

Abstract
The MCT-1 oncogene was originally identified from lymphoma cell lines. Herein we establish that MCT-1 is highly expressed in 85% of human diffuse large B-cell lymphomas (DLBCL) and that knocking down MCT-1 by a specific short hairpin RNA in DLBCL cells induces apoptosis, supporting a critical role for MCT-1 in DLBCL cell survival. However, the mechanism underlying MCT-1 regulation is largely unknown. We find that MCT-1 is phosphorylated and up-regulated by extracellular signal-regulated kinase (ERK). Furthermore, by using a small inhibitory molecule targeting ERK, we interrupted MCT-1 phosphorylation and stability. Significantly, cells with distinct levels of MCT-1 protein displayed differential sensitivity to ERK inhibitor–induced apoptosis. Treatment with the ERK inhibitor showed marked in vivo antitumor activity in a human DLBCL xenograft model. Our findings establish a functional molecular interaction between MCT-1 and the MEK/ERK signaling pathway and suggest that the activation of MCT-1 function by its upstream kinase ERK plays an important role in lymphomagenesis.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoid malignancy in adults, accounting for ~ 30,000 new cases each year and nearly 40% of all non–Hodgkin’s lymphomas (NHL; ref. 1). Despite recent advances in immunochemotherapy, long-term remission can only be achieved in ~ 50% of patients (2). Although some progress is being made, the fundamental abnormalities underlying DLBCL still remain elusive (2). Further research is required to identify relevant molecular targets to develop effective therapeutic approaches that will improve the clinical outcome of patients with DLBCL.
We have discovered a novel oncogene in a T-cell lymphoma cell line, multiple copies in T-cell lymphoma-1 (MCT-1), amplified in human T-cell lymphoma and mapped to chromosome Xq22-24 (3). The MCT-1 gene has an open reading frame that encodes a protein of 181 amino acids with a predicted molecular mass of 20 kDa (3). Constitutive expression of MCT-1 results in a strong proliferative signal and is associated with deregulation of the G1-S phase checkpoint (3). There is increasing evidence supporting a role for the MCT-1 oncogene in lymphomagenesis, including its ability to stimulate cell proliferation, suppress apoptosis, and promote angiogenesis (3-6). Importantly, MCT-1 has been shown to transform both human and murine immortalized cells (5, 6). The exact molecular mechanism(s) by which MCT-1 transforms cells is still evolving; however, there are data implicating MCT-1 in modulating the translation of cancer-related genes through its interaction with the cap complex (7, 8). MCT-1 protein forms a complex with DENR/DRP, a protein containing an SUI1 domain involved in recognition of the translation initiation codon (7). Recently, several lines of evidence indicate that abnormal control of translation contributes to lymphomagenesis (9-11). The deregulated function of those translational molecules associated with lymphomagenesis presents unique opportunities to target proteins critical to the malignant phenotype. Therefore, it may be beneficial to selectively block MCT-1 function and to diminish its involvement in abnormal cell functions such as cancer cell proliferation and transformation.
Currently, there are no available specific small inhibitory molecules that can directly modulate MCT-1 protein function. Phosphorylation of MCT-1 protein by extracellular signal-regulated kinase 1/2 (ERK1/2) is essential for protein stabilization and for its ability to promote cell proliferation (12). These data indicated that MCT-1 levels and function are dependent on the ERK signaling pathway. Therefore, targeting molecules upstream of MCT-1 could affect the stability and activity of MCT-1. Importantly, several reports linked unregulated activation of ERK proteins to cancer cell apoptosis, proliferation, and malignant transformation (13-15). Disruption of ERK1/2 activation by MEK1/2 inhibitors results in a dramatic increase in apoptosis of hematopoietic malignant cells (16, 17). Therefore, it seemed reasonable to attempt disruption of MCT-1 function by inhibiting its upstream kinase, ERK. Taking advantage of recently identified ERK docking domains and using computer-aided drug design, a novel small-molecule ERK inhibitor designated no. 76 has been identified (18). It binds to ERK2 with a KD of ~ 5 μmol/L and prevents its interaction with protein substrates. Targeting this inhibitor to individual ERK docking domains can potentially be used to disrupt ERK2 interactions with specific protein substrates (18).
Here, we report that MCT-1 is highly expressed in 85% of human DLBCLs, supporting the feasibility of therapeutic targeting of MCT-1 for DLBCL. Moreover, our data establish the functional interaction between MCT-1 and the MEK/ERK signaling pathway and the potential role of targeting MCT-1 and its upstream kinases in the therapy of DLBCL.
