; however, in the setting of chronic inflammation, tissue injury could result from repeated exposure to toxic neutrophil products. This mechanism of tissue injury is quite likely to occur in the gingival crevice, where neutrophils immigrate in large numbers in response to the resident tooth-associated biofilm bacteria.
Table 1.
Effects of aging on phagocytic cell function*
| Functional status | Phagocytes | |
|---|---|---|
| Neutrophils | Macrophages | |
 |
 |
|
| Reduced | Receptor recruitment to lipid rafts Signal transduction (e.g., Ca2+ influx, phosphorylation of ERK, p38, Akt, PLC-γ) Chemotaxis Phagocytosis |
Signal transduction (e.g., total levels and/or activation of STAT-1α, p38 & JNK MAPKs, MyD88, NF-κB) Chemotaxis Cytokine production (IL-6, TNF-α, MIP-1α, MIP-1β, MIP-2) Reactive oxygen species production |
| Maintained | Total cell number Receptor expression (GM-CSFR, TLR2, TLR4, CD14, CD11b) Apoptosis (spontaneous) |
Expression of IFN-γR and TLRs (TLR2, TLR4, TLR5, TLR6) Phagocytic receptor expression (CD14, CD11b, CD18, CD36, mannose receptor [CD206], dectin-1, scavenger receptor-AI) Phagocytosis (?) Expression of TLR negative regulators (e.g., SOCS-1, IRAK-M, A20, PPAR-γ) |
| Increased | Apoptosis (under priming conditions) | Receptors involved in inflammation amplification (C5aR, TREM-1) PGE2 production |
| Controversial or both increase/decrease observed | Reactive oxygen species production | Nitric oxide production and intracellular killing TLR1 (decreased in human monocytes; unaltered in mouse macrophages) |
This compilation is based on a great number of studies, mainly using mouse or human cells, and original work has been cited in excellent specialized reviews (Butcher et al., 2000; Fulop et al., 2004; Gomez et al., 2008; Kovacs et al., 2009; Plowden et al., 2004; Solana et al., 2006; Stout and Suttles 2005).
Interestingly, the above discussed age-associated functional alterations cannot be linked to significant changes in the expression of receptors mediating those responses. For example, the expression of the GM-CSF receptor (GM-CSFR), TLR2 and TLR4 (which respond to bacterial lipopeptides and LPS, respectively) is not affected as a function of age (Fulop et al., 2004; Solana et al., 2006). It should be noted, however, that intracellular signaling could be affected, even if surface receptor expression is preserved with age, as a consequence of age-associated alterations in the composition of membrane lipid rafts, which serve as cellular signaling platforms. For example, altered TLR4 signaling in aged human neutrophils was attributed to changes in the recruitability of this receptor to lipid rafts, which display age-related physicochemical changes (Fulop et al., 2004). Specifically, the cholesterol content of lipid rafts appears to increase with aging resulting in reduced membrane fluidity (Larbi et al., 2004). Similarly, altered signaling by the triggering receptor expressed on myeloid cells (TREM)-1 in aged neutrophils was explained by defective recruitment of TREM-1 to lipid rafts (Fortin et al., 2007). TREM-1 participates in the amplification of the inflammatory response and its ligation promotes neutrophil phagocytosis and degranulation (Gomez et al., 2008). Therefore, defective TREM-1 signaling, manifested by reduced phosphorylation of Akt and phospholipase C-γ (PLC-γ) (Fortin et al., 2007), would be expected to compromise neutrophil immunosurveillance in old age. This is consistent with the fact that the elderly are more susceptible to microbial infections (Solana et al., 2006). Moreover, although neutrophil phagocytosis declines with aging, no significant age-dependent differences were observed in the expression of important phagocytic receptors such as CD11b and CD14 (Butcher et al., 2000), implying that the defect could likewise lie in signal transduction. However, it cannot be ruled out that other phagocytic receptors may actually be affected.
Additional age-associated defects in signal transduction, such as decreased FMLP-induced Ca2+ responses and GM-CSF- or FMLP-induced activation of extracellular receptor-activated kinase (ERK) or p38 mitogen-activated protein kinase (MAPK) have also been documented (Table 1) and could similarly contribute to impaired neutrophil function in elderly individuals (Gomez et al., 2008).
Age-related molecular alterations in macrophages
Studies in humans suggest that a number of monocyte/macrophage functions become compromised with advancing age; these including chemotaxis, phagocytosis, production of reactive oxygen species, and induction of certain cytokine responses (reviewed by Gomez et al., 2008) (Table 1). However, discrepant results have often been reported owing to differences in experimental conditions and/or in the health status or possible medical treatment of the donors (Gomez et al., 2008). To control for these factors, many aging studies on macrophage function have been performed on experimental mice and rats. Macrophage chemotaxis appears to be reduced in old mice, which may be related, at least in part, to reduced inducible production of several chemokines, such as the macrophage inflammatory protein-1α (MIP-1α), MIP-1β, and MIP-2 (Stout and Suttles 2005). Variable results regarding the impact of aging on phagocytosis were obtained using aged and young mouse or rat macrophages. Indeed, this function was found to be up- or down-regulated or even remain unaltered with age (reviewed by Gomez et al., 2008 and Plowden et al., 2004). Our own results that age has no significant effects on mouse macrophage expression of important phagocytic receptors (CD14, CD11b, CD18, CD36, mannose receptor [CD206], dectin-1, and scavenger receptor-AI) (Liang et al., 2009a) is consistent with the notion that macrophage phagocytosis may probably be a well-preserved function. In this context, macrophages from young and old mice exhibit comparable capacities for uptake of Porphyromonas gingivalis, a major periodontal pathogen (Liang et al., 2009a).
