

Summary
Tribbles homolog 2 (Trib2) was identified as a down-regulated transcript in leukemic cells undergoing growth arrest. To investigate the effects of Trib2 in hematopoietic progenitors, mice were reconstituted with hematopoietic stem cells retrovirally expressing Trib2. Trib2-transduced bone marrow cells exhibited a growth advantage ex vivo and readily established factor-dependent cell lines. In vivo, Trib2-reconstituted mice uniformly developed fatal transplantable acute myelogenous leukemia (AML). In mechanistic studies, we found that Trib2 associated with and inhibited C/EBPα. Furthermore, Trib2 expression was elevated in a subset of human AML patient samples. Together, our data identify Trib2 as an oncogene that induces AML through a mechanism involving inactivation of C/EBPα.
Significance
AML, a heterogenous disease characterized by increased proliferation and a block in differentiation, is commonly associated with chromosomal translocations and transcription factor perturbations. We identified Trib2 as an oncogene that efficiently produces AML in mice. Retroviral Trib2 expression in hematopoietic stem cells perturbed myeloid development, enhanced progenitor proliferation, and directly inhibited the function of C/EBPα, a critical transcription factor that is frequently dysregulated in AML. Trib2 expression was upregulated in a subset of primary patient-derived AML samples, suggesting that it also contributes to the development of human AML. The identification of Trib2 as a potent leukemogen points to alternative pathogenic mechanisms and possible therapeutic opportunities in this aggressive cancer, which is not currently curable in the majority of patients.
Introduction
Acute Myelogenous Leukemia (AML) is a genetically and phenotypically heterogenous disease that is characterized by a block in myeloid differentiation, as well as enhanced proliferation and survival (Kelly and Gilliland, 2002). AML is frequently associated with chromosomal translocations that target transcription factors such as members of the core binding factor (CBF) family, resulting in fusion proteins that include AML1/ETO (t{8;21}), CBFβ/SMMHC (inv{16}) and TEL/AML1 (t{12;21}). Mutations in transcription factors such as PU.1, C/EBPα, AML1 and GATA-1 are also associated with AML, as are other types of oncogenic perturbations that lead to functional inactivation of critical transcription factors (Mueller and Pabst, 2006; Rosenbauer et al., 2005; Tenen, 2003). Recurrent involvement of a limited set of transcription factors suggests that disruption of these genes is rate limiting for leukemia development.
Tribbles was first identified in Drosophila as a gene that is required for gastrulation, oogenesis, and viability (Grosshans and Wieschaus, 2000; Mata et al., 2000; Seher and Leptin, 2000). Tribbles coordinates cell division during gastrulation by promoting turnover of the cell cycle protein String/CDC25, thereby inhibiting premature mitosis. In this context, loss of Tribbles function was associated with increased proliferation whereas over-expression of Tribbles slowed the cell cycle (Mata et al., 2000). Tribbles also promotes the degradation of slbo, the Drosophila homolog of the C/EBP family of basic region-leucine zipper transcription factors, during oogenesis (Rorth et al., 2000). Based on amino acid sequence, Tribbles resembles a serine-threonine kinase; however, it has a variant catalytic core that lacks a canonical ATP binding site and how it functions is unknown.
Mammals have three homologs, Trib1, Trib2 and Trib3. During fasting conditions, Trib3 negatively regulates AKT in the liver (Du et al., 2003) and binds the E3 ligase COP1 to degrade ACC in adipose tissue (Qi et al., 2006). It is also a transcriptional target of the nuclear hormone receptor PPARα (Koo et al., 2004). These findings are consistent with a function downstream of the insulin receptor. Trib3 is also upregulated in tumor cells and in hypoxic conditions (Bowers et al., 2003). Less is known about the other two mammalian Trib family members. When overexpressed, Trib1 can inhibit MEKK-1 mediated activation of AP-1 and stress kinase signaling (Hegedus et al., 2006; Kiss-Toth et al., 2004). Molecular data for Trib2, originally identified as a phosphoprotein in canine thyroid cells (Wilkin et al., 1997), are limited to a study identifying it as a possible autoantigen in autoimmune uveitis (Zhang et al., 2005) and as a differentially expressed gene in kidney mesenchymal cells and in a prostate cancer cell line (Bisoffi et al., 2004; Takasato et al., 2004).
To investigate its function in hematopoiesis, Trib2 was retrovirally expressed in murine hematopoietic progenitors, which were studied for changes in growth and differentiation in vivo and in vitro. This led to long-term growth of myeloid progenitors in vitro and induction of AML in vivo. Our biochemical studies suggest an important function of Trib2 in transformation is to inactivate C/EBPα. Furthermore, an analysis of microarray data generated from 285 AML patient samples identified elevated Trib2 expression in a distinct subset of patients in a cluster with a high frequency of C/EBPα mutations. Together, these data identify Trib2 as a leukemogenic oncogene with the capacity to dysregulate C/EBPα signaling.
Results
Trib2 perturbs myeloid development in vivo
We identified Trib2 as a transcript that was down-regulated after treatment of Notch-dependent murine T-ALL cell lines with gamma secretase inhibitors (manuscript in preparation). As little was known about Trib2 function, we initiated studies to determine its effects on hematopoiesis. Lethally irradiated C57BL/6 (B6) mice were reconstituted with Trib2 or retroviral control vector (MigR1) transduced hematopoietic progenitors as described (Pui et al., 1999). At 9–14 weeks post bone marrow transplant (BMT), MigR1 and Trib2 chimeric mice were similar in terms of bone marrow (BM) and splenic cellularity, the overall percentage of GFP+ cells, and the peripheral white blood cell (WBC) counts (Figure 1A–B, left panels). However, granulocytes (CD11b+Gr-1hi) were reduced in the GFP+ BM population of Trib2 chimeric mice (Figure 1A–B, center panels), and monocytes (CD11b+F4/80+) were significantly increased in the GFP+ population of both the BM and spleen of Trib2 mice (Figure 1A–B, right panels), suggesting that enforced expression of Trib2 promotes monocytic differentiation and inhibits granulocytic differentiation. Further characterization of myeloid subsets showed the presence of increased numbers of CD11b+CD11c+MHCII+ dendritic cells (DCs) and CD11b+F4/80+MHCII+ macrophages in the GFP+ population of Trib2 chimeric mice as compared to MigR1 control mice (Figure S1A and S1B). In contrast to myeloid development, Trib2-GFP+ lymphoid cells obtained from the BM, lymph nodes and thymus did not reveal developmental abnormalities (data not shown). These experiments suggest that enforced Trib2 expression perturbed myeloid development.
Figure 1. Trib2 perturbs myeloid development in vivo and promotes self-renewal in vitro.
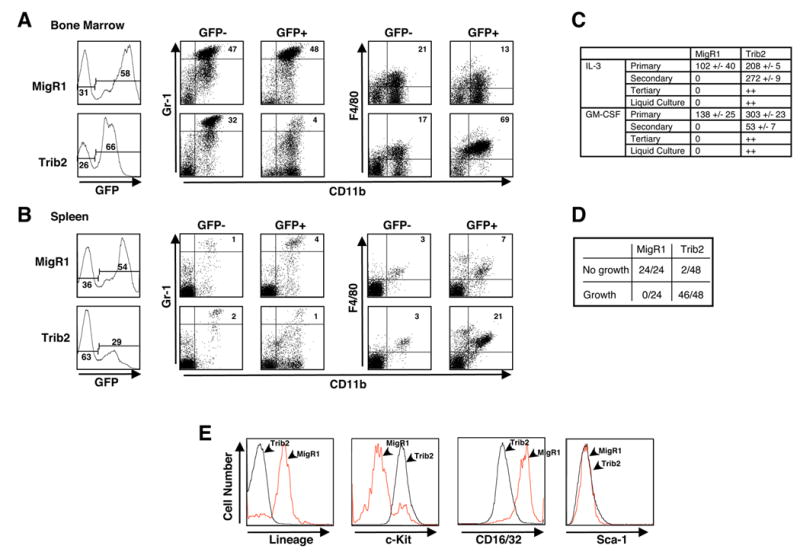
B6 mice were lethally irradiated and reconstituted with BM cells transduced with control MigR1 vector or Trib2. Flow cytometric analysis of MigR1 and Trib2 chimeric mice at 9–14 weeks post BMT.
(A) BM and (B) spleen showing GFP positivity and Gr-1hiCD11b+ neutrophils (percentages given) in the GFP+ and GFP− fractions (left panel). In the right panel, F4/80+CD11b+ monocytes (percentages given) in the GFP+ and GFP− fractions are shown. Results are representative of 3 independent experiments using 6 mice.
