

Abstract
Low doses of psychostimulants produce beneficial behavioral effects in ADHD patients but the mechanisms underlying the response are not understood. Here we use the hyperactive mouse mutant coloboma to identify D2-like dopamine receptor subtypes that mediate the hyperactivity and response to amphetamine; we have previously demonstrated that D1-like dopamine receptors are not involved. Targeted deletion of the D2, but not the D3 or the D4, dopamine receptor in coloboma mice eliminated the hyperactivity; depleting D2 dopamine receptors also restored the excess dopamine overflow that may drive the hyperactivity to normal concentrations. Similar to its effects on ADHD patients, amphetamine reduced the hyperactivity of coloboma mice. The D2 dopamine receptor-selective antagonist L-741,626, but not D3 or D4 dopamine receptor-selective antagonists, blocked the amphetamine-induced reduction in locomotor activity. Thus, the D2 dopamine receptor subtype mediates the both the hyperactivity and response to amphetamine, suggesting a specific target for novel therapeutics in ADHD.
Keywords: hyperactivity, coloboma, D2 dopamine receptor, D3 dopamine receptor, D4 dopamine receptor, amphetamine, ADHD, microdialysis, SNAP-25, knockout, S33084, L-745, 870, L-741, 626
A familiar feature of Attention Deficit Hyperactivity Disorder (ADHD) is the response to psychostimulants such as methylphenidate and amphetamine. In ADHD patients, stimulants reduce excess motor activity and enhance concentration. The reduction in physical activity in ADHD patients after psychostimulant treatment is supported by studies using subjective rating scales and objective measures such as actometers, respiration calorimetry and microwave motor activity detectors (Arnold et al., 1978; Arnold et al., 1972; Butte et al., 1999; Elia et al., 1991; Evans et al., 1986; Rapoport et al., 1978).
Because the primary treatment for ADHD is stimulant medication, research has focused on dopamine. Imaging experiments have identified dopamine transporters predominantly in the caudatoputamen as methylphenidate's site of action (Wang et al., 1995). SPECT and PET studies in ADHD patients have also demonstrated decreased metabolic activity in the basal ganglia (Lou et al., 1990; Lou et al., 1989), a region that contains high concentrations of dopamine and dopamine receptors. Assessments of catecholamine metabolites in cerebral spinal fluid of ADHD children support the imaging studies, demonstrating a positive correlation between the dopamine metabolite homovanillic acid and the degree of hyperactivity (Castellanos et al., 1994).
Although dopamine is implicated in the pathophysiology and treatment of ADHD, there is little evidence implicating specific dopamine receptor subtypes. The lack of identified receptor targets is due primarily to the entirely non-selective action of psychostimulants, which increase the extracellular concentration of monoamines, resulting in the broad activation of many receptor subtypes. In the absence of an unambiguous therapeutic mechanism of action, specific treatment strategies are not forthcoming. Although determining the receptors mediating the positive behavioral effects of psychostimulants in humans would require years of correlative experiments, animal models of ADHD provide an opportunity to explore directly both pathogenic and therapeutic mechanisms.
It is not feasible to reproduce the entire spectrum of a neuropsychiatric disorder in an animal because of the complexity of the behavioral pathology and because some phenotypes are not credibly mimicked in animals. Therefore, animal models of psychiatric disorders, including ADHD, focus on specific quantifiable behavioral features. The pathophysiological insight gained through the analysis of discrete quantifiable behaviors then provides a foundation for understanding more complex features of the disorder. For ADHD, hyperactivity and the response to psychostimulants are features of the disorder convincingly modeled in rodents. All models of ADHD exhibit hyperactivity, including coloboma mice. These mice exhibit inattention, impulsivity and hyperactivity attributable to a hemizygous deletion of Snap25 (Bruno et al., 2007; Hess et al., 1996), a gene that is also associated with ADHD in humans (Barr et al., 2000a; Brookes et al., 2006; Faraone et al., 2005). Similar to its effects on ADHD patients, amphetamine reduces the excess motor activity in coloboma mice. We have previously determined that the broad class of D2-like dopamine receptors, but not D1-like dopamine receptors, mediates the effects of amphetamine in coloboma mice (Fan and Hess, 2007). Here we dissect the D2 dopamine receptor subtypes (D2, D3 and D4) to define receptor-specific regulation of hyperactivity and response to amphetamine.
Materials and methods
Mice
All mouse strains were bred and group-housed at Johns Hopkins University. Coloboma (Cm/+) mice on the C3H/HeSnJ strain were originally obtained from JAX (Bar Harbor, ME). All dopamine receptor knockout mice were originally generated using 129/SV-derived embryonic stem cells and subsequently backcrossed onto the C57BL/6J strain. Dr. Greg Elmer (University of Maryland) generously provided D2 dopamine receptor knockout mice (Kelly et al., 1997). D3 dopamine receptor knockout mice were as described previously ((Xu et al., 1997). D4 dopamine receptor knockout mice (Rubinstein et al., 1997) were obtained from Dr. David Grandy (Oregon Health & Science University).
The coloboma mutation is semidominant. Therefore, coloboma mice lacking D2, D3 or D4 dopamine receptors were bred in a 2-generation cross. First, male coloboma mice were mated with female mice carrying the D2, D3 or D4 null alleles. F1 progeny were genotyped and then male coloboma mice that were also heterozygous for the knockout allele were bred with heterozygous knockout females. The F2 generation therefore consisted of all experimental control and mutant genotypes. For all experiments, mutant and control mice were 2-4 months of age. Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health.
Genotyping
Mice were screened for the D2, D3 and D4 dopamine receptor targeted deletions by PCR of genomic DNA extracted from tail samples. Neo primers were used to identify the null allele. For D2 and D4 receptor PCRs, a single reverse neo primer was embedded in the reaction to differentiate normal from null alleles. The primer sequences were: D2, forward 5′-TGATGACTGGGAATGTTGGTGTGC-3′, reverse 5′-CCGAGCCAAGCTAACACTGCAGAG-3′ and NEO reverse 5′- AGGATTGGGAAGACAATAGCAG-3′ (Diaz-Torga et al., 2002); D3, forward 5′-GCTCACCACTAGGTAGTTG-3′, reverse 5′-ACCTCTGAGCCAGATAAGC-3′, Neo forward 5′-CAAGATGGATTGCACGCAGG-3′, Neo reverse 5′-AGCAAGGCGAGATGACAGGA-3′ (Pritchard et al., 2003); D4, forward 5′-TCTCACATAACCAAAGAAGA -3′ reverse 5′-: CACTGGCGAAGCCACCGCGG -3′, Neo forward 5′-CAAGATGGATTGCACGCAGG-3′. PCR was performed in a volume of 12.5 μl containing 0.2 mM dNTP, 4 mM MgCl2, 0.4 μM – 0.8 μM primer, 50 ng template, and 1.25 U Taq polymerase. Reactions were denatured at 94°C for 5 min prior to 15 cycles consisting of 94°C for 1 min, 67°C (decreasing 1° per cycle) for 2 min, and 72°C for 3 min followed by 25 cycles of 94°C for 1 min, 52°C for 2 min, and 72°C for 3 min with a final 7 min extension at 72°C.
Mice were screened for the Snap25 gene dose by semi-quantitative PCR using the Il1b gene (interleukin 1β), which is also located on mouse chromosome 2, as a within reaction reference. Primers for Snap25 were: forward 5′-CGAAGAAGGCATGAACCATATCAACC-3′ and reverse 5′-GCCCGCAGAATTTTCCTAGGTCCG-3′. Primers for Il1b were: forward 5′ -CCTGAACTCAACTGTGAAATGCCAC-3′ and reverse 5′-GTCCGTCAACTTCAAAGAACAGGTC-3′. PCR was performed in a volume of 12.5 μl containing 0.2 mM dNTP, 5mM MgCl2, 0.5 μM primer, 50 ng template and 1.25 U Taq polymerase. Reactions were denatured at 94°C for 3 min, prior to 23 – 29 cycles of 15 sec at 94°C, 30 sec at 68°C, 30 sec at 72°C, plus a final 7min at 72°C extension. Each sample was subjected to at least 3 PCRs, each with a different number of cycles (23-29). Il1b and Snap25 PCR products from normal (+/+) mice were comparable in intensity on an agarose gel with ethidium bromide visualization; the Snap25 PCR product from coloboma mice was considerably less intense than the Il1b product.
Drugs
Drugs were injected intraperitoneally in a volume of 10 ml/kg. S33084 was a generous gift from Dr. Mark Millan (Servier, France). L-745,870 and L-741,626 were purchased from Tocris (Ellisville, MS). All other drugs were purchased from Sigma (St. Louis, MO).
Locomotor activity
Mice were tested in photocell activity cages (29 × 50 cm) equipped with 12 infrared beams arranged in a 4 × 8 grid (San Diego Instruments; San Diego, CA). Beam breaks were recorded every 10 min. Control and mutant mice were tested simultaneously and were habituated to the cages for at least 4 hr prior to start of the test. Mice had access to food and water ad lib during the entire habituation and test period. Test sessions started 3 hr after the start of the dark cycle.
For amphetamine-antagonist challenge experiments, mice were tested in a repeated measures design. The order of drug doses and vehicle was pseudorandom with each mouse receiving every dose only once within an experiment. Mice were given a 4-day drug holiday between challenges to avoid supersensitivity, as described previously (Fan and Hess, 2007). For dopamine receptor knockout experiments, vehicle and amphetamine were delivered in pseudorandom order.