An early study on the influence of aging on TLR expression found that aged mouse macrophages display reduced mRNA expression of TLR1-9, although at the protein level the results were confirmed for TLR4 only (Renshaw et al., 2002). Accordingly, LPS-induced cytokine responses were found to decline with age (Renshaw et al., 2002). This observation was confirmed by an independent study, although the observed age-associated reduction in cytokine responses was attributed to decreased expression of MAPK, rather than decreased TLR4 expression which in fact remained unaffected (Boehmer et al., 2004). Similarly, although macrophage induction of IL-6 in response to P. gingivalis declines with age, this finding cannot be explained by reduced expression of the signaling receptors involved (TLR2 and TLR1), since these molecules are not affected at either the mRNA or the protein level (Liang et al., 2009a). Normal levels of TLR2 expression are similarly maintained in monocytes from elderly humans, although the expression of monocytic TLR1 was reported to decline in old age (van Duin et al., 2007) (Table 1). Since TLR2 signals in obligatory cooperation with either TLR1 or TLR6 (Medzhitov 2007), it is likely that TLR2-mediated responses are affected at least in part, although signaling in response to TLR2/TLR6 ligands (e.g., yeast zymosan) should remain unaltered.
In addition to IL-6, reduced production was also seen for several other (e.g., TNF-α, IL-12, IFN-γ) (Boehmer et al., 2004; Gomez et al., 2008) but not all inflammatory mediators, some of which may actually increase as a function of age. For instance, activated macrophages from aged mice or humans produce higher levels of prostaglandin E2 (PGE2) than younger controls (Plowden et al., 2004). The property of PGE2 to inhibit induction of IL-12, which stimulates cell-mediated immunity, may compromise the capacity of older individuals to effectively clear infections (Plowden et al., 2004). Moreover, PGE2 is heavily implicated in periodontal tissue destruction (Noguchi and Ishikawa 2007) and its elevated levels in old age could be one of the mechanisms accounting for the increased prevalence and severity of this oral disease in the elderly.
As alluded to above, age-dependent changes in macrophage responses may involve post-receptor alterations in signaling pathways. In this context, we investigated the expression and inducibility of 14 key intracellular or transmembrane negative regulators of TLR signaling. With a few exceptions (see below), no significant differences were observed between young and aged macrophages in the basal or inducible expression of these molecules. Although P. gingivalis could induce significant up- or down-regulation of most of the investigated regulators (e.g., the suppressor of cytokine signaling-1 [SOCS-1], the interleukin-1 receptor-associated kinase M [IRAK-M], the A20 ubiquitin-editing enzyme, and the peroxisome proliferative activated receptor-γ [PPAR-γ]), the expression of most of these genes was similar between young and aged activated macrophages (Liang et al., 2009a). However, we observed age-related differential mRNA expression of the single immunoglobulin interleukin-1-related receptor and the mitogen-activated protein kinase phosphatase-1, which can downregulate TLR signal transduction, and could thereby impact on the way macrophages respond to certain microbial TLR ligands. However, additional studies are warranted to specifically address this possibility.
Regarding signaling intermediates that positively regulate the macrophage response, a number of studies using mouse macrophages or human monocytes have demonstrated age-dependent decline in the total levels and phosphorylation capacity of the IFN-γ-mediated signal transducer and activator of transcription-1α (STAT-1α), the MAPKs (p38 and Jun N-terminal kinase [JNK]), and the nuclear levels of NF-κB (reviewed by Gomez et al., 2008 and Kovacs et al., 2009) (Table 1). The reduced activity of STAT-1α may affect the ability of macrophages to become effectively activated by IFN-γ, even though the expression of the IFN-γ receptor (IFN-γR) remains unaltered with aging (Kovacs et al., 2009). It is conceivable that age-associated alterations in lipid raft composition and function could contribute to defective intracellular signaling in macrophages as seen in neutrophils (Fortin et al., 2007; Fulop et al., 2004). The findings for age-associated reduction in the levels of MAPK and NF-κB, as well as of MyD88, a major TLR signaling adaptor (Medzhitov 2007), similarly suggest that aging may negatively affect macrophage activation. However, these observations have not been adequately correlated with impaired functions other than reduced cytokine responses (Gomez et al., 2008; Kovacs et al., 2009).