(C) GFP+ BM cells (25,000) sorted from MigR1 and Trib2 chimeric mice at week 9 post-BMT were plated in methylcellulose in the presence of the indicated cytokines. Colonies with >50 cells were scored after primary plating (9 days), secondary replating (15,000 cells, 10 days), tertiary replating (15,000 cells, 8 days), and growth in liquid culture (8 days) in triplicate. The mean numbers of colonies, +/− S.E.M, for each condition are tabulated. “++” indicates colony growth or growth in liquid culture.
(D) 5,000 sorted GFP+, lin− BM cells from Trib2 and MigR1 chimeras were plated in triplicate in methylcellulose containing IL-3, IL-6, SCF and GM-CSF. Single colonies from primary MigR1 and Trib2 plates at day 9 were replated in liquid culture plus cytokines (IL-3, IL-6, SCF) in 24 well plates and assessed for growth after 11 days (growth = cell expansion and yellow media).
(E) FACS analysis of MigR1 and Trib2 cells from a representative liquid culture in (D).
Trib2 transduced progenitors exhibit a growth advantage in vitro and establish long-term myeloid progenitor cell lines
To assay the proliferative potential of Trib2 transduced cells in vitro, sorted GFP+ cells from the BM of MigR1 and Trib2 chimeras at 9–10 weeks post-BMT were plated in methylcellulose in the presence of IL-3 or GM-CSF. Significant differences in colony numbers were observed in both IL-3 and GM-CSF (Figure 1C). When replated, Trib2 cells formed secondary colonies in both IL-3 and GM-CSF, whereas MigR1 primary colonies did not. Cells from Trib2 secondary colonies formed tertiary colonies, and cells transferred from these colonies to liquid culture exhibited factor-dependent (IL-3 and GM-CSF) long-term growth (Figure 1C). Lineage-depleted GFP+ BM progenitor cells sorted from MigR1 and Trib2 chimeras were then assayed for proliferation. Single colonies were randomly picked from primary methylcellulose plates and replated in liquid media containing IL-3, IL-6 and SCF. Cells from MigR1 colonies did not proliferate in liquid medium, whereas cells from 46/48 Trib2 colonies exhibited factor-dependent long-term growth in liquid cultures (Figure 1D). These Trib2 expressing cell lines remained factor-dependent and did not produce leukemia when injected into recipient mice (data not shown). The proliferating Trib2 cells did not express lineage markers (Gr-1, Ter119, CD3, CD8, CD4, B220, CD19, CD11c, CD11b) or Sca-1, but did express c-Kit and CD16/32 (FcγIII/II) as compared to MigR1 control cells (Figure 1E). These data suggest that Trib2 expression in myeloid progenitors promotes replating and self-renewal activities.
Mice reconstituted with Trib2 bone marrow cells develop AML
Trib2 chimeric mice died prematurely at a median of 179 days post-BMT (Figure 2A and Table 1, one outlier mouse of N=26). These data include mice from multiple independent BMT experiments performed with different Trib2 retroviral constructs (MigR1-Trib2, MigR1-Trib2-Flag-tagged, tNGFR-Trib2-Flag-tagged). The peripheral blood (PB) prior to death exhibited a leukocytosis consisting of blasts and immature myelomonocytic cells (Figure 2C and Table 1). At necropsy, all mice displayed splenomegaly and lymphadenopathy (Figure 2B and Table 1). Morphologically, the infiltrating cells in these tissues resembled those seen in the PB; mature granulocytes were notably absent (Figure 2C). Histologic sections revealed extensive (>80% in all cases) involvement of the BM by myeloblasts and immature myelomonocytic cells (Figure 2D, top panels). Blasts made up >20% of the BM cellularity in all affected animals, supporting the diagnosis of AML. Leukemic cells found in the spleen, liver, lymph nodes and blood had similar morphologic and immunophenotypic characteristics (data not shown). By immunohistochemical staining, these cells were positive for myeloperoxidase (a marker of myeloblasts and more mature granulocytes) and were negative for terminal deoxytransferase (a marker of lymphoblasts) (Figure 2D, lower panels). Cytochemical stains for non-specific esterase, a marker of monocytes, were negative (data not shown).
Figure 2. Trib2 induces AML.
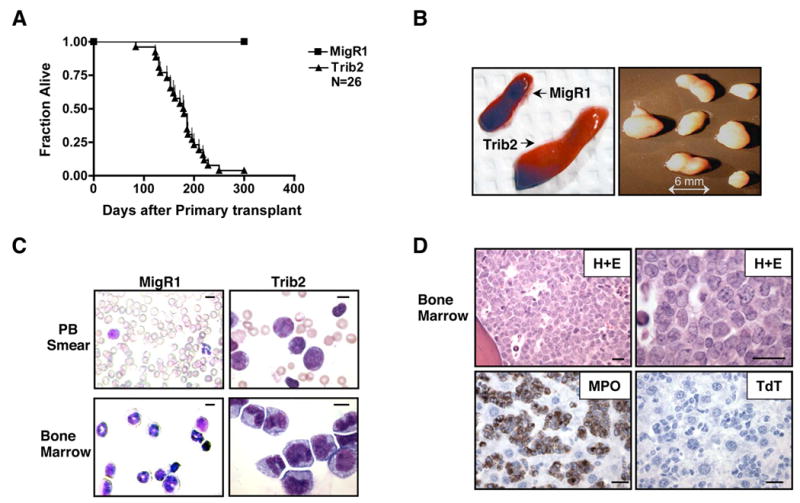
(A) Kaplan-Meier survival curve of mice receiving Trib2 transduced BM compared to MigR1 control. The median survival of Trib2 mice was 179 days. Results are derived from seven independent experiments.
(B) Representative photographs of splenomegaly in Trib2 mice compared to control MigR1 spleen, and lymphadenopathy in Trib2 mice.
(C) Wright-Giemsa stained PB and BM single cell suspensions from MigR1 and leukemic Trib2 mice. Scale bars (upper right) represent 10 μm. The percentage of GFP+ cells in Trib2 BM and spleen was approximately 90–100% and 65–80% respectively.
(D) Histopathology of BM sections from Trib2-induced AML. Hematoxylin and eosin (H+E) section showing hypercellularity (top left) due to the presence of sheets of immature cells and blasts (top right). The tumor cells stain positively for myeloperoxidase (MPO, bottom left) and negatively for terminal deoxytransferase (TdT, bottom right). Scale bars (lower right) represent 20 μm.
Table 1.
Summary of Trib2 primary bone marrow transplants
| Primary Trib2 BMT | Days post transplant (N=25 median=179) | WBC x106 (N=25, range=11–150, median 50) | Spleen WT (g) (N=25, range=0.12–0.66, median 0.41) | Leukemia | Secondary Transplant |
|---|---|---|---|---|---|
| 1 | 123 | 148 | 0.35 | AML | N/D |
| 2 | 146 | 75 | 0.45 | AML | N/D |
| 3 | 180 | 150 | 0.66 | AML | Yes |
| 4 | 153 | 129 | 0.63 | AML | Yes |
| 5 | 153 | 52 | 0.41 | AML | Yes |
| 6 | 162 | 122 | 0.50 | AML | N/D |
| 7 | 179 | 28 | 0.21 | AML | N/D |
| MigR1 | 146 | 7 (N=10, range=3–10) | 0.09 (N=10, range 0.06–0.1) | NO | N/D |
Results from a representative BMT cohort are shown. Days post transplant indicates time of death or time to onset of terminal symptoms consisting of cachexia, decreased activity, and increased WBC counts determined by tail bleeding. A representative MigR1 control mouse (cohort) is shown for comparison. N/D=not done.
Flow cytometric analysis revealed that the leukemic cells were GFP+, relatively large in size (as judged by forward scatter, Figure 3A) and expressed intermediate levels of Gr-1 and CD11b (Figure 3B), as well as c-Kit and F4/80 (Figure 3C). The co-expression of Gr-1 and CD11b at low/intermediate levels is characteristic of immature myeloid precursors and murine myeloid leukemias (Cozzio et al., 2003; de Guzman et al., 2002). The overall combination of morphologic and immunophenotypic findings is consistent with AML.
Figure 3. Analysis of Trib2-induced AML.