Stereotypy
During the locomotor activity tests, mice were rated for stereotypy under red light illumination every 10 min for 30 sec. A 0-5 behavioral scale was used: 0 = sleeping; 1 = awake, inactive; 2 = active or exploring; 3 = hyperactive; 4 = hyperactive with bursts of stereotypic behavior; and 5 = continuous persistent stereotypy.
Microdialysis
No-net-flux microdialysis was performed in alert, freely moving mice, as previously described (Fan and Hess, 2007). Briefly, concentric microdialysis probes were constructed as described (Kasim et al., 2006). Mice were anesthetized with Avertin and a microdialysis probe was implanted in the striatum (+0.6 AP, +1.7 ML, 4.5 DV). The probe was perfused overnight with artificial cerebrospinal fluid (aCSF: 147 mM NaCl, 3.5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1 mM NaH2PO4, 25 mM NaHCO3, pH 7.0-7.4) at a flow rate of 0.6 μl/min. No-net-flux microdialysis commenced the following morning. The probe was perfused with aCSF plus 250 μM ascorbic acid and 0, 2, 10 or 20 nM dopamine (Cin) presented in pseudo-random order. After a 25 min equilibration period for each concentration of dopamine, 3 samples (20 min each) were collected (Cout). After completion of the experiment, brains were removed and probe location was confirmed; only animals with probes in the striatum were included.
Samples were stored at -80°C until HPLC analysis. Dopamine concentrations (Cout) were determined by HPLC consisting of an MD-150 column, (150 mm length; 3.2 mm I.D.; ESA, Chelmsford, MA) a 5014B coulometric microdialysis cell plus guard cell (5020) with a mobile phase of 1.7 mM 1-octanesulfonic acid sodium salt, 25 μM EDTA, 75 mM NaH2PO4 and 8% acetonitrile (pH 2.9) at a flow rate of 0.6 ml/min. Extracellular dopamine concentrations were determined using a linear regression analysis of the gain or loss of dopamine from the perfusate (Cin − Cout) versus Cin (Shippenberg et al., 1999; Smith and Justice, 1994).
gamma-Butyrolactone (GBL)
Two groups of mice, each composed of 8 coloboma and 8 control mice, were injected with either saline or 0.5 mg/kg quinpirole. After 5 min, all mice were injected with 750 mg/kg GBL. Five min after GBL treatment, all mice received 100 mg/kg NSD 1015 to inhibit aromatic amino acid decarboxylase. Mice were killed 30 min after NSD 1015 injection and the heads were immediately submerged in dry ice-cooled 2-methylbutane for 1 min. Striata were dissected on ice and frozen at −80°C until HPLC analysis (Koeltzow et al., 1998). Striata were homogenized by sonication at 4°C in 0.4 ml of 100 mM perchloric acid, 2.5 mM EDTA and 250 μM ascorbic acid. After centrifugation at 14,000g for 10 min, the supernatants were filtered through microspin centrifuge filters (0.45 μm). 3,4-dihydroxyphenylalanine (l-DOPA) accumulation was determined by HPLC as described above.
Data analysis
StatView® software version 5.0.1 (SAS Institute, Inc.) was used for all statistical analyses. All data are expressed as means ± SEM. A 2-factor analysis of variance (ANOVA) was used to analyze DOPA accumulation data (genotype × quinpirole treatment) and no-net-flux microdialysis data (genotype × D2 dopamine receptor allele) with post hoc analyses using Student's t tests where appropriate. A 2-factor ANOVA with repeated measures was used to analyze amphetamine-antagonist challenge data (genotype × drug treatment with drug treatment as the repeated measure) and the effect of dopamine receptor knockout on response to amphetamine (dopamine receptor allele × drug treatment with drug treatment as the repeated measure) with post hoc analyses using paired Student's t tests.
Results
D2, but not D3 or D4, dopamine receptors mediate hyperactivity
Coloboma mice exhibit spontaneous hyperactivity that is several-fold greater than the activity of control littermates, although circadian rhythm is normal (Hess et al., 1992). Targeted deletions of the D2, D3 or D4 dopamine receptor were used to determine if specific D2-like dopamine receptor subtypes mediate the hyperactivity in coloboma mice. Baseline locomotor activity of control mice lacking the D2 dopamine receptor (+/+;D2-/-) was comparable to control mice carrying intact D2 dopamine receptor alleles (+/+;D2+/+; Fig. 1a). In contrast, deletion of the D2 dopamine receptor in coloboma mice (Cm/+;D2-/-) significantly reduced the hyperactivity compared to normal coloboma mice (Cm/+;D2+/+; Fig. 1a; ##p < 0.001). Indeed, locomotor activity in coloboma mice lacking D2 dopamine receptors did not differ from control mice. D3 dopamine receptor deficiency had no significant effect on baseline locomotor activity of either control or coloboma mice (Fig. 1b), although there was a trend toward an increase in baseline activity in control mice lacking the D3 dopamine receptor (p = 0.06). Likewise, D4 dopamine receptor deficiency did not affect the baseline locomotor activity of either control or coloboma mice (Fig. 1c).
Fig. 1.
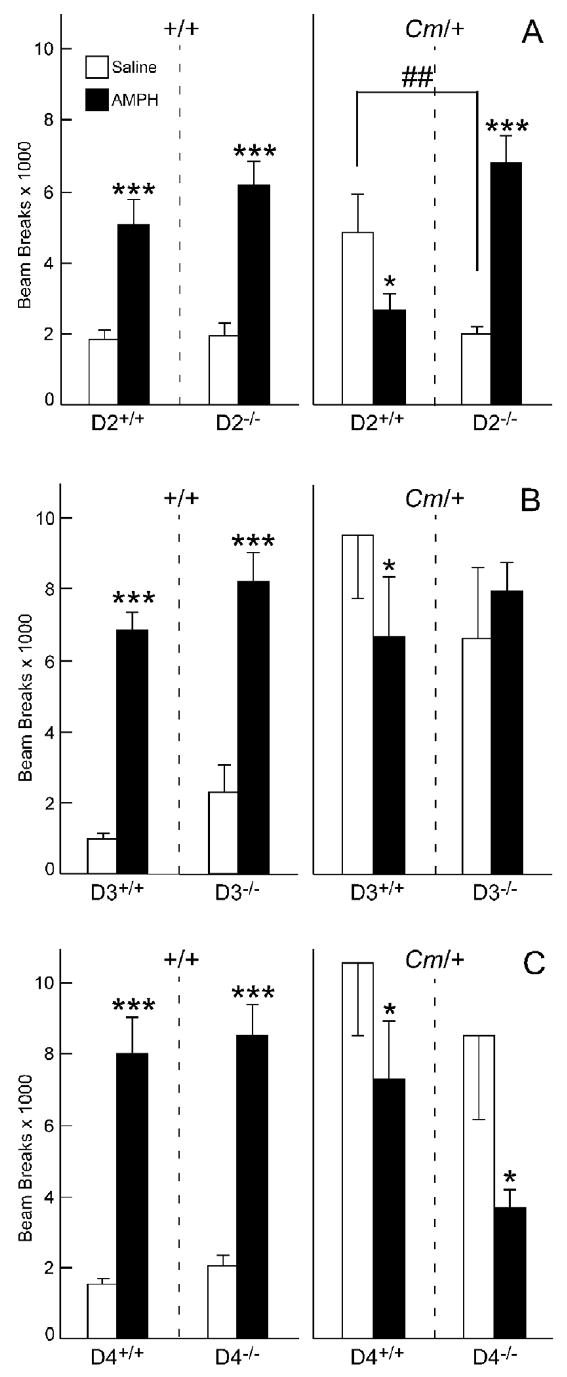
Effect of D2, D3 or D4 dopamine receptor knockout on baseline and amphetamine-mediated locomotor activity in control (+/+) and coloboma (Cm/+) mice. Amphetamine (4 mg/kg) significantly increased locomotor activity (***p < 0.001) in control mice (+/+) with normal (D2+/+, D3+/+, D4+/+) and null (D2-/-, D3-/-, D4-/-) dopamine receptor alleles (panels A, B, and C). Amphetamine significantly reduced locomotor activity (*p < 0.05) in coloboma mice (Cm/+) with normal dopamine receptor alleles (panels A, B, and C). A. D2 dopamine receptor deficiency. In control mice, there was a significant effect of amphetamine treatment (F1,21 = 67.6, p < 0.0001) without significant effect of D2 allele or D2 allele × treatment interactions. In coloboma mice, a significant genotype × treatment interaction effect was observed (F1,19 = 28.2, p < 0.0001). Post hoc analyses demonstrated that baseline locomotor activity was significantly lower in coloboma mice lacking the D2 dopamine receptor than normal coloboma mice (##p < 0.01) and amphetamine significantly increased activity in coloboma mice lacking the D2 dopamine receptor (***p < 0.001). B. D3 dopamine receptor deficiency. There were main effects of D3 allele (F1,22 = 6.7, p < 0.05) and amphetamine treatment (F1,22 = 99.2, p < 0.0001) for control mice, but not coloboma mice. C. D4 dopamine receptor deficiency. For both control and coloboma mice, there was a significant main effect of treatment (+/+:F1,22 = 94.9, p < 0.0001; Cm/+: F1,17 = 10.0, p < 0.01) without significant effects of genotype or genotype × treatment interactions. Locomotor activity in coloboma mice lacking the D4 dopamine receptor was significantly reduced by amphetamine (*p < 0.05, paired Student's t test). Data are beam breaks accumulated 1 hr after saline or drug treatment and are mean ± SEM (n = 9-14/genotype/dose).