In this context, we found that young and aged macrophages display comparable intracellular killing of P. gingivalis, consistent with similar induction of inducible nitric oxide synthase (iNOS) and production of nitric oxide by both age groups (Liang et al., 2009a). However, our data are not in accordance with another report that the ability of activated mouse macrophages to express iNOS and produce nitric oxide declines with age (Kissin et al., 1997). This study used purified Escherichia coli LPS as stimulus, whereas we used whole cells of P. gingivalis. It is conceivable that the outcome may be stimulus-dependent, although this possibility has yet to be addressed. Although aged macrophages exhibit reduced production of reactive oxygen species (Sebastian et al., 2005), this would not be a factor regarding their ability to handle P. gingivalis, which is exquisitely resistant to oxygen-dependent killing (Hajishengallis 2009). In contrast to our in vitro findings suggesting that age does not affect the macrophages in terms of P. gingivalis killing, we have demonstrated age-associated impaired killing of this oral pathogen in vivo (S. Liang, H. Domon, and G. Hajishengallis; unpublished data). This adds another layer of complexity in understanding the impact of aging on immunity, in that aged phagocytes may not simply be affected by age-dependent, cell-intrinsic changes but also by age-dependent changes in their tissue environment (discussed in more detail in the next section).
Aging and the impact of the in vivo environment on phagocyte function
It has recently been proposed that the various tissues may have greater control over the initiation of immunity than originally believed. Accordingly, the function of macrophages may be largely dictated by the type and condition of the tissue they reside (Stout and Suttles 2005). It is thus likely that the aged local microenvironment may influence macrophage function in ways that cannot be anticipated by in vitro experiments, which cannot faithfully replicate the in vivo setting. For example, at least some of the innate immune dysfunction seen in old age may be due to inefficient communication between macrophages (or other innate immune cells) and the tissues, which appear to express lower levels of adhesion molecules and display reduced responsiveness to growth factors (Stout and Suttles 2005).
In fact, although the production of proinflammatory cytokines by macrophages and other inflammatory cells appears to decline with aging, the same cytokines are detected at higher levels in the plasma of aged mice or humans (Krabbe et al., 2004; Stout and Suttles 2005), consistent with the concept of “inflamm-aging”, i.e., the heightened chronic inflammatory status of the elderly (Franceschi et al., 2000). A plausible interpretation is that, in vivo, the macrophages may have the opportunity to also interact with tissue stromal cells and other factors or inflammatory cells. Alternatively, or in addition, the heightened inflammation in aged individuals may be a secondary effect arising from inability to elicit an appropriate antimicrobial response that would effectively control infectious pathogens. Something similar may occur in periodontitis. Thus, according to in vitro studies, periodontal macrophages and neutrophils are expected to exhibit reduced inflammatory and antimicrobial responses on a per-cell basis. However, the resulting inability of these phagocytes to control microbial overgrowth in periodontal tissues could lead to chronic persistence of the pathogens and unresolved destructive inflammation. In other words, elevated inflammatory responses may occur in elderly individuals even if their inflammatory cells are not intrinsically more proinflammatory than their younger counterparts. These considerations raise reasonable concerns on the transferability of in vitro data to in vivo situations. However, in vitro studies are warranted for better understanding cell-intrinsic alterations on phagocyte function as a result of aging.
Although it is generally uncertain whether experimental mice undergo “inflamm-aging” like humans do, it is possible that mouse “inflamm-aging” may be manifested at least in certain tissues. Indeed, we have recently shown that mice develop inflammatory periodontitis as a function of age, induced by their indigenous oral microbiota and characterized by increased expression of proinflammatory cytokines and bone loss in the periodontium (see below). This supports the suitability of animal models for aging research.
Insights from aging models of animal periodontitis
Mice constitute a convenient and useful model of periodontal disease and induction of inflammatory bone loss is typically achieved within a few weeks following oral infection with human periodontal pathogens (Graves et al., 2008). Mice used for experimental periodontitis are typically 8–12 week-old and sham-infected mice do not typically develop appreciable periodontal bone loss. This may have given the erroneous impression that mice do not develop naturally occurring periodontitis. To the contrary, this observation is simply consistent with the fact that chronic periodontitis is normally associated with advanced age (van der Velden 1991). Moreover, certain reports have suggested that the indigenous oral microbiota of mice contains potential periodontal pathogens. Specifically, young mice with P/E-selectin deficiency resulting in impaired leukocyte migration to sites of infection, exhibit massive oral bacterial colonization and induction of periodontal inflammation and bone loss (Niederman et al., 2001). Similarly, lysosomal-associated membrane protein-2 knockout mice are unable to control their oral flora, due to impaired neutrophil killing function, and develop periodontitis at young age (Beertsen et al., 2008). Indigenous bacterial involvement in both periodontitis models is further supported by findings that the induction of inflammation and periodontal bone loss is prevented by antibiotic treatment (Beertsen et al., 2008; Niederman et al., 2001).