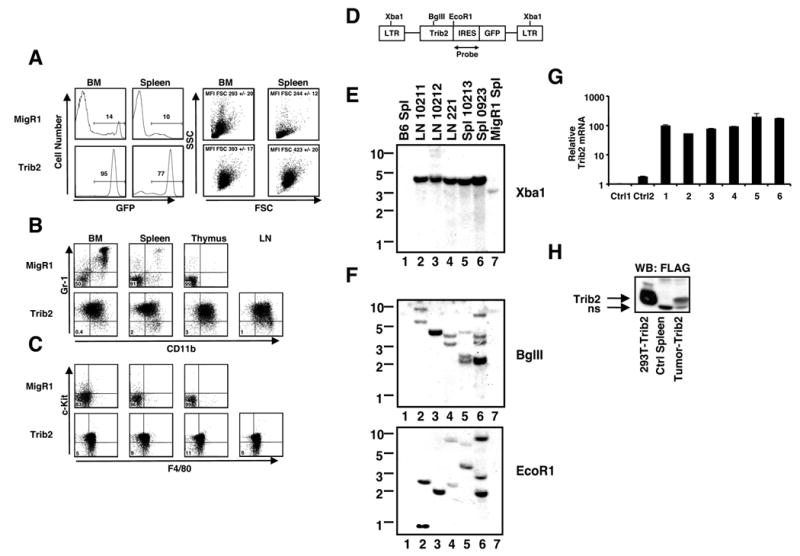
(A) Flow cytometric analysis of GFP expression (percentages given), forward scatter (FSC) and side scatter (SSC) in cells obtained from BM and spleens of leukemic mice. Mean Fluorescence Intensity (MFI) of FSC of the GFP+ cells are shown +/− S.D.
(B–C) Immunophenotypes of primary Trib2-induced AML cells from lymph nodes (LN), thymus, spleen, and BM compared to control MigR1 cells. (B) Flow cytometric analysis of Gr-1 and CD11b expression and (C) c-Kit and F4/80 expression, in the GFP+ fraction of MigR1 (top) and Trib2 (bottom panels) mice. The percentages of cells Gr-1−CD11b− are shown in (B), and the percentages of cells c-Kit-F4/80− are shown in (C). Results are representative of 7 independent experiments.
(D) Schematic of Trib2 provirus and probe used for Southern blotting.
(E–F) Southern blot dectection of proviruses in Trib2 mice. DNA preparations were digested with (E) Xba1 to detect intact provirus (~4kb) and (F) BglII or EcoR1, which cleaved once in the provirus, to detect different proviral integrants. All leukemic samples were obtained from different primary mice; lanes 1 and 7 contain control DNAs from B6 mouse spleen (Spl) and MigR1 control spleen, respectively.
(G) Real-time RT-PCR analysis of Trib2 expression in cDNAs from six primary Trib2-induced AML tumor spleen samples (1–6), using murine Trib2-specific primers. The results are presented as Trib2 mRNA in AML samples relative to expression in control MigR1 spleen cells (Ctrl1, Ctrl2), normalized for 18s rRNA content. Error bars denote +/− SD of each sample measured in triplicate.
(H) Analysis of Trib2 protein expression by western blot. Extracts of primary Trib2-induced AML cells obtained from spleen (>90%GFP, MigR1-Trib2-FLAG) were compared to extracts prepared from control MigR1 spleen cells and 293T cells transfected with pcDNA-Trib2-FLAG. Trib2-FLAG polypeptides were detected on the blot with the FLAG antibody (M2, Sigma). ns=non-specific band.
To detect provirus, DNA was isolated from the lymph nodes and spleens of chimeric mice and analyzed by digestion with restriction endonucleases (Figure 3D). These studies revealed that all tumors contained intact provirus (Figure 3E) and that individual tumors contained one to several distinct proviral insertions (Figure 3F). The intensity of the bands indicated that the tumors were either monoclonal (Figure 3F, lanes 2, 3, 4) or biclonal (Figure 3F, lanes 5, 6), suggesting that additional genetic events are necessary to generate Trib2-induced AML. In addition to GFP (Figure 3A), the tumor cells expressed both Trib2 mRNA (Figure 3G) and protein (38kDa, Figure 3H). Because antibodies against Trib2 are not available, the latter was detected in a cohort of tumors induced with epitope-tagged Trib2 retrovirus.
To further establish the malignancy of Trib2 leukemia, cells (2 x 106) from the BM and spleens of primary leukemic mice were transplanted into sublethally irradiated secondary recipients. All secondary recipients developed AML with an average latency of 36 days (Figure 4A and Table 2). Secondary leukemias were associated with leukocytosis and extensive involvement of the BM, liver, spleen and other tissues (Figure 4B and Table 2). The immunophenotype of the leukemic cells in secondary recipients resembled that of the primary disease, with most cells retaining intermediate expression of Gr-1 and CD11b (Figure 4B) and expression of F4/80 (Figure 4C). In addition, the leukemic cells were c-Kit+, Sca-1−, CD34intermediate and CD16/32+ (FcγIII/II) (Figure 4D). Together, these data indicate that Trib2 is a potent leukemogen.
Figure 4. Trib2-induced AML is 100% transplantable.
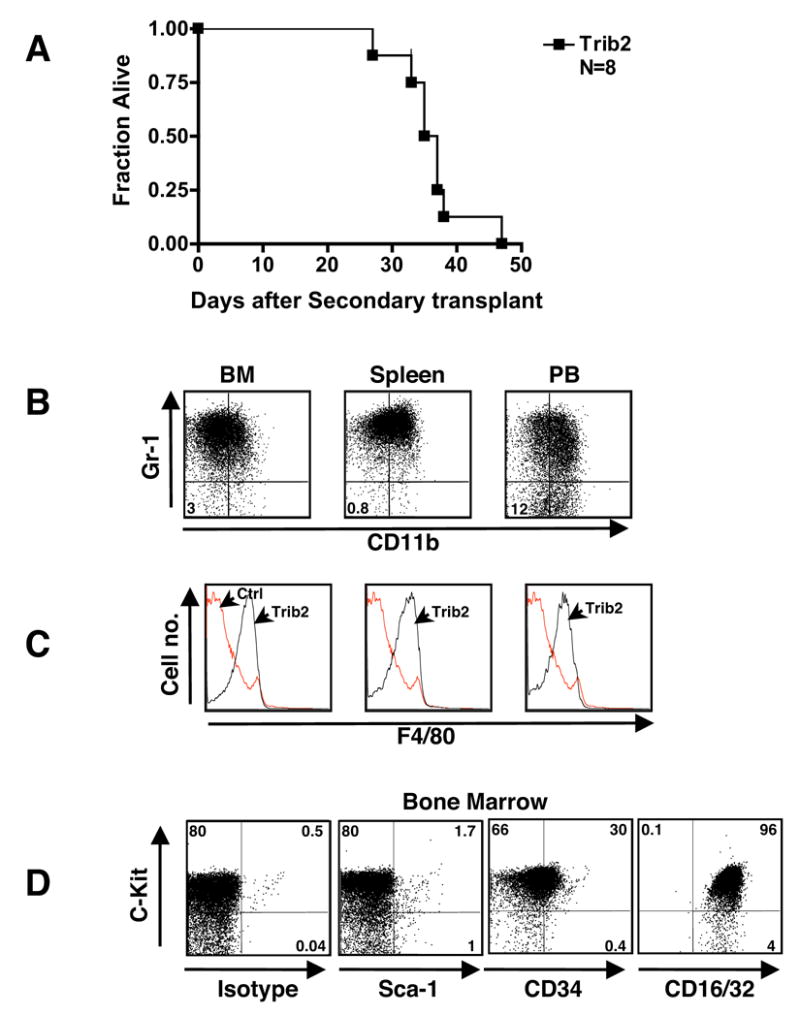
(A) Kaplan-Meier survival curve of secondary transplants. Sublethally irradiated mice (600 rads) received 2 x 106 BM or spleen cells from eight primary leukemic mice.
(B–D) Immunophenotype of Trib2-induced AML after secondary transplant. Cells from BM, spleen and PB were assessed by flow cytometry. (B) The Gr-1 and CD11b profile of the GFP+ population is shown; percentages given are cells negative for both markers. (C) F4/80 profile of the GFP+ Trib2 cells (black line) are overlayed on the normal F4/80 staining profile of B6 control BM cells (red line). Results shown are representative of N=8 mice. (D) The c-Kit and isotype/Sca-1/CD34/CD16/32 profile of the GFP+ population is shown.
Table 2.
Summary of Trib2 secondary transplants
| Source of transferred cells (primary mouse # from Table 1) | Days post transplant (median=36) | WBC x106 | Spleen WT (g) | Leukemia |
|---|---|---|---|---|
| BM (3) | 37 | 20 | 0.70 | Yes |
| Spleen (3) | 27 | 7 | 0.54 | Yes |
| Spleen (4) | 33 | N/A | 0.78 | Yes |
| BM (4) | 37 | 15 | 0.47 | Yes |
| BM (5) | 47 | 17 | 0.61 | Yes |
| Spleen | 35 | 21 | 0.53 | Yes |
| Spleen | 35 | 142 | 0.46 | Yes |
| Spleen | 38 | 25 | 0.53 | Yes |
Days post transplant indicates time of death or time to onset of terminal symptoms consisting of cachexia and/or decreased activity. Leukemia was determined by the presence of greater than 60% myeloblasts in the BM and spleen.