It is interesting to note the variability in locomotor activity among the mouse lines in this experiment. Coloboma mice that are inbred on the C3H/HeSnJ strain exhibit more variability in locomotor activity than normal C3H mice, but the overall level of activity is similar between experiments and groups of mice (e.g., compare Figs. 4-6). After breeding coloboma mice with the knockout mice, which are a mix of 129/SV and C57BL/6J, there were clear differences in locomotor activity between experiments, whereby mice bred onto the D2 dopamine receptor knockout line exhibit lower levels of locomotor activity than the other knockout lines. We attribute these differences to background strain effects as the within experiment variability is relatively low and typical for coloboma mice. Regardless of strain effects, the basic pattern of locomotor activity is consistent among all the lines; coloboma mice are clearly hyperactive in all experiments and respond to amphetamine with a significant decrease in locomotor activity.
Fig. 4.
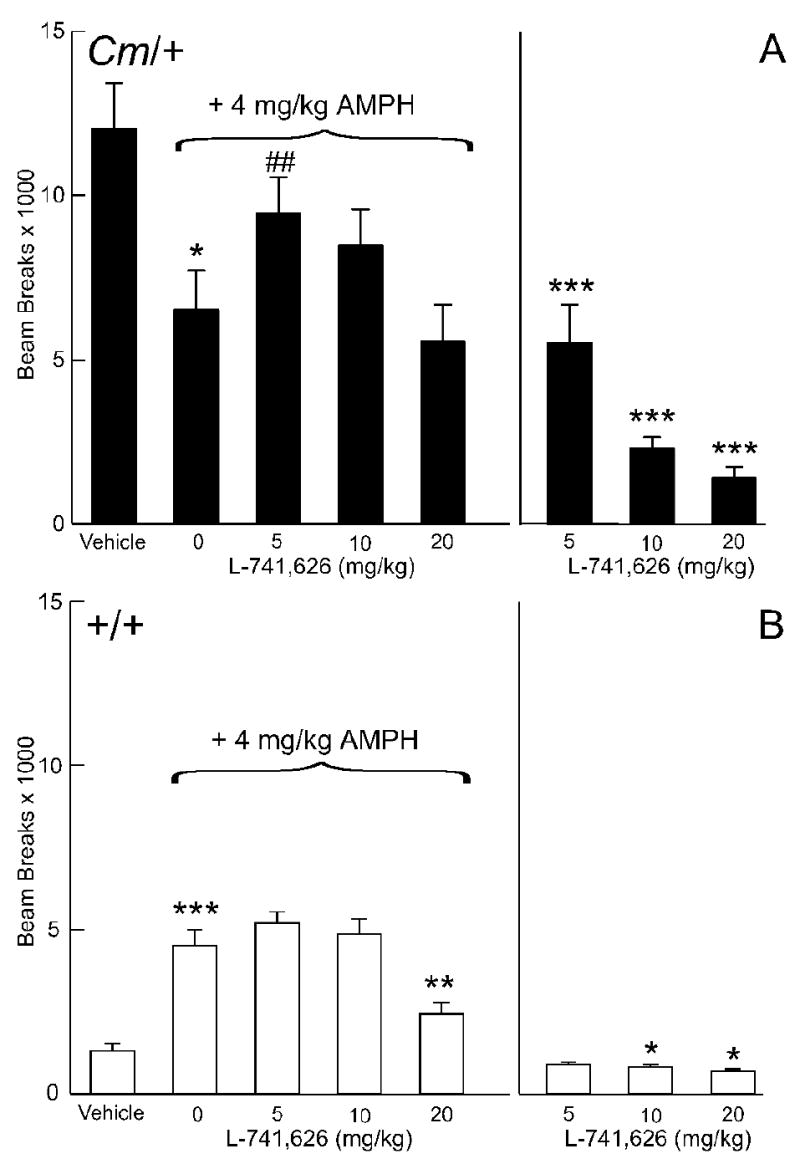
Effect of the D2-selective dopamine receptor antagonist L-741,626 on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with L-741,626. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (***p < 0.001) but significantly reduced locomotor activity in coloboma mice (*p<0.05). There was a significant effect of genotype (F1,14 = 16.5, p < 0.01) and dose of L-741,626 (F3,42 = 10.6, p < 0.0001) on amphetamine-mediated locomotor activity. Post hoc analyses demonstrated a significant increase in amphetamine-mediated locomotor activity after treatment with 5 mg/kg L-741,626 in coloboma mice (## p < 0.01); 20 mg/kg L-741,626 significantly reduced amphetamine-mediated locomotor activity in control mice (**p < 0.01). Treatment with L-741,626 alone produced a significant genotype × dose interaction effect (two-factor ANOVA with repeated measures; F3,42 = 31.7, p < 0.0001). Compared to vehicle treatment, all doses of L-741,626 significantly reduced locomotor activity in coloboma mice (***p < 0.001); 10 and 20 mg/kg significantly reduced the locomotor activity of control mice (*p < 0.05). Data are beam breaks accumulated in 1 hr following drug treatment and are expressed as mean ± SEM (n = 8/genotype/dose).
Fig. 6.
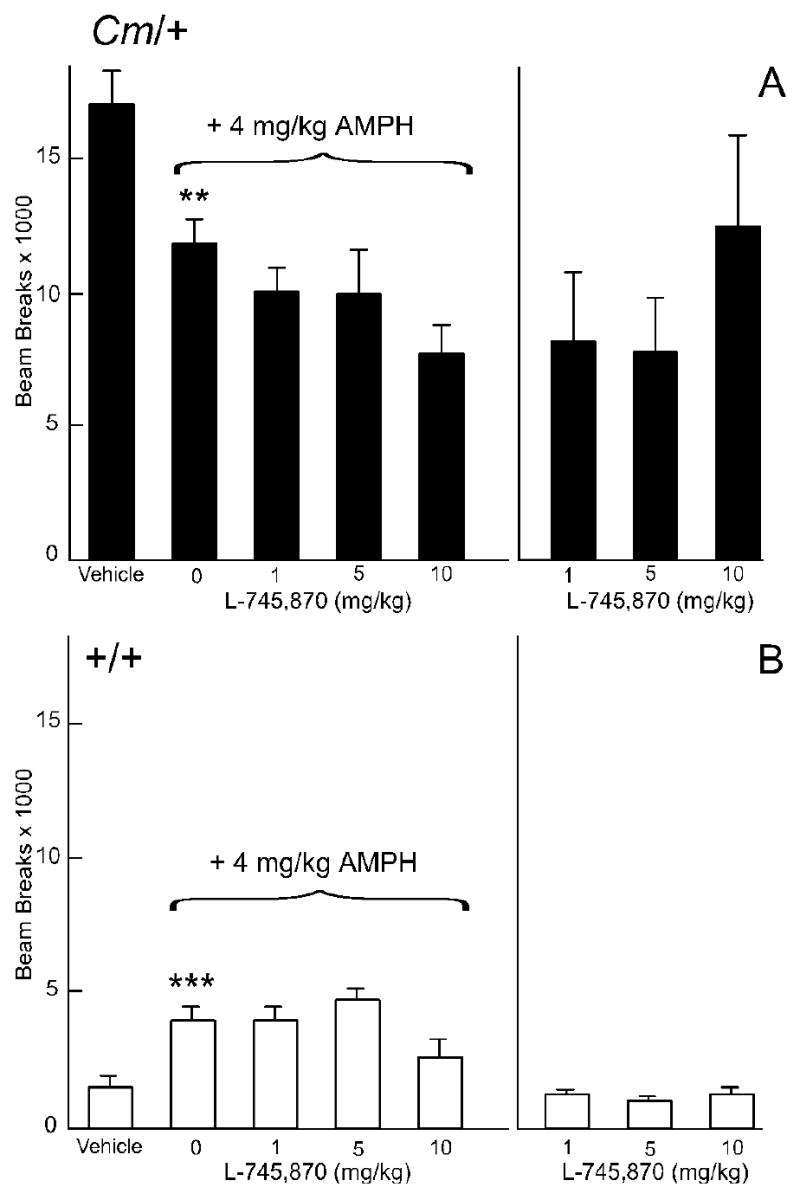
Effect of the D4-selective dopamine receptor antagonist L-745,870 on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with L-745,870. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (***p < 0.001) but significantly reduced locomotor activity in coloboma mice (**p<0.005). There was a significant effect of genotype (F1,12 = 54.9, p < 0.0001) and dose of L-745,870 (F3,36 = 4.1, p < 0.05) on amphetamine-mediated locomotor activity whereby L-745,870 reduced amphetamine-mediated locomotor activity in both control and coloboma mice overall. Post hoc analyses demonstrated a significant decrease in amphetamine-mediated locomotor activity after treatment with 1 mg/kg L-745,870 in coloboma mice (*p < 0.05). There was a significant effect of genotype after treatment with L-745,870 alone (F1,12 = 55.0, p < 0.0001) without significant effect of dose or genotype × dose interactions. However, post hoc analyses demonstrated a significant reduction in locomotor activity of coloboma mice at 5 mg/kg L-745,870 (*p < 0.05). Data are beam breaks accumulated in 1 hr following drug treatment and are expressed as mean ± SEM (n = 7/genotype/dose).