On the basis of these pivotal observations, we hypothesized that mice can develop inflammatory periodontal bone loss as a function of age. Indeed, examination of mice of various ages (2 to 18 months of age) revealed an age-associated increase in periodontal bone loss which became quite dramatic after 9 months of age (Liang et al., 2009b) (Fig. 1). The oldest mice (≥ 18 month-old) often display increased mobilityof molar teeth or even missing molars. The age-dependent increase of periodontal bone loss is accompanied by elevated expression of proinflammatory cytokines (IL-1β, TNF-α, and IL-17A) and of innate immune receptors involved in the induction or amplification of inflammation (TLR2, CD14, CD11b, CD18, the complement C5a receptor [C5aR], and TREM-3) (Liang et al., 2009b).
Figure 1. Periodontal inflammation and bone loss as a function of age in BALB/c mice.

(A): Increased periodontal bone loss with increasing mouse age, measured as the distance between the cementoenamel junction (CEJ) and the alveolar bone crest (ABC) (B–E): Representative images from the maxillae of young (B, right; D, left) and old (C, right; E, left) mice. Young mice were 8–10 week-old and old mice were ≥ 18 months of age. (F–G): Maxillary molar blocks seen from the occlusal surfaces of young (F) and old (G) mice. Note that all three molars in young mice are in line with each other, in contrast to overt migration of molar teeth in old mice. Reprinted with modifications from Liang et al., 2009b. Copyright, Wiley-Blackwell.
The augmented expression of IL-1β and TNF-α in the aged mouse periodontium is significant in that both cytokines are major mediators of destructive bone resorption in periodontitis (Gaffen and Hajishengallis 2008; Graves et al., 2008). However, not all inflammatory mediators were upregulated in the gingivae of old mice (e.g., IL-6 and the high-mobility group box-1 protein remained unaltered) (Liang et al., 2009b). In the same study, examination of “signature” molecules (IFN-γ, IL-4, IL-17A, and forkhead box P3) indicative of specific T helper (Th) cell responses (Th1, Th2, Th17, and T regulatory cells, respectively) showed that only IL-17A was upregulated in the aged mouse gingivae. This is an intriguing finding in view of the recent emergence of the Th17 lineage as a specialized osteoclastogenic T cell subset with a prominent role in chronic inflammatory or autoimmune diseases (Gaffen and Hajishengallis 2008).
We moreover found that six out of fifteen investigated innate immune receptors are differentially expressed in the gingivae of young and old mice. These include C5aR and TREM-3 which may contribute to heightened periodontal inflammation due to their involvement in the amplification of the host inflammatory response (reviewed by Liang et al., 2009a). Other upregulated receptors, specifically TLR2, CD14, and the CD11b/CD18 heterodimer (also known as complement receptor-3; CR3) constitute critical components of the TLR2/CR3 inside-out signaling pathway (Harokopakis and Hajishengallis 2005). This pathway is exploited by P. gingivalis for evading immune elimination and promoting its persistence (reviewed by Hajishengallis 2009), but it is probably a universal immune evasion pathway since it was subsequently shown to be exploited also by Mycobacterium tuberculosis and Bacillus anthracis (Oliva et al., 2009) (Fig. 2). Therefore, the exploitability of the TLR2/CR3 pathway by P. gingivalis and possibly other periodontal pathogens, and the impact of this pathway on periodontitis, could increase with aging.
Figure 2. Activation of the TLR2/CR3 pathway by microbial pathogens.
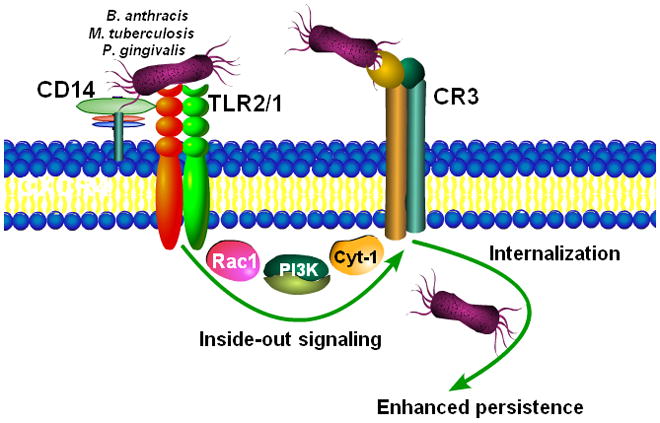
Certain bacteria (P. gingivalis, M. tuberculosi, B. anthracis) bind CD14 and induce TLR2-dependent inside-out signaling, mediated by Rac-1, phosphatidylinositol-3 kinase (PI3K), and cytohesin-1 (Cyt-1), leading to activation of the ligand-binding capacity of CR3. The interaction of activated CR3 with bacteria leads to a relatively safe internalization and persistence of the bacteria within the cells (reviewed by Hajishengallis 2009). On the basis of age-related alterations in the CD14/TLR2/CR3 pathway (Liang et al., 2009b), the exploitability of CR3 by periodontal pathogens like P. gingivalis could be enhanced in advanced age, contributing to increased susceptibility to periodontal disease.