Trib2 reduces normal C/EBPα levels and inhibits C/EBPα function
A deficiency of the transcription factor C/EBPα inhibits granulocyte differentiation and is associated with AML (Tenen, 2003), features similar to the consequences of Trib2 expression in hematopoietic progenitors. Furthermore, Drosophila Tribbles can promote the degradation of Slbo, the Drosophila homolog of C/EBP, during oocyte border cell migration (Rorth et al., 2000). To determine whether Trib2 alters C/EBPα protein levels in myeloid cells, western blot analysis was performed on whole cell extracts prepared from 32D and U937 cells transduced with MigR1 or Trib2. In both cell lines, Trib2 expression reduced levels of the C/EBPαp42 full-length protein and concomitantly increased levels of C/EBPαp30 (Figure 5A), a dominant negative variant of C/EBPαp42 that is thought to arise from an internal translation initiation site (Calkhoven et al., 2000).
Figure 5. Trib2 reduces normal C/EBPα levels and inhibits its activity.

(A) Sorted GFP+ 32D and U937 cells transduced with either MigR1 or Trib2 were assessed for C/EBPα protein expression by western blot. C/EBPαp42 is the full-length protein and C/EBPαp30 is the truncated protein. Actin is the protein loading control.
(B) Analysis of C/EBPαp42 and p30 protein expression in primary leukemic samples from BM (93% GFP+), spleen (63% GFP+) and lymph node (88% GFP+) compared to normal levels expressed in CMPs and GMPs from B6 BM (left panel). Levels of C/EBPαp42 and p30 proteins were also compared in unfractionated normal B6 mouse BM cells and primary (94% GFP+) and secondary (98% GFP+) leukemic BM samples (right panel).
(C) Schematic of the IL-12 promoter containing the C/EBPα binding site.
(D) RAW264.7 macrophages were co-transfected with i) the IL-12 promoter firefly luciferase constructs containing the C/EBP WT or mutant binding sites, ii) either empty vector, Trib2, C/EBPα or both Trib2 and C/EBPα, and iii) a pRL-TK Renilla luciferase internal control plasmid. Luciferase activity was measured following LPS (100 ng/ml) treatment for 8 hrs, 24 hrs post-transfection. Reporter luciferase activity for each sample was normalized to the Renilla luciferase activity of the same sample. Data presented are mean +/− SD of triplicate cultures.
(E) C/EBPα DNA binding activity was assessed by EMSA using a double-stranded oligonucleotide probe containing the C/EBP binding site from the human G-CSF receptor. Nuclear extracts from sorted GFP+ U937 cells transduced with MigR1 (lanes 1, 2), Trib2 (lanes 3, 4), or C/EBPα (lanes 5, 6), were incubated with 32P-labeled probe. In lanes 2, 4, and 6, 2 μL of C/EBPα antibody was added. SS indicates the supershifted C/EBPα complex. The extracts used in lanes 1, 3, and 5 were tested with an OCT-1 probe in a second EMSA assay as a control for the integrity and quantity of nuclear binding proteins.
Human AML is associated with mutations in C/EBPα that lead to decreased levels of normal C/EBPαp42 and increased levels of C/EBPαp30 (Leroy et al., 2005; Pabst et al., 2001b). C/EBPαp30 dominantly inhibits C/EBPαp42 function when the ratio of C/EBPαp42 to C/EBPαp30 is less than one (Calkhoven et al., 2000). In contrast to normal CMPs and GMPs (Akashi et al., 2000), high levels of C/EBPαp30 and low ratios of C/EBPαp42 to C/EBPαp30 were present in all Trib2 primary leukemic samples (Figure 5B, left panel). The inverted ratio of C/EBPαp42 to C/EBPαp30 protein persisted in serially passaged leukemic cells (Figure 5B, right panel). The identity of C/EBPαp30 was confirmed by performing additional western blots with an antibody specific for the N-terminal C/EBPα epitope, which detected only C/EBPαp42 (data not shown).
To show that the relative decrease in C/EBPαp42 translated into diminished functional activity, we investigated whether Trib2 inhibited activation of an IL-12 promoter luciferase reporter by C/EBPα. This reporter contains a single C/EBPα binding site, which is involved in the induction of IL-12 transcription in RAW cells following LPS exposure (Figure 5C). Trib2 inhibited the stimulation of an IL-12 reporter construct by both C/EBPαp42 and LPS (Figure 5D). In both experiments, the stimulation of reporter gene activity by C/EBPα or LPS required an intact C/EBPα binding site (Figure 5D). Trib2 had no effect on an NFκB-sensitive luciferase reporter gene (Figure S2A), suggesting that the effect of Trib2 was specific for C/EBPα. Furthermore, the IL-6 promoter also contains a functional C/EBPα binding site, and BM-DCs and macrophages derived from Trib2 chimeric mice released less IL-12 and IL-6 in response to LPS than did MigR1 control cells (Figure S2B–E). To further characterize the relationship between C/EBPα and Trib2, the C/EBPα DNA binding activity was assayed in extracts prepared from transduced U937 cells. The C/EBPα-DNA-binding activity was markedly reduced by Trib2 (Figure 5E, lanes 1 and 2), with the specificity of the observed gel shift being confirmed by production of a "supershift" upon addition of an antibody against C/EBPα (Figure 5E, lane 2 and 4). These data demonstrate that Trib2 expression inhibits the DNA binding function of C/EBPαp42.
As Trib2 over-expression decreased C/EBPαp42, we investigated whether siRNA-mediated knockdown of endogenous Trib2 would increase endogenous C/EBPαp42. Indeed, human U937 cells transfected with siRNA against human Trib2 had reduced Trib2 mRNA expression (Figure 6A) and a concomitant increase in C/EBPαp42 protein (Figure 6B). C/EBPαp30 is not detectable in these cells (see Figure 6D). Moreover, siRNA-mediated knockdown of retrovirally expressed murine Trib2 in U937 cells reduced murine Trib2 expression (Figure 6C) and abrogated its inhibitory effect on C/EBPαp42 (Figure 6D). Trib2 knockdown restored the C/BPαp42:C/EBPαp30 ratio by increasing C/EBPαp42 expression. The failure to observe a concomitant decrease in C/EBPαp30 may stem from a long C/EBPαp30 half-life, residual Trib2 activity or other unknown factors, and is a point that requires further investigation. Nevertheless, these data complement the Trib2 overexpression studies and further support the hypothesis that Trib2 opposes C/EBPαp42 function.
Figure 6. Trib2-dependent inhibition of C/EBPα expression and function is abrogated by siRNA-mediated knockdown of Trib2 and over-expression of C/EBPα.
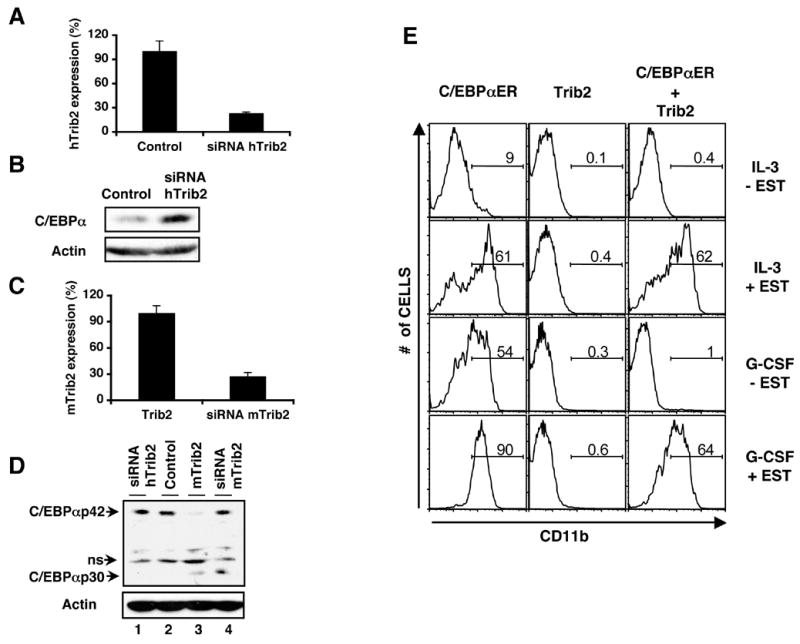
(A–B) siRNA designed to knockdown human Trib2 (hTrib2) was electroporated into U937 cells and analyzed 24 hr later. (A) hTrib2 expression assessed by real-time RT-PCR. Error bars denote +/− S.E.M of each sample measured in triplicate. (B) Western blot of C/EBPα protein expression. Lane 1=negative control siRNA and lane 2=hTrib2 siRNA. C/EBPαp30 is undetectable in parental U937 cells.