D2 dopamine receptors mediate dopamine overflow in coloboma mice
Extracellular dopamine concentrations in coloboma mice are nearly twice that of normal mice, which may, at least in part, account for the hyperactivity (Fan and Hess, 2007). Because the D2 dopamine receptor null mutation eliminated the hyperactivity in coloboma mice, we investigated whether D2 dopamine receptor deficiency also restored extracellular dopamine concentrations to normal concentrations. Extracellular dopamine concentrations were determined using the no net flux method of microdialysis, which provides an unbiased estimate of transmitter concentration. Consistent with previous results, the extracellular concentration of dopamine in coloboma mice exceeded that of control mice by more than 80% (p < 0.05) whereas the extraction fraction (slope) was comparable in control and coloboma mice (Fig. 2). The extraction fraction is an indirect measure of dopamine reuptake (Smith and Justice, 1994). D2 dopamine receptor deficiency had no effect on either the extracellular dopamine concentration or extraction fraction in control mice. However, the extracellular dopamine concentration in coloboma mice lacking D2 dopamine receptors was significantly reduced compared to coloboma mice with normal D2 dopamine receptors (p < .001). In fact, D2 dopamine receptor deficiency restored the extracellular dopamine concentration in coloboma mice to control levels without changing the extraction fraction (Fig. 2).
Fig. 2.
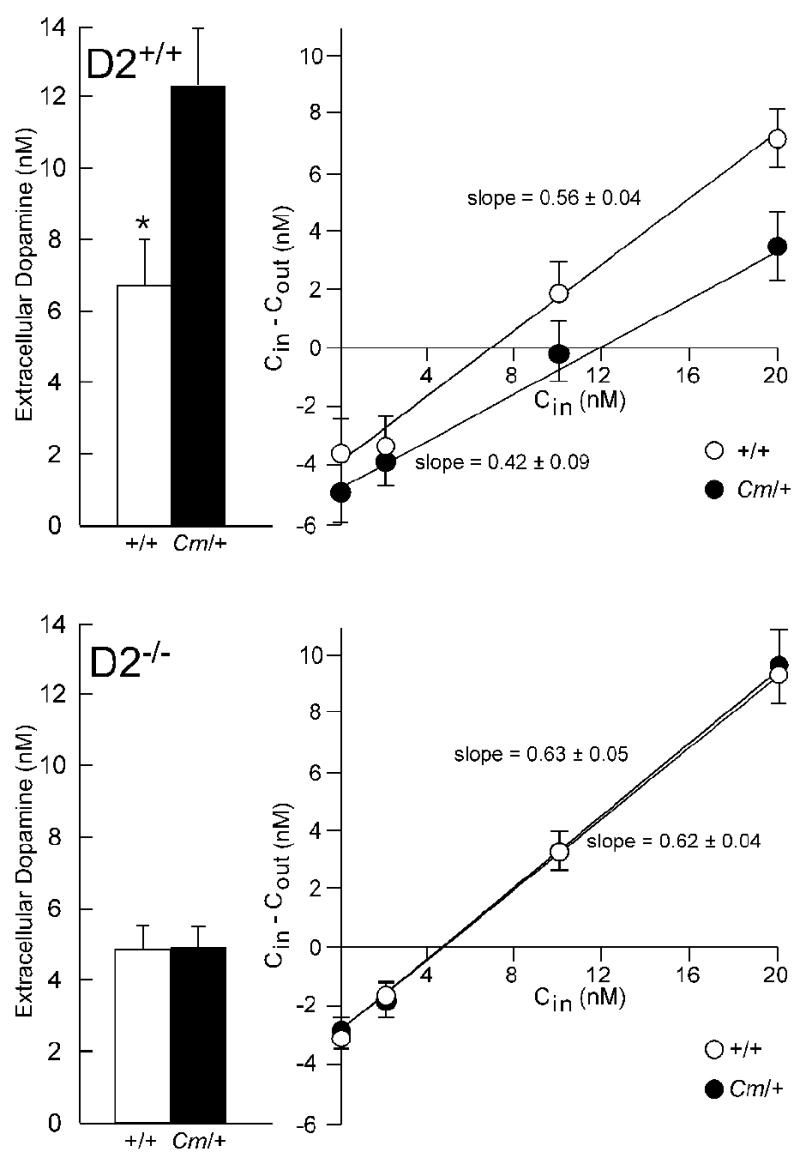
Effect of D2 dopamine receptor deficiency on extracellular dopamine concentrations in control and coloboma mice. A. Striatal dopamine concentrations were measured by no net flux microdialysis in alert freely moving control (+/+) or coloboma mice (Cm/+) carrying normal (D2+/+) or knockout (D2-/-) alleles of the D2 dopamine receptor gene (n=7-8/genotype). There was a significant genotype (+/+ vs Cm/+) × D2 allele interaction effect (F1,27 = 6.0, p < 0.05). Post hoc analyses using Student's t tests demonstrated that dopamine overflow in normal coloboma mice (Cm/+;D2+/+) was significantly higher normal control mice (+/+;D2+/+). No significant difference in dopamine overflow was observed between coloboma mice lacking the D2 dopamine receptor and control mice with or without D2 dopamine receptors (***p < 0.001). B. The extraction fraction, an indirect measure of dopamine reuptake, is the slope of the linear regression analysis of the concentration of dopamine perfused into the microdialysis probe (Cin) versus the perfused dopamine concentration minus the dialysate dopamine concentration (Cin - Cout). No significant differences were observed among the extraction fractions. Values are means ± SEMs.
D2 dopamine autoreceptor function
Dopamine receptors located at the presynaptic terminals of midbrain dopamine neurons regulate dopamine release and synthesis. It is thought that these autoreceptors are D2, but not D3, dopamine receptors (Koeltzow et al., 1998). To determine whether the abnormal dopamine overflow in coloboma mice is attributable to aberrant dopamine autoreceptor function, GBL was used to isolate autoreceptor regulation of dopamine synthesis in vivo. GBL blocks long-loop dopaminergic feedback pathways by inhibiting dopamine neuronal impulses thereby restricting dopamine agonist-induced modulation of dopamine synthesis to terminal autoreceptors (Walters and Roth, 1976). In this method, dopamine autoreceptor regulation of tyrosine hydroxylation is determined via DOPA accumulation in response to D2 dopamine receptor agonists. In the presence of GBL, baseline DOPA accumulation did not differ between control and coloboma mice (Fig. 3). Quinpirole significantly reduced GBL-induced DOPA accumulation in both control and coloboma mice (p < 0.0001) with no difference in the response to quinpirole treatment between control and coloboma mice.
Fig. 3.
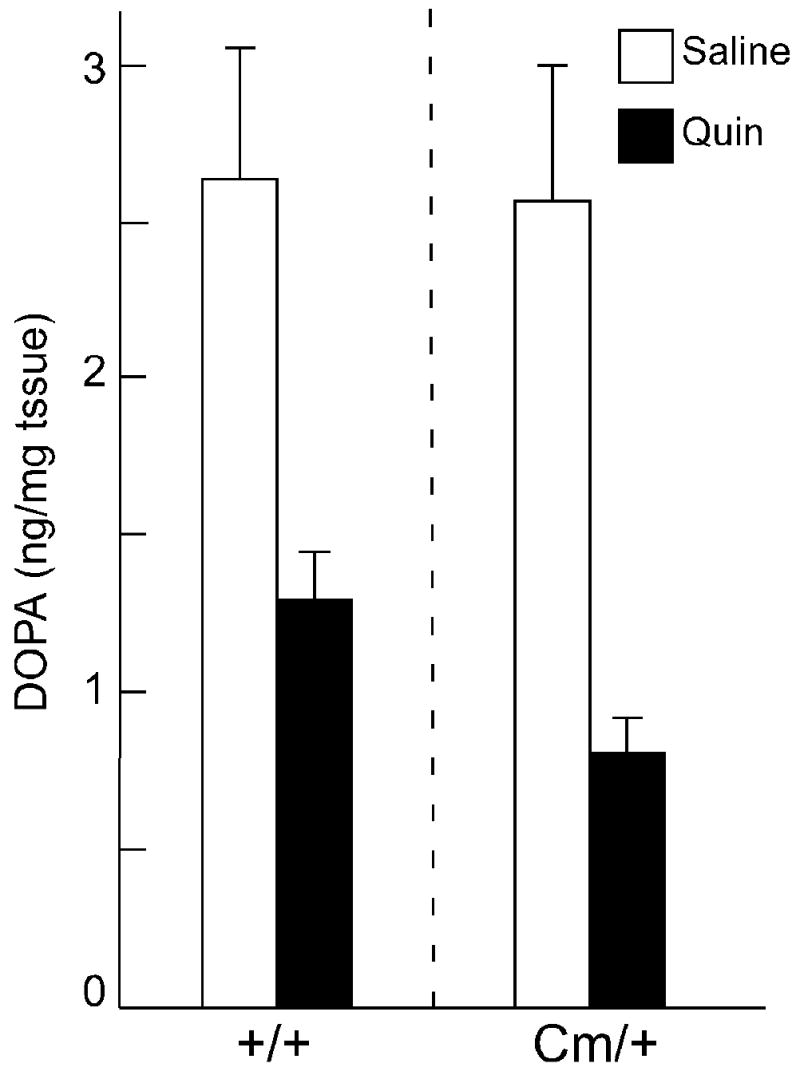
D2 dopamine autoreceptor function in control and coloboma mice. Striatal DOPA accumulation was assessed in the presence and absence of quinpirole (Quin). Basal levels of DOPA accumulation were comparable in control and coloboma mice. Two-factor ANOVA revealed a significant main effect of quinpirole treatment (F1,28 = 24.3, p < 0.0001) without significant effects of genotype or of treatment × genotype interactions. Values are means ± SEMs (n = 8/group).