This genuinely chronic model of periodontitis can facilitate the investigation of the impact of aging on innate periodontal immunity and pathogenesis, in a way that cannot be achieved in humans, as it is difficult to control for confounding factors, in addition to limitations associated with ethical considerations. In fact, very little is known regarding age-associated changes in human periodontal innate immunity. Chronic inflammatory periodontal bone loss may be an example of “inflamm-aging” that is shared by humans and mice. Importantly, the parallel raising and aging of wild-type mice and mice with defined genetic defects may lead to the identification of specific recognition and signaling pathways that protect against or exacerbate periodontal tissue destruction. This knowledge may in turn be exploited for therapeutic purposes. Additional insights may be obtained from nonhuman primate models. In a preliminary study to this direction, it was established that the prevalence and severity of periodontitis increases as a function of age in rhesus monkeys, accompanied by elevated levels of several systemic inflammatory mediators (Ebersole et al., 2008).
Aging and respiratory infections
In addition to causing periodontitis, the tooth-associated biofilm (dental plaque) forms a persistent reservoir for respiratory infections, and oral pathogens can be aspirated into the lung to cause pneumonia (Paju and Scannapieco 2007). Indeed, certain gram-negative anaerobic bacteria including P. gingivalis are common isolates from aspiration pneumonia and lung abscesses (Finegold 1991). More recently, periodontitis was epidemiologically implicated as a mortality risk factor for aspiration pneumonia in the elderly (Awano et al., 2008). Oropharyngeal aspiration of microorganisms is a major cause of pneumonia (12% of all cases) in old individuals, although the disease is additionally seen in immunocompromised individuals regardless of age (Finegold 1991; Paju and Scannapieco 2007). Pneumonia, in general, is a leading cause of death in the elderly and is attributed, at least in part, to declined pulmonary innate immunity (Meyer 2005). In addition to increased susceptibility to lung infections, the elderly also display delayed resolution of pulmonary inflammation (Meyer 2005). In this regard, the association of aging with low-grade chronic systemic inflammation (Franceschi et al., 2000) is also reflected in the bronchoalveolar lavage fluid from old individuals, which contains increased levels of proinflammatory cytokines compared to young adults (Krabbe et al., 2004; Meyer et al., 1996).
In the absence of lung infection or inflammation, neutrophils are essentially absent and alveolar macrophages (AM) account for more than 98% of the total leukocyte numbers in the alveolar space. Although the impact of aging on neutrophil function in pulmonary defense can be predicted by findings on neutrophils isolated from peripheral blood (Table 1), the AM may need to be studied in their own right. This is due to their special characteristics, such as the propensity to exert immunosuppressive function for maintaining respiratory tract homeostasis, although they retain the ability to become activated should a microbial threat appears (Holt et al., 2008). Moreover, AM express little or no CR3 (Stokes et al., 1998) which, at least in principle, could render them quite resistant to pathogens like P. gingivalis that exploit this receptor for escaping intracellular killing (reviewed by Hajishengallis 2009) (Fig. 2). Indeed, mouse AM are significantly more potent than their peritoneal or splenic counterparts in killing P. gingivalis, in a way attributable to limited AM CR3 expression (Hajishengallis et al., 2008).
First-line pulmonary defense by AM at the air-tissue interface is mainly exerted through phagocytosis and killing of invading microorganisms and by coordinating the innate response (Holt et al., 2008). Thus, when the infection becomes overwhelming or the pathogen is too virulent to be contained by the AM alone, AM-generated chemokines and other inflammatory mediators recruit and activate large numbers of neutrophils from the pulmonary vasculature into the alveolar space (Zhang et al., 2000). Besides chemokines/cytokines, phagocyte-produced reactive oxygen and nitrogen intermediates are also important for early bacterial clearance (Strieter et al., 2003).
There are very few mechanistic studies on aging and lung innate immunity in humans, although it was shown that the relative proportion of AM in the bronchoalveolar lavage is reduced as a function of age relative to the lymphocytes (Zissel et al., 1999). Moreover, the accessory function of human AM is compromised with aging, although weak associations were found between age and proinflammatory cytokine production (Zissel et al., 1999). Reduced AM numbers were also noted in the bronchoalveolar lavage fluid of old rats (Antonini et al., 2001), which have been extensively used as a model (more than mice) for aging studies on pulmonary innate immunity (Table 2). AM from old rats, moreover, display reduced production of TNF-α in response to LPS, resulting from defective protein kinase C activation relative to young controls (Corsini et al., 1999). Rat AM also display reduced production of IL-10 with aging, attributable to reduced activity of the cAMP-dependent signaling pathway (Corsini et al., 2005). The in vivo impact of this observation is that old rats display exacerbated inflammatory tissue damage in a model of sterile inflammation (Corsini et al., 2005). Impaired production of both proinflammatory and anti-inflammatory cytokines may predispose to both increased susceptibility to infections and inability to resolve inflammation, although additional studies in independent systems would need to confirm these results for cross-species extrapolation. In a related context regarding age-associated changes in homeostatic mechanisms, it was shown that mouse AM express reduced levels of heme oxygenase-1 with aging, in response to in vitro or in vivo LPS challenge (Ito et al., 2009). This defect is likely to affect the ability of the lung tissue for protection against oxidative stress in old age.
Table 2.