(C–D) siRNA designed to knockdown murine Trib2 (mTrib2) was electroporated into U937 cells retrovirally expressing murine Trib2 and analyzed 24 hr later. (C) mTrib2 expression assessed by real-time RT-PCR. Error bars denote +/− S.E.M of each sample measured in triplicate. (D) Western blot of C/EBPαp42 and C/EBPαp30 protein expression. Lanes 1 & 2=U937 cells + human Trib2 siRNA and negative control siRNA respectively as in (B); lanes 3 & 4=U937-mTrib2 cells + negative control siRNA and siRNA mTrib2 respectively. ns=non-specific band
(E) 32D cells transduced with C/EBPαER (GFP)α Trib2 (tNGFR), or C/EBPαER+Trib□ were sorted for GFP and/or tNGFR expression. Sorted cells were plated in equal numbers (1x105) (day 0, 48 hrs post-transduction) in IL-3 or G-CSF +/− 1μM β-estradiol (EST). CD11b expression was assessed after 2 days. Data are representative of 3 independent experiments.
To further investigate the link between Trib2's inhibitory effects on granulopoiesis (Figure 1A) and C/EBPαp42 activity, we co-expressed Trib2 and C/EBPαp42 in 32D cells, which require IL-3 to survive and undergo C/EBPαp42-dependent granulocytic differentiation in response to G-CSF (Wang et al., 1999). To control C/EBPαp42 activity, we utilized an estradiol-inducible C/EBPαp42 fusion gene (C/EBPαER) cloned in MigR1 (Cammenga et al., 2003). In these experiments, Trib2 was expressed in a MSCV retrovirus that also bears a tNGFR gene as a surrogate marker (Izon et al., 2001). 32D cells were sorted for retroviral expression of GFP (C/EBPαER), tNGFR (Trib2), or both, treated with either estradiol or vehicle in the presence of either IL-3 or G-CSF, and then assayed for granulocytic differentiation by both flow cytometry (Figure 6E) and morphology (data not shown). As expected, vehicle-treated cells transduced with C/EBPαER differentiated only in the presence of G-CSF (induction of endogenous C/EBPα activity), whereas estradiol (induction of exogenous C/EBPα activity) induced the differentiation of these cells in the presence of either G-CSF or IL-3 (Figure 6E). Further, cells transduced with Trib2 failed to differentiate in response to either G-CSF or IL-3, as did cells that were co-transduced with Trib2 and C/EBPαER in the presence of vehicle. In contrast, the inhibitory effect of Trib2 in doubly transduced cells was overcome by estradiol treatment, indicating that high doses of C/EBPα are dominant to the inhibitory effects of Trib2.
Cell proliferation and survival were also assayed in these cultures by cell counting and trypan blue exclusion. 32D-C/EBPαER cells proliferated in IL-3, and this proliferation was inhibited by either endogenous or exogenous C/EBPα activity (Figure S3, left bars). 32D-Trib2 cells proliferated in the presence of IL-3 but died in the presence of G-CSF (Figure S3, middle bars), an effect that was similar to transduction of 32D cells with a C/EBPα DNA binding mutant that was unable to induce differentiation (Keeshan et al., 2003). Cell numbers/viability were restored following estradiol induction of C/EBPα in the Trib2 + C/EBPαER expressing cells (Figure S3, compare 2 far right bars). Similar to the CD11b expression data, these results also show that high doses of C/EBPα are dominant to Trib2’s inhibitory effects.
We next sought to determine the mechanism by which Trib2 decreased normal C/EBPαp42 levels. In 32D and U937 cells, C/EBPαp42 levels in Trib2 expressing cells were restored by pre-treatment with the proteasome inhibitor MG132 (Figure 7A). In co-immunoprecipitation experiments using 293T cells, Trib2 association with C/EBPαp42 was detected only in the presence of MG132, whereas C/EBPαp30 co-immunoprecipitated in the presence and absence of MG132 (Figure 7B, lane 4 and 5). Expression of Trib2 in U937 cells decreased the half-life of endogenous C/EBPα protein as determined by 35S metabolic pulse labeling and immunoprecipitation chase of labeled C/EBPα (Figure 7C). These data suggest that Trib2 acts by associating with C/EBPαp42 and promoting its degradation via the proteasome.
Figure 7. Trib2 forms a complex with C/EBPα and results in its proteosomal-dependent degradation.
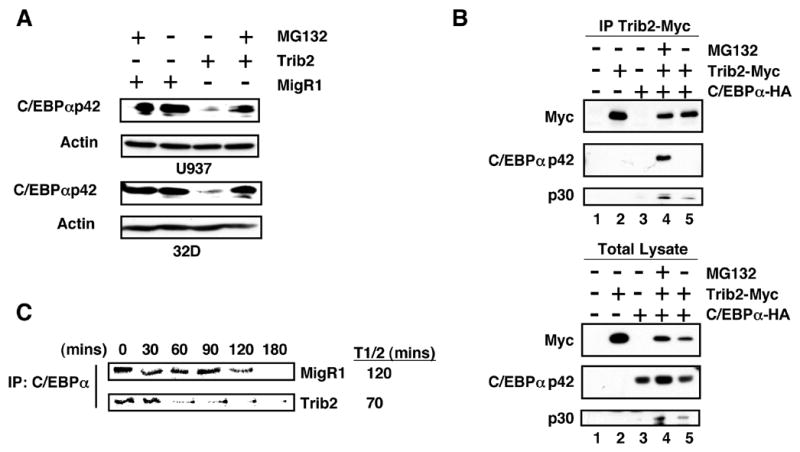
(A) Sorted GFP+ U937 (top panel) and 32D (lower panel) cells transduced with either MigR1 or Trib2, treated +/− 10 μM MG132 for 2 hrs were assessed for C/EBPα expression by western blot.
(B) 293T cells were transfected with empty vector (lane 1), myc-tagged Trib2 (lane 2), HA-tagged C/EBPα (lane 3), or co-transfected with both (lanes 4 and 5), and treated with 10 μM MG132 for 2 hrs (lane 4). Trib2 was immunoprecipitated using a Myc 9E10 antibody and western blotting performed with HA and Myc antibodies on immunoprecipitates (top panel) and total lysates (lower panel).
(C) U937 cells transduced with MigR1 or Trib2 were metabolically labeled with 35S-methionine for 60 min and chased for the indicated times. C/EBPα was immunoprecipitated and radiolabeled protein detected by SDS-PAGE. The half-life (T1/2) was calculated using ImageJ software.
Elevated Trib2 expression is found in a subset of AML patients exhibiting C/EBPα defects
Since Trib2 is a potent inducer of AML in our experimental model, we investigated Trib2 expression in a mRNA expression database derived from 285 AML patient samples. Previous analysis of gene expression in this data set identified 16 groups of AML with distinct gene expression profiles (Valk et al., 2004). Based on signals from two different Trib2 probe sets, we noted that elevated Trib2 expression was observed in cluster #4 (Figure 8), which is one of two clusters that are associated with C/EBPα mutations (Valk et al., 2004). Although cluster 4 contains AML samples with wild-type (green) or mutated (red) C/EBPα, Trib2 expression was primarily elevated in tumors with wild-type C/EBPα (Figure 8, enlarged area). Thus, in an unbiased screen of AML samples, high levels of Trib2 expression were preferentially found in a cohort associated with C/EBPα defects. This association again links Trib2 to tumors with altered C/EBPα function, and suggests that elevated Trib2 may have a pathogenic role in a subset of human AML.
Figure 8. Trib2 is elevated in a subset of human AML.
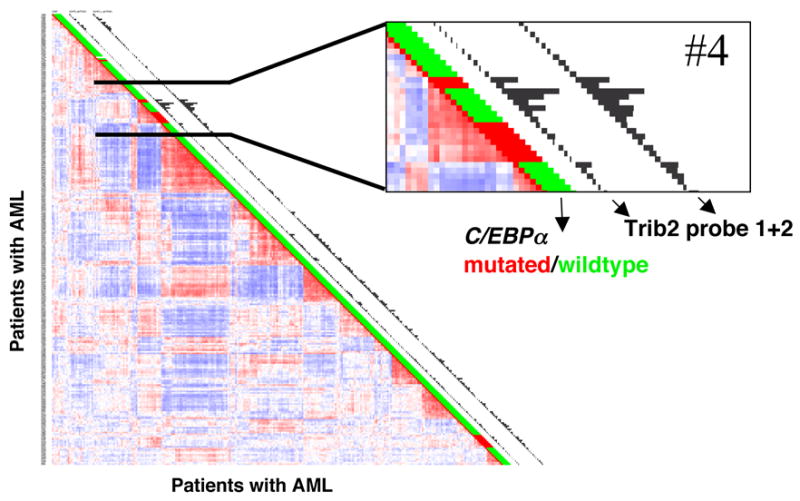
Correlation view of 285 AML patients (Valk et al. 2004). Colors of cells relate to Pearson’s correlation coefficient values, red indicates higher positive and blue indicates higher negative correlation between samples. Sixteen clusters represented by red blocks along the diagional can be identified. C/EBPα mutation status is indicated next to each tumor (red = mutant, green = wild type). Histograms next to each tumor represent expression levels of the two probe sets for Trib2. Cluster 4, one of the two clusters harboring most patients with C/EBPα mutations, has a significantly elevated expression of Trib2 relative to other clusters.