D2 dopamine receptors mediate the amphetamine-induced reduction in locomotor activity
To determine if the response to amphetamine is dependent on a specific D2-like dopamine receptor subtype, mice were either challenged with amphetamine in the presence of D2, D3 or D4 dopamine receptor subtype-selective antagonists or challenged with amphetamine after breeding D2, D3 or D4 dopamine receptor null mutations onto the coloboma mouse strain. In all experiments, 4 mg/kg amphetamine significantly reduced the locomotor hyperactivity exhibited by coloboma mice (p < 0.05; Figs. 4-6A), consistent with previous results (Hess et al., 1996). Stereotypic behavior was not observed at this dose of amphetamine with any drug treatment or with any null dopamine receptor allele in either control or coloboma mice (data not shown).
In coloboma mice, amphetamine reduces locomotor activity. The D2 dopamine receptor-selective antagonist L-741,626 blocked the amphetamine-induced decrease in locomotor activity in coloboma mice (Fig. 4a). Specifically, 5 mg/kg L-741,626 caused a significant 45% attenuation in the amphetamine-induced reduction in locomotor activity in coloboma mice (p < 0.01). By itself, this dose of L-741,626 significantly reduced locomotor activity in coloboma mice.
In control mice, amphetamine induced an increase in locomotor activity (p < 0.001). Doses of 5 or 10 mg/kg L-741,626 in combination with amphetamine had no significant effect on motor activity in control mice, whereas 20 mg/kg significantly decreased amphetamine-induced locomotor activity (Fig. 4b). Treatment with L-741,626 alone dose-dependently reduced locomotor activity in both control and coloboma mice with a significant genotype × dose interaction effect (p < 0.0001) suggesting that there was a differential response to L-741,626 between genotypes.
Although L-741,626 is selective for D2 dopamine receptors, it is not exquisitely specific. L-741,626 also exhibits some affinity for D3 and D4 dopamine receptor subtypes with ∼40-fold selectivity for D2 over D3 dopamine receptors and ∼100-fold selectivity for D2 over D4 dopamine receptors (Kulagowski et al., 1996). Therefore, the role of D2 dopamine receptors in the response to amphetamine was also tested in null mutants (Fig. 1). Amphetamine induced a significant increase in the locomotor activity of both normal control mice (+/+;D2+/+) and control mice lacking the D2 dopamine receptor (+/+;D2-/-), consistent with a previous report (Chen et al., 2001). There was no significant difference in the response to amphetamine between +/+;D2+/+ and +/+;D2-/- mice. In coloboma mice, the response to amphetamine was more complex. Even on this mixed strain background (C3H/HeSnJ × C57Bl/6J), amphetamine induced a reduction in locomotor activity in normal coloboma mice (Cm/+;D2+/+), similar to that observed in coloboma mice inbred on the C3H/HeSnJ strain (see for example Figs. 4-6). D2 dopamine receptor deficiency in coloboma mice restored baseline locomotor activity to normal levels. In this context, amphetamine produced an increase in locomotor activity in Cm/+;D2-/- mice, whereby the amphetamine response in coloboma mice lacking the D2 dopamine receptor was similar to normal mice (Fig. 1a).
To determine if D3 dopamine receptors play a role in the amphetamine-induced reduction in locomotor activity observed in coloboma mice, the D3 dopamine receptor-selective antagonist S33084 was used in a similar series of amphetamine challenge experiments (Figs. 5a & b). S33084 has good specificity with >1000-fold selectivity for D3 over D4 dopamine receptors and >100-fold selectivity over D2 dopamine receptor (Millan et al., 2000a; Millan et al., 2000b; Millan et al., 2004). S33084 did not change amphetamine-mediated locomotor activity in either control or coloboma mice. Administration of 5 mg/kg S33084 alone significantly reduced basal locomotor activity in coloboma mice (p < 0.001), demonstrating that this dose-range was motorically active. Similar results were obtained with the D3 dopamine receptor-selective antagonist U99194 (not shown).
Fig. 5.
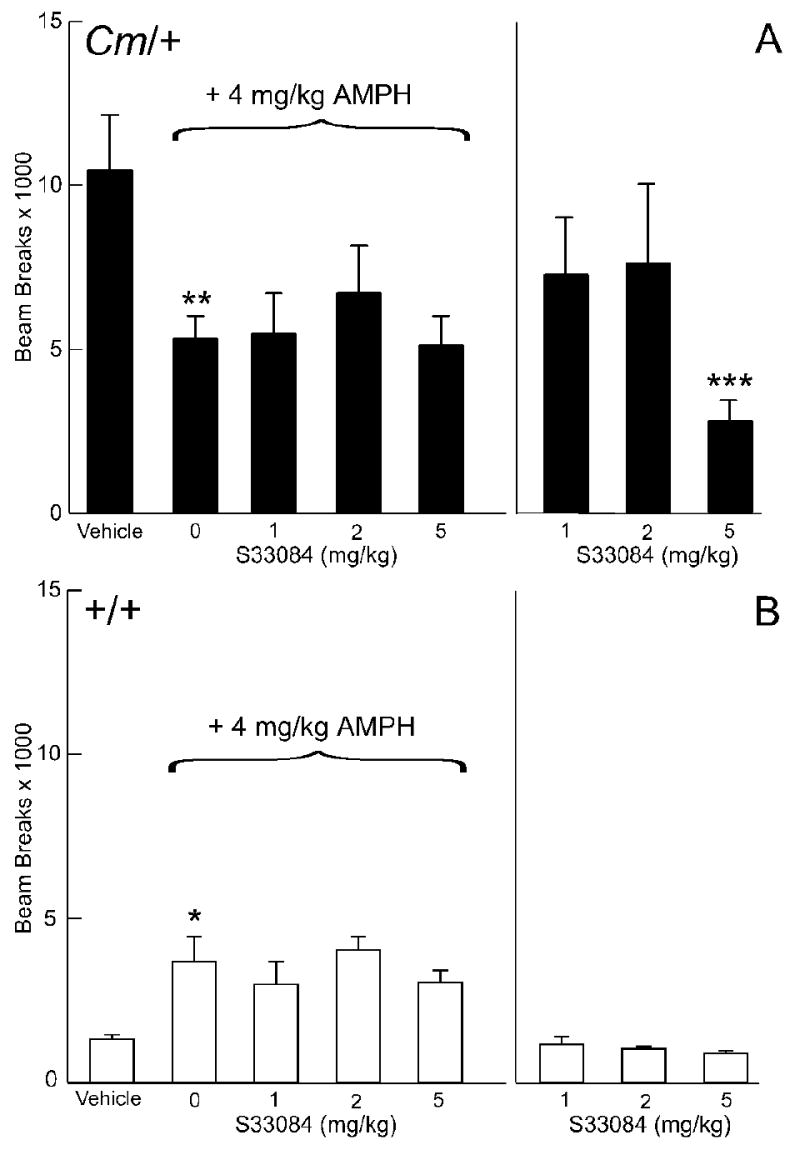
Effect of the D3-selective dopamine receptor antagonist S33084 on amphetamine-mediated locomotor activity. Coloboma (A) and control mice (B) were treated with saline or 4 mg/kg amphetamine and challenged with S33084. Compared to vehicle treatment, amphetamine significantly increased locomotor activity in control mice (*p < 0.05) but significantly reduced locomotor activity in coloboma mice (**p<0.01). There was a significant effect of genotype (F1,14 = 11.2, p < 0.01) without significant effect of dose or genotype × dose interactions. Treatment with S33084 alone produced a significant genotype × dose interaction effect (F3,42 = 8.3, p < 0.001). Compared to vehicle treatment, 5 mg/kg S33084 significantly reduced locomotor activity in coloboma mice (***p < 0.001); the locomotor activity of control mice was unaffected by any dose. Data are beam breaks accumulated in 1 hr following drug treatment and are expressed as mean ± SEM (n = 8/genotype/dose).
D3 dopamine receptor deficiency abrogated the response to amphetamine in coloboma mice; amphetamine neither increased nor decreased locomotor activity in coloboma mice lacking D3 dopamine receptors. In control mice, the response to amphetamine was apparently unaffected by the D3 dopamine receptor deletion (Fig. 1b), although there was an overall trend for an increase in locomotor activity in D3 dopamine receptor null mice (+/+;D3-/-) when the data were collapsed over treatment (p <0.05), consistent with a previous report (McNamara et al., 2006).