Age-dependent alterations in immune functions of alveolar macrophages
| Age-associated alteration | Experimental model | Refs |
|---|---|---|
| Reduced numbers in bronchoalveolar lavage fluid | rats | (Antonini et al., 2001) |
| Defective LPS-induced protein kinase C activation and TNF-α production | rats | (Corsini et al., 1999) |
| Decreased cAMP-dependent signaling and IL-10 production | rats | (Corsini et al., 2005) |
| Reduced induction of heme oxygenase-1 in response to LPS | mice | (Ito et al., 2009) |
| Reduced production of nitric oxide in response to LPS or bacteria | rats | (Antonini et al., 2001; Tasat et al., 2003) |
| Increased induction of reactive oxygen species in response to phorbol myristate acetate or bacteria | rats | (Antonini et al., 2001; Tasat et al., 2003) |
| Increased phagocytosis | rats | (Mancuso et al., 2001) |
| Preserved ability to produce leukotrienes and prostaglandins | rats | (Mancuso et al., 2001) |
| Reduced TLR expression (TLR2, TLR1, TLR4) | mice | (Hinojosa et al., 2009) |
In terms of effector functions, AM from old rats induce higher production of reactive oxygen species but lower production of nitric oxide in response to phorbol myristate acetate or LPS, respectively (Tasat et al., 2003). These basic findings are consistent with an independent study, which compared the relative ability of aged and young rats to handle a pulmonary infection with Listeria monocytogenes. Aged rats were found to display increased susceptibility to infection accompanied by enhanced tissue damage and mortality, owing to dramatically reduced AM production of nitric oxide, despite a surprising increase in the generation of reactive oxygen species (Antonini et al., 2001). Its potential antimicrobial effects notwithstanding, it appears that the oxidative burst might actually contribute to lung tissue injury in old age rather than to protect against pathogen infection.
In another report, it was surprisingly found that the ability of rat AM to phagocytose Klebsiella pneumoniae increases with aging, whereas their capacity to release arachidonic acid and produce leukotrienes or prostaglandins is preserved (Mancuso et al., 2001). On the other hand, in the same study, lung-recruited neutrophils from old rats exhibited reduced phagocytosis of K. pneumoniae (Mancuso et al., 2001). This raises the possibility that impaired phagocytosis by recruited neutrophils may be one of the factors accounting for increased susceptibility to respiratory infections in old age. However, as shown by other investigators, the numbers of resident AM decline in aged rats (Antonini et al., 2001). Therefore, AM may not be quite capable to perform immunosurveillance in old age, despite preservation (or even enhancement) of certain immune functions at the cellular level (Table 2).
Consistent with the findings in rats and with clinical observations on the increased susceptibility of the elderly to aspiration pneumonia, we found that old mice exhibit reduced ability to clear acute pulmonary infection with P. gingivalis, relative to young controls (S. Liang, H. Domon, and G. Hajishengallis; unpublished data). The underlying mechanisms are currently under investigation. However, since the immune clearance of P. gingivalis from the mouse lung is TLR2-dependent (Hajishengallis et al., 2008) and TLR expression (TLR2, TLR1, and TLR4) is reduced in the mouse lung tissue with aging (Hinojosa et al., 2009), the age-associated impaired killing of this pathogen could be due to defective TLR2 immunity in the lung.
Additional studies are warranted to better characterize functional alterations in AM, and in lung innate immunity in general, as a result of the aging process. It should be borne in mind that species-specific age-associated changes in pulmonary innate immunity cannot be ruled out. It is encouraging, nonetheless, that data from experimental animals and clinical observations concur that old age predisposes to increased susceptibility to respiratory infectious and inflammatory diseases, thus validating the use of animal models in aging research.
Conclusions and future perspectives
The analysis of available literature indicates substantial age-associated abnormalities in the function of neutrophils and macrophages (also seen in other innate immune cells, such as dendritic cells, for which the reader is referred to excellent recent reviews (Gomez et al., 2008; Solana et al., 2006)). The observed cellular abnormalities seem to be selective in that certain signaling pathways and functions appear to be preserved in advanced age. However, the role of extrinsic factors associated with a particular tissue environment cannot be underestimated, as they can exert profound influence on phagocytic cell function. Thus, aging studies should place great emphasis on extrinsic/microenvironmental factors in addition to innate immune cell-intrinsic changes that affect innate immunity. Experimental and clinical observations also suggest that periodontal and pulmonary innate immunity become dysregulated with aging, which may contribute, at least in part, to the increased susceptibility of aged individuals to periodontitis and oral aspiration pneumonia. By determining the molecular causes of innate immune dysfunction and understanding their impact on these diseases, it would be possible to develop appropriate therapeutic strategies that would benefit the elderly.
Acknowledgments
Studies performed in author’s laboratory and cited in this review were supported by U.S. Public Health Service Grants DE018292 and DE015254. The author regrets that a number of important studies could only indirectly be cited through comprehensive reviews, due to space and reference number limitations.