Discussion
Our data provide strong evidence that dysregulated Trib2 expression contributes to the pathogenesis of AML. Retroviral expression of Trib2 immortalized hematopoietic progenitors in vitro and induced fatal transplantable AMLs in murine recipients with a mean latency of 179 days. In accordance with the central role of Trib2 in the development of these tumors, we have never observed AML in several hundred mice that have received MigR1-transduced BM cells using identical protocols. Microarray analysis of a cohort of primary human AMLs identified elevated Trib2 mRNA expression in a cluster of tumors associated with a high frequency of C/EBPα mutations. Although we have not yet determined the mechanism of Trib2 upregulation in these leukemias, our data linking Trib2 expression to inhibition of C/EBPα function suggests that Trib2 over-expression is likely to be an important pathogenic mechanism in a subset of human AML.
Decreased C/EBPαp42 levels are observed in diverse molecular subtypes of human AML (Helbling et al., 2004; Helbling et al., 2005; Pabst et al., 2001a; Perrotti et al., 2002; Westendorf et al., 1998) and are sometimes associated with point mutations in C/EBPα (Pabst et al., 2001b). A number of observations suggest that Trib2-mediated alterations of C/EBPα function are likely to contribute to AML induction. We found that Trib2 forms a complex with C/EBPαp42 that results in its destabilization through proteosome-dependent degradation. We further demonstrated an inverse functional relationship between Trib2 and C/EBPα through both gain-and loss-of-function approaches. The effects of C/EBPα on myeloid cell differentiation are opposed by enforced Trib2 expression, and we found high Trib2 expression in a specific subset of human AMLs that is associated with C/EBPα mutations. We hypothesize that Trib2-expressing AMLs lacking C/EBPα mutations share a similar expression profile due to functional inactivation of C/EBPα. It is unlikely, however, that Trib2 causes AML only through C/EBPα inactivation. Mice bearing knockout and mutated C/EBPα alleles have abnormalities of myeloid differentiation, but do not develop AML (Heath et al., 2004; Zhang et al., 1997; Zhang et al., 2004). Furthermore, despite its effect on C/EBPα function, Trib2 is not sufficient for AML, as the tumors arising in Trib2 mice were either mono- or biclonal.
In addition to C/EBPα, Trib2 may influence other signaling pathways. In Drosophila, Tribbles degrades String/CDC25 to inhibit the cell cycle. Although it seems counterintuitive that decreased String/CDC25 would be oncogenic, Tribbles antagonism of String is associated with enhanced proliferation in certain contexts in the fly (Mata et al., 2000). Other Trib family members have been associated with Akt and MAPK signaling (Hegedus et al., 2006) and it remains to be determined whether Trib2 influences these pathways in AML cells.
It is also possible that proviral integration by the Trib2-expressing retroviral vector created lesions in genes that cooperate with Trib. We have begun to address this by cloning proviral integrations in Trib2 tumors. In at least one Trib2 AML, the provirus was found adjacent to HoxA9, whose mRNA was markedly elevated in this tumor (K.K. unpublished observations). HoxA9 has been shown to synergize with other oncogenes to accelerate AML development (Kawagoe and Grosveld, 2005). We are currently investigating the functional significance of this finding.
An additional novel property of Trib2 is its association with increased C/EBPαp30 levels in cell lines and leukemic samples. C/EBPα is an intronless gene and it has been suggested that the two major C/EBPα isoforms stem from alternative translational start sites. Other possibilities exist, however, as the homologous protein C/EBPβ is proteolytically processed into full-length and N-terminal truncated dominant negative isoforms (Baer and Johnson, 2000; Lin et al., 1993; Ossipow et al., 1993; Welm et al., 1999). In our co-immunoprecipitation experiments, the C/EBPα expression construct lacked the uORF (upstream open reading frame) and contained a C-terminal HA tag that was detected with a HA antibody and verified with a specific C/EBPα antibody. This suggests that the 30 kDa protein is an N-terminal truncation of the full length C/EBPα induced by Trib2 expression. However, this construct retains the alternative ATG start codon and therefore translational control cannot be ruled out. In favor of this possibility, we were unable to detect C/EBPαp30 in the pulse chase experiment.
Although Trib2 potently induced AML in our BMT model, it is not clear whether the diminution of C/EBPαp42, the enhancement of C/EBPαp30, or both were critical to the phenotypes that we observed. Decreased C/EBPαp42 levels are associated with human AML (Mueller and Pabst, 2006), and mice engineered to express a C/EBPαp42 mutant that fails to block the cell cycle develop a transplantable myeloproliferative disease that progresses to an AML-like phenotype but does not affect survival (Porse et al., 2005). Murine BM cells transduced with the dominant negative C/EBPαp30 showed poor long-term engraftment (Schwieger et al., 2004). However, in CFU assays, the C/EBPαp30 dominant negative was shown to block murine and human myeloid differentiation (Iwama et al., 2002; Schwieger et al., 2004; Wang and Friedman, 2002). Thus, both decreased C/EBPαp42 and increased C/EBPαp30 have been associated with myeloid transformation. Additional studies will be required to determine which of these isoforms is most relevant to Trib2-induced AML.
We initially identified Trib2 by screening for transcripts that were down-regulated when Notch1-dependent T cell tumor cell lines were induced to growth arrest by gamma secretase inhibitor treatment (Weng et al., 2003). Although we find that Trib2 expression is modulated by Notch1 via CSL binding sites in the Trib2 promoter (manuscript in preparation), mice that retrovirally express activated Notch1 do not develop AML or exhibit major defects in granulocyte differentiation. Trib2 regulation by Notch1 may be context dependent and non-operational in myeloid progenitors. However, Notch signaling promotes macrophage/DC differentiation in some assays (Cheng et al., 2003; Ohishi et al., 2001) and it will be interesting to determine if this is Trib2 dependent. In addition, the subset of human AMLs associated with increased Trib2 expression are very immature and express T cell markers, such as TCRδ (Valk et al., 2004). Trib2 is highly expressed during several stages of T cell development (data not shown), and it is possible that Trib2 normally contributes to the expression of genes associated with T cell development.
Our data predict that Trib2 would functionally inactivate C/EBPα by causing its degradation. Thus, one expectation would be that it would associate with tumors in which C/EBPα is inactivated. Analysis of microarray data obtained from 285 AMLs showed that elevated Trib2 expression preferentially associated with a cluster of AMLs characterized by C/EBPα defects. High Trib2 expression was predominantly seen in tumors without C/EBPα mutations, however, it was also high in two tumors with these mutations (Figure 8). It may be that induction of this molecular class of AML requires C/EBPα function to be suppressed below a certain threshold, and that this may occur by mutation alone, Trib2 expression, or a combination of these two factors.
In summary, our studies identify Trib2 as an oncoprotein that contributes to the pathogenesis of AML through the inhibition of C/EBPα function. This occurs through the interaction of Trib2 with C/EBPα, resulting in the proteasomal-dependent degradation of C/EBPα. Our observations suggest that multiple leukemogenic mechanisms target C/EBPα in AML. Understanding the precise role of Trib2 in AML may lead to new diagnostic and therapeutic strategies for this aggressive human malignancy.
Experimental Procedures
Constructs and retroviruses
A 1032 bp fragment encoding the entire murine Trib2 cDNA was subcloned (+/− N-terminus FLAG tag) into pcDNA3.1, pcDNA3.1/myc-HIS plasmid or one of two different versions of MigR1 (one co-expressing GFP; the other truncated human nerve growth factor receptor, tNGFR). C/EBPα rat cDNA (Keeshan et al., 2003) was cloned into the pcDNA3.1/myc-HIS plasmid. The MigR1-C/EBP□ER construct was a kind gift of Dr. S. Nimer. The IL-12 p40 promoter containing the B6 genomic fragment −700 to +54 of the IL-12 p40 gene was cloned into the pGL3-basic vector (Promega) and site directed mutagenesis of the C/EBP binding site (−93 to −89) was performed using the QuickChange kit (Stratagene) according to manufacturers instructions (kind gifts from Dr. R Carmody).