The D4 dopamine receptor-selective antagonist L-745,870 was used to test the role of D4 dopamine receptors in the amphetamine-induced reduction in locomotor activity in coloboma mice. L-745,870 has excellent specificity with >1000-fold selectivity for D4 over both D2 and D3 dopamine receptors (Kulagowski et al., 1996). There was a significant overall reduction in amphetamine mediated locomotor activity in response to L-745,870 in both control and coloboma mice (Figs. 6a& b; p < 0.05); post hoc analyses demonstrated that 1 mg/kg L-745,870 significantly reduced amphetamine-mediated locomotor activity in coloboma mice (p < 0.05), with trends toward a reduction in activity at other doses. By itself, 5 mg/kg L-745,870 significantly reduced locomotor activity in coloboma mice, but did not change baseline locomotor activity of control mice. Consistent with the antagonist response, the lack of D4 dopamine receptors had little effect on the response to amphetamine in either control or coloboma (Fig. 1c).
Discussion
Converging lines of evidence from both pharmacologic and genetic manipulation demonstrated that D2 dopamine receptors mediate the hyperactivity, elevated extracellular dopamine concentrations and the response to amphetamine in coloboma mice. The contribution of D3 and D4 dopamine receptors appears minor or irrelevant in this mouse model. Further, we have previously demonstrated that D1-like dopamine receptors do not contribute to the amphetamine-induced decrease in hyperactivity in coloboma mice (Fan and Hess, 2007). Thus, a single dopamine receptor subtype plays a dominant role in mediating this complex neurobehavioral phenotype.
D2 dopamine receptor deficiency reduced the hyperactivity of coloboma mice but did not affect the activity of normal mice. The D2 dopamine receptor subtype is the most abundant form of D2-like dopamine receptors with expression in the basal ganglia far exceeding that of D3 or D4 dopamine receptors. It is therefore not surprising that D2 dopamine receptors have a profound influence on the coloboma phenotype. The failure of D2 dopamine receptor deficiency to reduce locomotor activity in normal mice was surprising as pharmacological blockade of D2 dopamine receptors causes akinesia. Previous reports suggest that D2 dopamine receptor-deficient mice are indeed hypoactive, but the hypoactivity is observed only in novel environments (Baik et al., 1995; Kelly et al., 1998). After extended periods of habituation, similar to our experimental conditions, the locomotor activity of D2 dopamine receptor-deficient mice does not differ from wild type mice, consistent with our results (Clifford et al., 2001; Clifford et al., 2000). Therefore, the reduction in locomotor activity observed in coloboma mice lacking D2 dopamine receptors is a genotype-specific phenomenon, reflecting the combination of Snap25 plus D2 dopamine receptor gene deficiency.
The restoration of normal locomotor activity to coloboma mice after elimination of D2 dopamine receptors is likely mediated by the restoration of normal extracellular dopamine concentrations. However, D2 dopamine receptor deficiency had no effect on extracellular dopamine concentration in otherwise normal mice, consistent with previous reports (Dickinson et al., 1999; Rouge-Pont et al., 2002). The abnormally high extracellular dopamine concentration in coloboma mice is caused by an increase in dopamine release, not by a reduction in reuptake, as determined by no-net-flux microdialysis, suggesting that D2 dopamine receptors are necessary for the release abnormalities caused by the reduction in SNAP-25 expression. As yet, there is no evidence suggesting that D2 dopamine receptors interact directly with SNAP-25, so it is likely that either short-loop or long-loop dopaminergic feedback pathways are involved.
The increase in extracellular dopamine in coloboma mice in the context of a decrease in the expression of SNAP-25, a protein essential for exocytotic transmitter release, is paradoxical. Experiments using GBL excluded abnormalities in dopamine autoreceptor regulation of dopamine synthesis as a source of the increase in extracellular dopamine. Dopamine autoreceptors also mediate the release of dopamine (Galloway et al., 1986; Talmaciu et al., 1986; Wolf and Roth, 1987). We have previously demonstrated that the behavioral response to low doses of apomorphine, which is an indirect measure of autoreceptor-mediated dopamine release, is also intact in coloboma mice (Jones et al., 2001). Taken together, these data suggest that the increase in extracellular dopamine is not likely attributable to local presynaptic (short-loop) abnormalities within the striatum.
It is more likely that the increase in extracellular dopamine results from abnormal long-loop feedback pathways. SNAP-25 is critical for evoked GABA release (Tafoya et al., 2006). In mice, stimulation of afferents arising from the striatum, globus pallidus or substantia nigra pars reticulata causes long-lasting inhibition of dopaminergic neurons (Brazhnik et al., 2008). Deficits in GABAergic signaling in these long-loop feedback pathways caused by a reduction in SNAP-25 expression may disinhibit dopaminergic neurons thereby increasing extracellular dopamine concentrations. This hypothesis, which can be tested in future studies, not only explains the increase in striatal extracellular dopamine concentrations, it also explains the discrepancy between in vitro and in vivo measures of striatal dopamine release in coloboma mice. In contrast to the results of the in vivo microdialysis experiments, dopamine release is attenuated in striatal synaptosomes from coloboma mice (Raber et al., 1997). In synaptosomes, where long-loop feedback and firing rates are irrelevant, the direct biochemical consequence of the SNAP-25 deficit is reflected by the expected reduction in dopamine release. Eliminating D2 dopamine receptors may reduce the inhibitory drive imposed on striatonigral neurons (Bamford et al., 2004; Brown and Arbuthnott, 1983; Cepeda et al., 1993; O'Donnell and Grace, 1994; West and Grace, 2002) to enhance GABAergic signaling in substantia nigra neurons, therefore normalizing dopamine transmission. This hypothesis is likely too simplistic because it ignores important caveats in the interpretation of knockout mouse results including compensatory mechanisms, developmental idiosyncrasies and the complexity of the homeostatic mechanisms regulating basal ganglia signaling. However, because both pharmacological and genetic manipulations resulted in similar outcomes, it is reasonable to conclude that D2 dopamine receptors are integral to the phenotype.
D3 dopamine receptors are thought to damp or inhibit spontaneous and psychostimulant-mediated locomotor activity (De Boer et al., 1997; McNamara et al., 2006; Ouagazzal and Creese, 2000), suggesting a role for these receptors in hyperactivity. There was an overall increase in locomotor activity of D3 dopamine receptor null mutants consistent with previous reports (Accili et al., 1996; McNamara et al., 2006; Xu et al., 1997). However, the D3 dopamine receptor antagonist S33084 had little effect on baseline and amphetamine-stimulated locomotor activity in normal mice. D3 dopamine receptor deficiency in coloboma mice clearly did not enhance spontaneous or amphetamine-mediated locomotor activity. In fact, the trend was toward a reduction in locomotor activity. Further, S33084 did not modify the amphetamine-induced decrease in hyperactivity indicating that D3 dopamine receptors do not directly mediate the response to amphetamine. In contrast, deletion of D3 dopamine receptors in coloboma mice abolished the response to amphetamine suggesting the possibility for an auxiliary role for these receptors. Overall, this receptor does not appear to play a dominant role in the phenotype in these mice nor is the D3 dopamine receptor implicated in ADHD based on genetic association studies in humans (Barr et al., 2000b).
It is surprising that the D4 dopamine receptor apparently did not contribute to either the hyperactivity or the response to amphetamine. The D4 dopamine receptor gene is the only D2-like dopamine receptor associated with ADHD in a meta-analysis of candidate genes (Faraone et al., 2005) and D4 dopamine receptor antagonists reduce locomotor activity in the neonatal 6OHDA model (Zhang et al., 2002; Zhang et al., 2001), suggesting that these receptors are important modulators of locomotor hyperactivity. However, others have demonstrated that D4 dopamine receptor null mutants do not exhibit abnormalities of impulsivity, baseline locomotor activity or overall amphetamine-mediated locomotor activity (Helms et al., 2008; Kruzich et al., 2004), consistent with the results presented here. Although the D4 dopamine receptor may be important for neuronal plasticity as it plays a role in behavioral sensitization to amphetamine (Kruzich et al., 2004) and in the neonatal 6OHDA model (Avale et al., 2004), these receptors appear to have minimal influence on baseline motor activity or acute responses to psychostimulants in normosensitive animals. In light of the very high dopamine overflow elicited by amphetamine and the low abundance of D4 dopamine receptors, it is possible that the influence of the D2 dopamine receptor subtype, which is far more abundant than any other D2-like dopamine receptor, dominates the response and dilutes the effect of other dopamine receptors.
These experiments were designed to identify the specific receptor target(s) of psychostimulants in a model of ADHD. D2 dopamine receptor blockade attenuated the effects of amphetamine while D3 and D4 dopamine receptor antagonism did not modify the response. These results suggest that D2 dopamine receptor-selective agonists are good candidates for a specific therapeutic approach that provides greater mechanistic resolution than psychostimulants in the treatment of ADHD. There are a limited number of D2-like dopamine receptor agonists available for use in humans including pergolide (Permax), ropinirole (Requip), and pramipexole (Mirapex). None are particularly selective for a specific dopamine receptor subtype (Kvernmo et al., 2008). These drugs are most commonly used in the treatment of Parkinson disease (Foley et al., 2004), but are currently used only in exceptional circumstances to treat ADHD plus comorbid conditions. For example, ropinirole and pergolide improve the symptoms of ADHD in children treated with these agonists for restless leg syndrome (Konofal et al., 2005; Walters et al., 2000). Likewise, pergolide not only reduces tics in children, it also improves the ADHD (Gilbert et al., 2003). It appears that D2 dopamine receptor agonists have not been systematically tested for the treatment of ADHD per se, likely due to the occurrence of serious side effect. However, in light of the similarities between the mechanism of action of psychostimulants and dopamine agonists, and considering that other non-stimulant medications with similar mechanisms, including tricyclic antidepressants, norepinephrine reuptake inhibitors and adrenergic agonists, are options for managing ADHD, D2 dopamine receptor specific agonists are obvious candidates. It is likely that with the development of compounds with high specificity, side effects will become more manageable, making D2 dopamine receptor agonists a viable option for the treatment of ADHD.