References
- Antonini JM, Roberts JR, Clarke RW, Yang HM, Barger MW, Ma JY, Weissman DN. Effect of age on respiratory defense mechanisms: pulmonary bacterial clearance in Fischer 344 rats after intratracheal instillation of Listeria monocytogenes. Chest. 2001;120:240–249. doi: 10.1378/chest.120.1.240. [DOI] [PubMed] [Google Scholar]
- Awano S, Ansai T, Takata Y, Soh I, Akifusa S, Hamasaki T, Yoshida A, Sonoki K, Fujisawa K, Takehara T. Oral health and mortality risk from pneumonia in the elderly. J Dent Res. 2008;87:334–339. doi: 10.1177/154405910808700418. [DOI] [PubMed] [Google Scholar]
- Beertsen W, Willenborg M, Everts V, Zirogianni A, Podschun R, Schroder B, Eskelinen EL, Saftig P. Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J Immunol. 2008;180:475–482. doi: 10.4049/jimmunol.180.1.475. [DOI] [PubMed] [Google Scholar]
- Boehmer ED, Goral J, Faunce DE, Kovacs EJ. Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol. 2004;75:342–349. doi: 10.1189/jlb.0803389. [DOI] [PubMed] [Google Scholar]
- Boehmer ED, Meehan MJ, Cutro BT, Kovacs EJ. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev. 2005;126:1305–1313. doi: 10.1016/j.mad.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. 2000;100:411–416. doi: 10.1046/j.1365-2567.2000.00079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini E, Battaini F, Lucchi L, Marinovich M, Racchi M, Govoni S, Galli CL. A defective protein kinase C anchoring system underlying age-associated impairment in TNF-α production in rat aacrophages. J Immunol. 1999;163:3468–3473. [PubMed] [Google Scholar]
- Corsini E, Di Paola R, Viviani B, Genovese T, Mazzon E, Lucchi L, Marinovich M, Galli CL, Cuzzocrea S. Increased carrageenan-induced acute lung inflammation in old rats. Immunology. 2005;115:253–261. doi: 10.1111/j.1365-2567.2005.02148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole JL, Steffen MJ, Gonzalez-Martinez J, Novak MJ. Age and oral disease effects on systemic inflammatory and immune parameters in nonhuman primates. Clin Vaccine Immunol. 2008;15:1067–1075. doi: 10.1128/CVI.00258-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT. Seeking wisdom in innate immunity. Nature. 1997;388:323–324. doi: 10.1038/40967. [DOI] [PubMed] [Google Scholar]
- Finegold SM. Aspiration pneumonia. Rev Infect Dis. 1991;13(Suppl 9):S737–742. doi: 10.1093/clinids/13.supplement_9.s737. [DOI] [PubMed] [Google Scholar]
- Fortin CF, Lesur O, Fulop T., Jr Effects of aging on triggering receptor expressed on myeloid cells (TREM)-1-induced PMN functions. FEBS Lett. 2007;581:1173–1178. doi: 10.1016/j.febslet.2007.02.029. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Fransson C, Berglundh T, Lindhe J. The effect of age on the development of gingivitis. Clinical, microbiological and histological findings. J Clin Periodontol. 1996;23:379–385. doi: 10.1111/j.1600-051x.1996.tb00561.x. [DOI] [PubMed] [Google Scholar]
- Fulop T, Larbi A, Douziech N, Fortin C, Guerard KP, Lesur O, Khalil A, Dupuis G. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3:217–226. doi: 10.1111/j.1474-9728.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817–828. doi: 10.1177/154405910808700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez CR, Nomellini V, Faunce DE, Kovacs EJ. Innate immunity and aging. Exp Gerontol. 2008;43:718–728. doi: 10.1016/j.exger.2008.05.0168.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Fine D, Teng Y-TA, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. 2008;35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Bagby GJ, Nelson S. Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J Immunol. 2008;181:4141–4149. doi: 10.4049/jimmunol.181.6.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637–645. doi: 10.1016/j.micinf.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- Hinojosa E, Boyd AR, Orihuela CJ. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J Infect Dis. 2009;200:546–554. doi: 10.1086/600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- Huttner EA, Machado DC, de Oliveira RB, Antunes AG, Hebling E. Effects of human aging on periodontal tissues. Spec Care Dentist. 2009;29:149–155. doi: 10.1111/j.1754-4505.2009.00082.x. [DOI] [PubMed] [Google Scholar]
- Ito Y, Betsuyaku T, Moriyama C, Nasuhara Y, Nishimura M. Aging affects lipopolysaccharide-induced upregulation of heme oxygenase-1 in the lungs and alveolar macrophages. Biogerontology. 2009;10:173–180. doi: 10.1007/s10522-008-9164-4. [DOI] [PubMed] [Google Scholar]
- Kissin E, Tomasi M, McCartney-Francis N, Gibbs CL, Smith PD. Age-related decline in murine macrophage production of nitric oxide. J Infect Dis. 1997;175:1004–1007. doi: 10.1086/513959. [DOI] [PubMed] [Google Scholar]
- Kornman KS. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am J Clin Nutr. 2006;83:475S–483S. doi: 10.1093/ajcn/83.2.475S. [DOI] [PubMed] [Google Scholar]