32D Differentiation assay
32D cells were transduced with C/EBPαER (GFP), Trib2 (tNGFR+FLAG), or both and sorted for GFP and/or tNGFR 48 hrs post-transduction, and cultured in DMEM without phenol red (GIBCO) containing 10% charcoal stripped FCS (Gemini Bioproducts) and 5 ng/ml rIL-3. 1 x 105 cells were plated in 5 ng/ml IL-3 or 25 ng/ml G-CSF +/− 1μM β-estradiol (Sigma, E2257) and assessed for granulocytic differentiation by FACS analysis and morphological criteria.
Methylcellulose Clonogenic Assays
Sorted GFP+ and GFP+/Lin− (CD3, CD4, CD8, B220, Gr-1, Ter119, CD19, MHCII, IL-7Rα) BM cells from MigR1 and Trib2 transplanted mice were plated in triplicate in methylcellulose media (Methocult™ M3231, Stem Cell Technologies) supplemented with cytokines (GM-CSF, 10 ng/ml, IL-3, 10 ng/ml, IL-6, 10 ng/ml, SCF, 50 ng/ml (Peprotech and BD Pharmingen). Cells (15,000) from primary plates (colonies scored at 9 days) were transferred to secondary (colonies scored at 10 days) and tertiary (colonies scored at 8 days) plates containing the indicated cytokines, then transferred to RPMI liquid culture supplemented with 10% FBS and cytokines. Single colonies were transferred from primary plates to media containing IL-3, IL-6 and SCF in 24 well plates and assessed for growth after 11 days.
Bone marrow transduction and transplantation
C57BL/6 mice (B6) were obtained from Taconic Farms. Experiments were performed according to guidelines from the National Institutes of Health and with an approved protocol from the University of Pennsylvania Animal Care and Use Committee. Retroviral Transduction of B6 BM cells and subsequent transfer of these cells into lethally irradiated recipients were performed as described (Pui et al., 1999) and are provided in supplementary experimental procedures. Seven independent transplant experiments were performed with different Trib2 retroviral contructs (MigR1-Trib2, MigR1-FLAG-Trib2, NGFR-FLAG-Trib2); each gave similar results and are summarized together in Figure 2A. Secondary transplants were performed by injecting 2 x 106 nucleated BM or spleen cells from the primary leukemic mice into sublethally irradiated (600 rads) B6 mice. Histology and immunohistochemical staining details are provided in supplementary experimental procedures.
Flow Cytometry
Cell suspensions were stained in PBS/2% FBS after blocking with non-specific Rat/Mouse IgG (Sigma). Cells were sorted on a MoFlo (Cytomation) cell sorter. Analytical flow cytometry was performed on a FACS Calibur (Becton Dickinson) and analyzed using FlowJo software (Treestar). Antibodies used are described in supplementary experimental procedures. Dead cells were excluded by a combination of forward and side scatter properties.
Immunoprecipitation, Western Blotting and 35S-Methionine pulse-chase
293T cells were transfected with 3 μg pcDNA3.1/myc-HIS-Trib2 and 2 μg MigR1-C/EBPα. After 36 hrs, co-transfected cells were treated with 10 μM MG132 for 1 hr and 10 mM N-ethylmaleimide (both Calbiochem) for 30 seconds, washed in 1xPBS containing N-ethylmaleimide and lysed with modified RIPA buffer (50 mM Tris, pH 8.0, containing 0.5% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3Vo4, 1 mM NaF and 20 mM N-ethylmaleimide) supplemented with protease inhibitors (Complete EDTA-free; Roche). 3 mg of precleared lysates were incubated overnight with 10 μl of protein A beads coated with 5 μg anti-MYC 9E10 antibody. To detect C/EBPα in leukemic samples, cells were lysed directly in 2xSDS buffer. To detect Flag-tagged Trib2 in leukemic samples, protein lysates were prepared with modified RIPA buffer. Antibodies used were anti-C/EBPα (Sc-61, Santa Cruz), anti-HA (HA.11, Covance), anti-FLAG (M2, Sigma) and anti-MYC (9E10). In pulse-chase experiments, U937 cells transduced with MigR1 and Trib2 were starved for 30 min, pulse-labeled for 60 min with 35S-methionine (1 mCi/ml), and then chased for up to 180 min. Precleared protein lysates were incubated overnight with protein G beads coated with 5 μg C/EBPα antibody (SC-61x). Western blotting was performed according to standard procedures.
Luciferase Reporter Assay
RAW264.7 macrophages were transfected with various combinations of pcDNA control vector, vectors encoding Trib2 (390 ng) or C/EBPα (10 ng), IL-12 p40 (100 ng) wild-type and mutant promoter-luciferase (firefly) constructs, and the internal control Renilla-luciferase plasmid pRL-TK (10 ng, Promega). Firefly and Renilla luciferase activities were measured in whole cell lysates using the Dual-Luciferase reporter assay kit (Promega), as per the manufacturer's instructions.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed as described (Keeshan et al., 2003) except that nuclear extracts were prepared using Nuclear Extract Kit (Active Motif) as per the manufacturer’s instructions. The G-CSF receptor promoter oligonucleotide (C/EBP site underlined) had the sequence 5’-AAGGTGTTGCAATCCCCAGC-3’. The Oct-1 consensus oligonucleotide was obtained from Santa Cruz. 2 μl of C/EBPα (SC-61x) was used for supershift experiments.
Quantitative RT-PCR
RNA, isolated using the RNEasy kit (Qiagen), was digested with DNaseI and used for reverse transcription according to the manufacturers instructions (Superscript II™ kit, Invitrogen). Validated human Trib2 and 18s rRNA primer/probe sets with TaqMan® Universal PCR Master Mix (Applied Biosystems), and murine Trib2 primers (forward AGCCCGACTGTTCTACCAGA, reverse AGCGTCTTCCAAACTCTCCA) with SYBR® GREEN PCR Master mix (Applied Biosystems) were used for qRT-PCR and analyzed on the ABI Prism 7900 sequence detection system (Applied Biosystems).
DNA analysis
Southern blotting was performed according to standard procedures using QuikHyb® (Stratagene) buffer and a 32P labeled IRES probe.
RNA Interference
Human Trib2 siRNA (Silencer™ Validated siRNA #1060), murine Trib2 siRNA (Silencer Pre-designed siRNA #169360-exon2) and control Silencer Negative Control #1 were obtained from Ambion. 2 x 106 U937 parental cells (expressing human Trib2) and U937 cells transduced with Trib2 (murine) in 100 μl of Amaxa solution (Nucleofector™ Kit V, AMAXA biosystems), mixed with 100 nM of siRNA, and electroporated using the AMAXA Nucleofector. Trib2 (human and murine) mRNA expression and C/EBPα protein expression were assessed 24 hr later.
Supplementary Material
Acknowledgments
We thank Ruaidhrí Carmody, Alan Diehl, Gary Koretzky, Jeff Kutok, Craig Thompson, and members of the Pear lab for important contributions to these studies. We are grateful to Bruno Calabretta and Stephen Nimer for reagents. We are also grateful to the UPenn John Morgan and Stemmler ASU, UPenn Abramson Cancer Center Flow Cytometry and Genomics cores, and AFCRI cores. Immunohistologic studies were carried out with the technical support of the Dana Farber/Harvard Cancer Center Hematopathology Core Laboratory. We apologize to authors whose work was not cited due to space limitations.