Acknowledgments
We thank Catherine Weisz for technical support and Dr. Mark Millan (Sevier, France) for the generous gift of S33084. We thank Drs. Elmer and Grandy for supplying dopamine receptor knockout breeder females. Supported by U.S. Public Health Service Grant R01 NS34845.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, Gauda EB, Lee EJ, Cool MH, Sibley DR, Gerfen CR, Westphal H, Fuchs S. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci U S A. 1996;93:1945–9. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold LE, Christopher J, Huestis R, Smeltzer DJ. Methylphenidate vs dextroamphetamine vs caffeine in minimal brain dysfunction. Arch Gen Psychiatry. 1978;35:463–473. doi: 10.1001/archpsyc.1978.01770280073008. [DOI] [PubMed] [Google Scholar]
- Arnold LE, Wender PH, McCloskey K, Snyder SH. Levoamphetamine and dextroamphetamine: comparative efficacy in the hyperkinetic syndrome. Assessment by target symptoms. Arch Gen Psychiatry. 1972;27:816–22. doi: 10.1001/archpsyc.1972.01750300078015. [DOI] [PubMed] [Google Scholar]
- Avale ME, Falzone TL, Gelman DM, Low MJ, Grandy DK, Rubinstein M. The dopamine D4 receptor is essential for hyperactivity and impaired behavioral inhibition in a mouse model of attention deficit/hyperactivity disorder. Mol Psychiatry. 2004;9:718–26. doi: 10.1038/sj.mp.4001474. [DOI] [PubMed] [Google Scholar]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–8. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–63. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Barr CL, Wigg KG, Bloom S, Schachar R, Tannock R, Roberts W, Malone M, Kennedy JL. - Further evidence from haplotype analysis for linkage of the dopamine D4 receptor gene and attention-deficit hyperactivity disorder. Am J Med Genet. 2000a;96:262–7. doi: 10.1002/1096-8628(20000612)96:3<262::aid-ajmg5>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Barr CL, Wigg KG, Wu J, Zai C, Bloom S, Tannock R, Roberts W, Malone M, Schachar R, Kennedy JL. Linkage study of two polymorphisms at the dopamine D3 receptor gene and attention-deficit hyperactivity disorder. Am J Med Genet. 2000b;96:114–117. [PubMed] [Google Scholar]
- Brazhnik E, Shah F, Tepper JM. GABAergic afferents activate both GABAA and GABAB receptors in mouse substantia nigra dopaminergic neurons in vivo. J Neurosci. 2008;28:10386–98. doi: 10.1523/JNEUROSCI.2387-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, Zhou K, Neale B, Lowe N, Aneey R, Franke B, Gill M, Ebstein R, Buitelaar J, Sham P, Campbell D, Knight J, Andreou P, Altink M, Arnold R, Boer F, Buschgens C, Butler L, Christiansen H, Feldman L, Fleischman K, Fliers E, Howe-Forbes R, Goldfarb A, Heise A, Gabriels I, Korn-Lubetzki I, Marco R, Medad S, Minderaa R, Mulas F, Muller U, Mulligan A, Rabin K, Rommelse N, Sethna V, Sorohan J, Uebel H, Psychogiou L, Weeks A, Barrett R, Craig I, Banaschewski T, Sonuga-Barke E, Eisenberg J, Kuntsi J, Manor I, McGuffin P, Miranda A, Oades RD, Plomin R, Roeyers H, Rothenberger A, Sergeant J, Steinhausen HC, Taylor E, Thompson M, Faraone SV, Asherson P, Johansson L. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry. 2006;11:934–53. doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- Brown JR, Arbuthnott GW. The electrophysiology of dopamine (D2) receptors: a study of the actions of dopamine on corticostriatal transmission. Neuroscience. 1983;10:349–55. doi: 10.1016/0306-4522(83)90138-0. [DOI] [PubMed] [Google Scholar]
- Bruno KJ, Freet CS, Twining RC, Egami K, Grigson PS, Hess EJ. Abnormal latent inhibition and impulsivity in coloboma mice, a model of ADHD. Neurobiol Dis. 2007;25:206–216. doi: 10.1016/j.nbd.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte NF, Treuth MS, Voigt RG, Llorente AM, Heird WC. Stimulant medications decrease energy expenditure and physical activity in children with attention-deficit/hyperactivity disorder. J Pediatr. 1999;135:203–7. doi: 10.1016/s0022-3476(99)70023-5. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Elia J, Kruesi MJ, Gulotta CS, Mefford IN, Potter WZ, Ritchie GF, Rapoport JL. Cerebrospinal fluid monoamine metabolites in boys with attention-deficit hyperactivity disorder. Psychiatry Res. 1994;52:305–316. doi: 10.1016/0165-1781(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci U S A. 1993;90:9576–80. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Moratalla R, Impagnatiello F, Grandy DK, Cuellar B, Rubinstein M, Beilstein MA, Hackett E, Fink JS, Low MJ, Ongini E, Schwarzschild MA. The role of the D(2) dopamine receptor (D(2)R) in A(2A) adenosine receptor (A(2A)R)-mediated behavioral and cellular responses as revealed by A(2A) and D(2) receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:1970–5. doi: 10.1073/pnas.98.4.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford JJ, Kinsella A, Tighe O, Rubinstein M, Grandy DK, Low MJ, Croke DT, Waddington JL. Comparative, topographically-based evaluation of behavioural phenotype and specification of D(1)-like:D(2) interactions in a line of incipient congenic mice with D(2) dopamine receptor ‘knockout’. Neuropsychopharmacology. 2001;25:527–36. doi: 10.1016/S0893-133X(01)00246-9. [DOI] [PubMed] [Google Scholar]
- Clifford JJ, Usiello A, Vallone D, Kinsella A, Borrelli E, Waddington JL. Topographical evaluation of behavioural phenotype in a line of mice with targeted gene deletion of the D2 dopamine receptor. Neuropharmacology. 2000;39:382–90. doi: 10.1016/s0028-3908(99)00150-1. [DOI] [PubMed] [Google Scholar]
- De Boer P, Enrico P, Wright J, Wise LD, Timmerman W, Moor E, Dijkstra D, Wikstrom HV, Westerink BH. Characterization of the effect of dopamine D3 receptor stimulation on locomotion and striatal dopamine levels. Brain Res. 1997;758:83–91. doi: 10.1016/s0006-8993(96)01438-2. [DOI] [PubMed] [Google Scholar]
- Diaz-Torga G, Feierstein C, Libertun C, Gelman D, Kelly MA, Low MJ, Rubinstein M, Becu-Villalobos D. Disruption of the D2 dopamine receptor alters GH and IGF-I secretion and causes dwarfism in male mice. Endocrinology. 2002;143:1270–9. doi: 10.1210/endo.143.4.8750. [DOI] [PubMed] [Google Scholar]
- Dickinson SD, Sabeti J, Larson GA, Giardina K, Rubinstein M, Kelly MA, Grandy DK, Low MJ, Gerhardt GA, Zahniser NR. Dopamine D2 receptor-deficient mice exhibit decreased dopamine transporter function but no changes in dopamine release in dorsal striatum. J Neurochem. 1999;72:148–56. doi: 10.1046/j.1471-4159.1999.0720148.x. [DOI] [PubMed] [Google Scholar]
- Elia J, Borcherding BG, Rapoport JL, Keysor CS. Methylphenidate and dextroamphetamine treatments of hyperactivity: Are there true nonresponders? Psychiatry Res. 1991;36:141–155. doi: 10.1016/0165-1781(91)90126-a. [DOI] [PubMed] [Google Scholar]
- Evans RW, Gualtieri T, Hicks RE. A neuropathic substrate for stimulant drug effects in hyperactive children. Clin Neuropharmacol. 1986;9:264–281. doi: 10.1097/00002826-198606000-00005. [DOI] [PubMed] [Google Scholar]
- Fan X, Hess EJ. D2-like dopamine receptors mediate the response to amphetamine in a mouse model. of ADHD Neurobiol Dis. 2007;26:201–211. doi: 10.1016/j.nbd.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, Sklar P. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–23. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Foley P, Gerlach M, Double KL, Riederer P. Dopamine receptor agonists in the therapy of Parkinson's disease. J Neural Transm. 2004;111:1375–446. doi: 10.1007/s00702-003-0059-x. [DOI] [PubMed] [Google Scholar]
- Galloway MP, Wolf ME, Roth RH. Regulation of dopamine synthesis in the medial prefrontal cortex is mediated by release modulating autoreceptors: studies in vivo. J Pharmacol Exp Ther. 1986;236:689–98. [PubMed] [Google Scholar]
- Gilbert DL, Dure L, Sethuraman G, Raab D, Lane J, Sallee FR. Tic reduction with pergolide in a randomized controlled trial in children. Neurology. 2003;60:606–11. doi: 10.1212/01.wnl.0000044058.64647.7e. [DOI] [PubMed] [Google Scholar]