- Kovacs EJ, Palmer JL, Fortin CF, Fulop T, Jr, Goldstein DR, Linton PJ. Aging and innate immunity in the mouse: impact of intrinsic and extrinsic factors. Trends Immunol. 2009;30:319–324. doi: 10.1016/j.it.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–699. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Larbi A, Douziech N, Dupuis G, Khalil A, Pelletier H, Guerard KP, Fulop T., Jr Age-associated alterations in the recruitment of signal-transduction proteins to lipid rafts in human T lymphocytes. J Leukoc Biol. 2004;75:373–381. doi: 10.1189/jlb.0703319. [DOI] [PubMed] [Google Scholar]
- Liang S, Domon H, Hosur KB, Wang M, Hajishengallis G. Age-related alterations in innate immune receptor expression and ability of macrophages to respond to pathogen challenge in vitro. Mech Ageing Dev. 2009a;130:538–546. doi: 10.1016/j.mad.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Hosur KB, Domon H, Hajishengallis G. Periodontal inflammation and bone loss in aged mice. J Periodont Res. 2009b doi: 10.1111/j.1600-0765.2009.01245.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso P, McNish RW, Peters-Golden M, Brock TG. Evaluation of phagocytosis and arachidonate metabolism by alveolar macrophages and recruited neutrophils from F344xBN rats of different ages. Mech Ageing Dev. 2001;122:1899–1913. doi: 10.1016/s0047-6374(01)00322-0. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Meyer KC, Ershler W, Rosenthal NS, Lu XG, Peterson K. Immune dysregulation in the aging human lung. Am J Respir Crit Care Med. 1996;153:1072–1079. doi: 10.1164/ajrccm.153.3.8630547. [DOI] [PubMed] [Google Scholar]
- Meyer KC. Aging. Proc Am Thorac Soc. 2005;2:433–439. doi: 10.1513/pats.200508-081JS. [DOI] [PubMed] [Google Scholar]
- Miller RA. The aging immune system: primer and prospectus. Science. 1996;273:70–74. doi: 10.1126/science.273.5271.70. [DOI] [PubMed] [Google Scholar]
- Niederman R, Westernoff T, Lee C, Mark LL, Kawashima N, Ullman-Culler M, Dewhirst FE, Paster BJ, Wagner DD, Mayadas T, Hynes RO, Stashenko P. Infection-mediated early-onset periodontal disease in P/E-selectin-deficient mice. J Clin Periodontol. 2001;28:569–575. doi: 10.1034/j.1600-051x.2001.028006569.x. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Ishikawa I. The roles of cyclooxygenase-2 and prostaglandin E2 in periodontal disease. Periodontol 2000. 2007;43:85–101. doi: 10.1111/j.1600-0757.2006.00170.x. [DOI] [PubMed] [Google Scholar]
- Oliva C, Turnbough CL, Jr, Kearney JF. CD14-Mac-1 interactions in Bacillus anthracis spore internalization by macrophages. Proc Natl Acad Sci U S A. 2009;106:13957–13962. doi: 10.1073/pnas.0902392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paju S, Scannapieco FA. Oral biofilms, periodontitis, and pulmonary infections. Oral Dis. 2007;13:508–512. doi: 10.1111/j.1601-0825.2007.1410a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell. 2004;3:161–167. doi: 10.1111/j.1474-9728.2004.00102.x. [DOI] [PubMed] [Google Scholar]
- Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–4701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- Sebastian C, Espia M, Serra M, Celada A, Lloberas J. MacrophAging: a cellular and molecular review. Immunobiology. 2005;210:121–126. doi: 10.1016/j.imbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–494. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Stokes RW, Thorson LM, Speert DP. Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J Immunol. 1998;160:5514–5521. [PubMed] [Google Scholar]
- Stout RD, Suttles J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev. 2005;205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieter RM, Belperio JA, Keane MP. Host innate defenses in the lung: the role of cytokines. Curr Opin Infect Dis. 2003;16:193–198. doi: 10.1097/00001432-200306000-00002. [DOI] [PubMed] [Google Scholar]
- Tasat DR, Mancuso R, O’Connor S, Molinari B. Age-dependent change in reactive oxygen species and nitric oxide generation by rat alveolar macrophages. Aging Cell. 2003;2:159–164. doi: 10.1046/j.1474-9728.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- van der Velden U. The onset age of periodontal destruction. J Clin Periodontol. 1991;18:380–383. doi: 10.1111/j.1600-051x.1991.tb02304.x. [DOI] [PubMed] [Google Scholar]
- van Duin D, Mohanty S, Thomas V, Ginter S, Montgomery RR, Fikrig E, Allore HG, Medzhitov R, Shaw AC. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178:970–975. doi: 10.4049/jimmunol.178.2.970. [DOI] [PubMed] [Google Scholar]
- Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- Zissel G, Schlaak M, Muller-Quernheim J. Age-related decrease in accessory cell function of human alveolar macrophages. J Investig Med. 1999;47:51–56. [PubMed] [Google Scholar]