This work was supported by the grants from the National Institutes of Health to J.C.A. (CA82308) and W.S.P. (CA93615, AI47833, 5P01CA119070-040002) and the Leukemia and Lymphoma Society SCOR Program. Individual support was provided by a Leukemia and Lymphoma Society Fellow Award to K.K., and a Fellow award from the Damon Runyon Cancer Research Foundation (DRG-102-05) to I.M. The authors have no conflicting financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Baer M, Johnson PF. Generation of truncated C/EBPbeta isoforms by in vitro proteolysis. J Biol Chem. 2000;275:26582–26590. doi: 10.1074/jbc.M004268200. [DOI] [PubMed] [Google Scholar]
- Bisoffi M, Klima I, Gresko E, Durfee PN, Hines WC, Griffith JK, Studer UE, Thalmann GN. Expression profiles of androgen independent bone metastatic prostate cancer cells indicate up-regulation of the putative serine-threonine kinase GS3955. J Urol. 2004;172:1145–1150. doi: 10.1097/01.ju.0000135117.40086.fa. [DOI] [PubMed] [Google Scholar]
- Bowers AJ, Scully S, Boylan JF. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene. 2003;22:2823–2835. doi: 10.1038/sj.onc.1206367. [DOI] [PubMed] [Google Scholar]
- Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Cammenga J, Mulloy JC, Berguido FJ, MacGrogan D, Viale A, Nimer SD. Induction of C/EBPalpha activity alters gene expression and differentiation of human CD34+ cells. Blood. 2003;101:2206–2214. doi: 10.1182/blood-2002-05-1546. [DOI] [PubMed] [Google Scholar]
- Cheng P, Nefedova Y, Miele L, Osborne BA, Gabrilovich D. Notch signaling is necessary but not sufficient for differentiation of dendritic cells. Blood. 2003;102:3980–3988. doi: 10.1182/blood-2003-04-1034. [DOI] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guzman CG, Warren AJ, Zhang Z, Gartland L, Erickson P, Drabkin H, Hiebert SW, Klug CA. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Mol Cell Biol. 2002;22:5506–5517. doi: 10.1128/MCB.22.15.5506-5517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- Grosshans J, Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101:523–531. doi: 10.1016/s0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- Heath V, Suh HC, Holman M, Renn K, Gooya JM, Parkin S, Klarmann KD, Ortiz M, Johnson P, Keller J. C/EBPalpha deficiency results in hyperproliferation of hematopoietic progenitor cells and disrupts macrophage development in vitro and in vivo. Blood. 2004;104:1639–1647. doi: 10.1182/blood-2003-11-3963. [DOI] [PubMed] [Google Scholar]
- Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: novel regulators of cell function; evolutionary aspects. Cell Mol Life Sci. 2006 doi: 10.1007/s00018-006-6007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling D, Mueller BU, Timchenko NA, Hagemeijer A, Jotterand M, Meyer-Monard S, Lister A, Rowley JD, Huegli B, Fey MF, Pabst T. The leukemic fusion gene AML1-MDS1-EVI1 suppresses CEBPA in acute myeloid leukemia by activation of Calreticulin. Proc Natl Acad Sci U S A. 2004;101:13312–13317. doi: 10.1073/pnas.0404731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling D, Mueller BU, Timchenko NA, Schardt J, Eyer M, Betts DR, Jotterand M, Meyer-Monard S, Fey MF, Pabst T. CBFB-SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16) Blood. 2005;106:1369–1375. doi: 10.1182/blood-2004-11-4392. [DOI] [PubMed] [Google Scholar]
- Iwama A, Osawa M, Hirasawa R, Uchiyama N, Kaneko S, Onodera M, Shibuya K, Shibuya A, Vinson C, Tenen DG, Nakauchi H. Reciprocal roles for CCAAT/enhancer binding protein (C/EBP) and PU.1 transcription factors in Langerhans cell commitment. J Exp Med. 2002;195:547–558. doi: 10.1084/jem.20011465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izon DJ, Punt JA, Xu L, Karnell FG, Allman D, Myung PS, Boerth NJ, Pui JC, Koretzky GA, Pear WS. Notch1 regulates maturation of CD4+ and CD8+ thymocytes by modulating TCR signal strength. Immunity. 2001;14:253–264. doi: 10.1016/s1074-7613(01)00107-8. [DOI] [PubMed] [Google Scholar]
- Kawagoe H, Grosveld GC. Conditional MN1-TEL knock-in mice develop acute myeloid leukemia in conjunction with overexpression of HOXA9. Blood. 2005;106:4269–4277. doi: 10.1182/blood-2005-04-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, Santilli G, Corradini F, Perrotti D, Calabretta B. Transcription activation function of C/EBPalpha is required for induction of granulocytic differentiation. Blood. 2003;102:1267–1275. doi: 10.1182/blood-2003-02-0477. [DOI] [PubMed] [Google Scholar]
- Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- Kiss-Toth E, Bagstaff SM, Sung HY, Jozsa V, Dempsey C, Caunt JC, Oxley KM, Wyllie DH, Polgar T, Harte M, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P, Preudhomme C. CEBPA point mutations in hematological malignancies. Leukemia. 2005;19:329–334. doi: 10.1038/sj.leu.2403614. [DOI] [PubMed] [Google Scholar]
- Lin FT, MacDougald OA, Diehl AM, Lane MD. A 30-kDa alternative translation product of the CCAAT/enhancer binding protein alpha message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci U S A. 1993;90:9606–9610. doi: 10.1073/pnas.90.20.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Curado S, Ephrussi A, Rorth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101:511–522. doi: 10.1016/s0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Mueller BU, Pabst T. C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7–14. doi: 10.1097/01.moh.0000190110.08156.96. [DOI] [PubMed] [Google Scholar]
- Ohishi K, Varnum-Finney B, Serda RE, Anasetti C, Bernstein ID. The Notch ligand, Delta-1, inhibits the differentiation of monocytes into macrophages but permits their differentiation into dendritic cells. Blood. 2001;98:1402–1407. doi: 10.1182/blood.v98.5.1402. [DOI] [PubMed] [Google Scholar]
- Ossipow V, Descombes P, Schibler U. CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc Natl Acad Sci U S A. 1993;90:8219–8223. doi: 10.1073/pnas.90.17.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, Hiddemann W, Zhang DE, Tenen DG. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 2001a;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001b;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, Iervolino A, Condorelli F, Gambacorti-Passerini C, Caligiuri MA, Calabretta B. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I, Jacobsen SE, Nerlov C. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med. 2005;202:85–96. doi: 10.1084/jem.20050067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC, Pear WS. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. 1999;11:299–308. doi: 10.1016/s1074-7613(00)80105-3. [DOI] [PubMed] [Google Scholar]
- Qi L, Heredia JE, Altarejos JY, Screaton R, Goebel N, Niessen S, Macleod IX, Liew CW, Kulkarni RN, Bain J, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006;312:1763–1766. doi: 10.1126/science.1123374. [DOI] [PubMed] [Google Scholar]
- Rorth P, Szabo K, Texido G. The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mol Cell. 2000;6:23–30. doi: 10.1016/s1097-2765(05)00008-0. [DOI] [PubMed] [Google Scholar]
- Rosenbauer F, Koschmieder S, Steidl U, Tenen DG. Effect of transcription-factor concentrations on leukemic stem cells. Blood. 2005;106:1519–1524. doi: 10.1182/blood-2005-02-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieger M, Lohler J, Fischer M, Herwig U, Tenen DG, Stocking C. A dominant-negative mutant of C/EBPalpha, associated with acute myeloid leukemias, inhibits differentiation of myeloid and erythroid progenitors of man but not mouse. Blood. 2004;103:2744–2752. doi: 10.1182/blood-2003-07-2280. [DOI] [PubMed] [Google Scholar]
- Seher TC, Leptin M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol. 2000;10:623–629. doi: 10.1016/s0960-9822(00)00502-9. [DOI] [PubMed] [Google Scholar]
- Takasato M, Osafune K, Matsumoto Y, Kataoka Y, Yoshida N, Meguro H, Aburatani H, Asashima M, Nishinakamura R. Identification of kidney mesenchymal genes by a combination of microarray analysis and Sall1-GFP knockin mice. Mech Dev. 2004;121:547–557. doi: 10.1016/j.mod.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Wang QF, Friedman AD. CCAAT/enhancer-binding proteins are required for granulopoiesis independent of their induction of the granulocyte colony-stimulating factor receptor. Blood. 2002;99:2776–2785. doi: 10.1182/blood.v99.8.2776. [DOI] [PubMed] [Google Scholar]
- Wang X, Scott E, Sawyers CL, Friedman AD. C/EBPalpha bypasses granulocyte colony-stimulating factor signals to rapidly induce PU.1 gene expression, stimulate granulocytic differentiation, and limit proliferation in 32D cl3 myeloblasts. Blood. 1999;94:560–571. [PubMed] [Google Scholar]
- Welm AL, Timchenko NA, Darlington GJ. C/EBPalpha regulates generation of C/EBPbeta isoforms through activation of specific proteolytic cleavage. Mol Cell Biol. 1999;19:1695–1704. doi: 10.1128/mcb.19.3.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC, Aster JC. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–664. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol. 1998;18:322–333. doi: 10.1128/mcb.18.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin F, Suarez-Huerta N, Robaye B, Peetermans J, Libert F, Dumont JE, Maenhaut C. Characterization of a phosphoprotein whose mRNA is regulated by the mitogenic pathways in dog thyroid cells. Eur J Biochem. 1997;248:660–668. doi: 10.1111/j.1432-1033.1997.t01-1-00660.x. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci U S A. 1997;94:569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21:853–863. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Davis JL, Li W. Identification of tribbles homolog 2 as an autoantigen in autoimmune uveitis by phage display. Mol Immunol. 2005;42:1275–1281. doi: 10.1016/j.molimm.2004.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.