- Helms CM, Gubner NR, Wilhelm CJ, Mitchell SH, Grandy DK. D4 receptor deficiency in mice has limited effects on impulsivity and novelty seeking. Pharmacol Biochem Behav. 2008;90:387–93. doi: 10.1016/j.pbb.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess EJ, Collins KA, Wilson MC. Mouse model of hyperkinesis implicates SNAP-25 in behavioral regulation. J Neurosci. 1996;16:3104–3111. doi: 10.1523/JNEUROSCI.16-09-03104.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess EJ, Jinnah HA, Kozak CA, Wilson MC. Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on Chromosome 2. J Neurosci. 1992;12:2865–2874. doi: 10.1523/JNEUROSCI.12-07-02865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MD, Williams ME, Hess EJ. Abnormal presynaptic catecholamine regulation in a hyperactive SNAP-25-deficient mouse mutant. Pharmacol Biochem Behav. 2001;68:669–676. doi: 10.1016/s0091-3057(01)00481-6. [DOI] [PubMed] [Google Scholar]
- Kasim S, Blake BL, Fan X, Chartoff E, Egami K, Breese GR, Hess EJ, Jinnah HA. The role of dopamine receptors in the neurobehavioral syndrome provoked by activation of L-type calcium channels in rodents. Dev Neurosci. 2006;28:505–17. doi: 10.1159/000095113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, Ben-Jonathan N, Grandy DK, Low MJ. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–13. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci. 1998;18:3470–9. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeltzow TE, Xu M, Cooper DC, Hu XT, Tonegawa S, Wolf ME, White FJ. Alterations in dopamine release but not dopamine autoreceptor function in dopamine D3 receptor mutant mice. J Neurosci. 1998;18:2231–8. doi: 10.1523/JNEUROSCI.18-06-02231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konofal E, Arnulf I, Lecendreux M, Mouren MC. Ropinirole in a child with attention-deficit hyperactivity disorder and restless legs syndrome. Pediatr Neurol. 2005;32:350–1. doi: 10.1016/j.pediatrneurol.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Kruzich PJ, Suchland KL, Grandy DK. Dopamine D4 receptor-deficient mice, congenic on the C57BL/6J background, are hypersensitive to amphetamine. Synapse. 2004;53:131–9. doi: 10.1002/syn.20043. [DOI] [PubMed] [Google Scholar]
- Kulagowski JJ, Broughton HB, Curtis NR, Mawer IM, Ridgill MP, Baker R, Emms F, Freedman SB, Marwood R, Patel S, Ragan CI, Leeson PD. 3-((4-(4-Chlorophenyl)piperazin-1-yl)-methyl)-1H-pyrrolo-2,3-b-pyridine: an antagonist with high affinity and selectivity for the human dopamine D4 receptor. J Med Chem. 1996;39:1941–2. doi: 10.1021/jm9600712. [DOI] [PubMed] [Google Scholar]
- Kvernmo T, Houben J, Sylte I. Receptor-binding and pharmacokinetic properties of dopaminergic agonists. Curr Top Med Chem. 2008;8:1049–67. doi: 10.2174/156802608785161457. [DOI] [PubMed] [Google Scholar]
- Lou HC, Henriksen L, Bruhn P. Focal cerebral dysfunction in developmental learning disabilities. Lancet. 1990;335:8–11. doi: 10.1016/0140-6736(90)90136-s. [DOI] [PubMed] [Google Scholar]
- Lou HC, Henriksen L, Bruhn P, Borner H, Nielsen JB. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch Neurol. 1989;46:48–52. doi: 10.1001/archneur.1989.00520370050018. [DOI] [PubMed] [Google Scholar]
- McNamara RK, Logue A, Stanford K, Xu M, Zhang J, Richtand NM. Dose-response analysis of locomotor activity and stereotypy in dopamine D3 receptor mutant mice following acute amphetamine. Synapse. 2006;60:399–405. doi: 10.1002/syn.20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Dekeyne A, Rivet JM, Dubuffet T, Lavielle G, Brocco M. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther. 2000a;293:1063–73. [PubMed] [Google Scholar]
- Millan MJ, Gobert A, Newman-Tancredi A, Lejeune F, Cussac D, Rivet JM, Audinot V, Dubuffet T, Lavielle G. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: I. Receptorial, electrophysiological and neurochemical profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther. 2000b;293:1048–62. [PubMed] [Google Scholar]
- Millan MJ, Seguin L, Gobert A, Cussac D, Brocco M. The role of dopamine D3 compared with D2 receptors in the control of locomotor activity: a combined behavioural and neurochemical analysis with novel, selective antagonists in rats. Psychopharmacology (Berl) 2004;174:341–57. doi: 10.1007/s00213-003-1770-x. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res. 1994;634:105–12. doi: 10.1016/0006-8993(94)90263-1. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, Creese I. Intra-accumbens infusion of D(3) receptor agonists reduces spontaneous and dopamine-induced locomotion. Pharmacol Biochem Behav. 2000;67:637–45. doi: 10.1016/s0091-3057(00)00406-8. [DOI] [PubMed] [Google Scholar]
- Pritchard LM, Logue AD, Hayes S, Welge JA, Xu M, Zhang J, Berger SP, Richtand NM. 7-OH-DPAT and PD 128907 selectively activate the D3 dopamine receptor in a novel environment. Neuropsychopharmacology. 2003;28:100–7. doi: 10.1038/sj.npp.1300018. [DOI] [PubMed] [Google Scholar]
- Raber J, Mehta PP, Kreifeldt M, Parsons LH, Weiss F, Bloom FE, Wilson MC. Coloboma hyperactive mutant mice exhibit regional and transmitter-specific deficits in neurotransmission. J Neurochem. 1997;68:176–186. doi: 10.1046/j.1471-4159.1997.68010176.x. [DOI] [PubMed] [Google Scholar]
- Rapoport JL, Buchsbaum MS, Zahn TP, Weingartner H, Ludlow C, Mikkelsen EJ. Dextroamphetamine: cognitive and behavioral effects in normal prepubertal boys. Science. 1978;199:560–3. doi: 10.1126/science.341313. [DOI] [PubMed] [Google Scholar]
- Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E. Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. J Neurosci. 2002;22:3293–301. doi: 10.1523/JNEUROSCI.22-08-03293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, He M, Chefer V. The use of microdialysis in the mouse: conventional versus quantitative techniques. Psychopharmacology (Berl) 1999;147:33–4. doi: 10.1007/s002130051138. [DOI] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Tafoya LC, Mameli M, Miyashita T, Guzowski JF, Valenzuela CF, Wilson MC. Expression and function of SNAP-25 as a universal SNARE component in GABAergic neurons. J Neurosci. 2006;26:7826–38. doi: 10.1523/JNEUROSCI.1866-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmaciu RK, Hoffmann IS, Cubeddu LX. Dopamine autoreceptors modulate dopamine release from the prefrontal cortex. J Neurochem. 1986;47:865–70. doi: 10.1111/j.1471-4159.1986.tb00691.x. [DOI] [PubMed] [Google Scholar]
- Walters AS, Mandelbaum DE, Lewin DS, Kugler S, England SJ, Miller M. Dopaminergic therapy in children with restless legs/periodic limb movements in sleep. and ADHD Dopaminergic Therapy Study Group Pediatr Neurol. 2000;22:182–6. doi: 10.1016/s0887-8994(99)00152-6. [DOI] [PubMed] [Google Scholar]
- Walters JR, Roth RH. Dopaminergic neurons: an in vivo system for measuring drug interactions with presynaptic receptors. Naunyn Schmiedebergs Arch Pharmacol. 1976;296:5–14. doi: 10.1007/BF00498834. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Fowler JS, Ding YS, Logan J, Gatley SJ, MacGregor RR, Wolf AP. Comparison of two pet radioligands for imaging extrastriatal dopamine transporters in human brain. Life Sci. 1995;57:187–91. doi: 10.1016/0024-3205(95)02099-5. [DOI] [PubMed] [Google Scholar]
- West AR, Grace AA. Opposite influences of endogenous dopamine D1 and D2 receptor activation on activity states and electrophysiological properties of striatal neurons: studies combining in vivo intracellular recordings and reverse microdialysis. J Neurosci. 2002;22:294–304. doi: 10.1523/JNEUROSCI.22-01-00294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Roth RH. Dopamine neurons projecting to the medial prefrontal cortex possess release-modulating autoreceptors. Neuropharmacology. 1987;26:1053–9. doi: 10.1016/0028-3908(87)90248-6. [DOI] [PubMed] [Google Scholar]
- Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:837–48. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]
- Zhang K, Davids E, Tarazi FI, Baldessarini RJ. Effects of dopamine D4 receptor-selective antagonists on motor hyperactivity in rats with neonatal 6-hydroxydopamine lesions. Psychopharmacology (Berl) 2002;161:100–6. doi: 10.1007/s00213-002-1018-1. [DOI] [PubMed] [Google Scholar]
- Zhang K, Tarazi FI, Baldessarini RJ. Role of dopamine D(4) receptors in motor hyperactivity induced by neonatal 6-hydroxydopamine lesions in rats. Neuropsychopharmacology. 2001;25:624–32. doi: 10.1016/S0893-133X(01)00262-7. [DOI] [PubMed] [Google Scholar]