Materials and Methods
Cell culture, treatment, and transfection
DLBCL (SUDHL4, SUDHL6, Farage), Burkitt lymphoma (Daudi and Raji), and T-cell leukemia/lymphoma (Jurkat) cells were grown in RPMI 1640 (Invitrogen) containing 10% fetal bovine serum. Farage-Vector (F-Vector) and Farage-overexpressing MCT-1 (F-MCT-1) cells were described previously (7). Normal donor peripheral blood lymphocytes (PBL) were isolated and cultured as described (4). MEK inhibitors PD98059 (40 μmol/L), U0126 (10 μmol/L), and ERK inhibitor no. 76 [3-(2-aminoethyl)-5-((4-ethoxyphenyl) methylene)-2,4-thiazolidinedione, HCl] were from Calbiochem. Wild-type MEK2 (WT MEK2) and constitutively active MEK2 (CA MEK2) constructs have been previously described (19). Specific oligo small interfering RNAs (siRNA) for MEK2, ERK1, ERK2, and the negative siRNA control were obtained from Qiagen. Transfection experiments were done using Amaxa Nucleofector kit V (Amaxa) as previously described (7).
MCT-1 knockdown in SUDHL4 and SUDHL6 cell lines
Mission-TRC short hairpin RNA (shRNA)–encoding lentiviruses targeting human MCT-1 (Sigma-Aldrich) were used for transduction in SUDHL4 and SUDHL6 cell lines according to the manufacturer’s protocol. These shRNA-encoding lentiviruses targeting human MCT-1 contained four individual clones: NM_014060.1-499s1c1 (defined as no. 1), NM_014060.1-374s1c1 (defined as no. 2), NM_014060.1-542s1c1 (defined as no. 3), and NM_014060.1-638s1c1 (defined as no. 4). We also used a control shRNA lentivirus (SHC-002V) encoding a shRNA targeted against no known mouse or human gene. For transient knockdown experiments, SUDHL4 and SUDHL6 cells were transduced, and 72 h after transduction, harvested and assayed for apoptosis. For stable knockdown experiments, SUDHL4 and SUDHL6 cells were transduced, and 72 h after transduction, cells were selected in a puromycin-containing medium (0.5 μg/mL) for 14 d and then expanded for further experimentation.
Cell apoptosis assays
SUDHL4, SUDHL6, and Farage cells (2 × 105/mL) were seeded at equal density and then treated with ERK inhibitor no. 76 in complete RPMI 1640 medium. Forty-eight hours after ERK inhibitor treatment, cells were harvested and apoptosis was analyzed by flow cytometry using Annexin V staining kit (Southern Biotech). The significance of differences between experimental conditions was determined using the Student’s t test.
Soft agar colony-forming assay
SUDHL4 and SUDHL6 cells transient expressing either control shRNA or shMCT-1 were assessed in vitro for their ability to form colonies in soft agar as previously described (8). The number of colonies in each well was counted. Colony formation (>50 cells) was examined under phase-contrast microscopy. Images were taken at room temperature using a Nikon Eclipse TE-2000S microscope. Each experiment was conducted several times and the significance of differences between experimental conditions was determined using the Student’s t test.
Immunoprecipitation, Western blot, and glutathione S-transferase fusion protein pull-down assay
After treatment with ERK inhibitor, cell lysates were prepared using standard methodology (20). Antibodies were added to lysates and incubated for 1 h at 4°C. Antibodies were collected with protein A or protein G–Sepharose beads, and protein complexes were washed three times at 4°C with the lysis buffer. Western blotting was performed as previously described (20). Anti–pElk-1 (Santa Cruz Biotechnology), anti–poly(ADP-ribose) polymerase (PARP; Santa Cruz Biotechnology), anti-ERK1/2 (Cell Signaling) and anti–phospho-threonine antibody (Chemicon) were used at 1:1,000, whereas anti–MCT-1 (7) and anti-actin (C2, Santa Cruz Biotechnology) were used at 1:5,000. Glutathione S-transferase (GST) fusion proteins were expressed and purified as previously described (21). The immobilized GST fusion proteins were incubated with lysates of Jurkat cells for 1 h at 4°C. The beads were washed with lysis buffer four times and then protein complexes were separated by SDS-PAGE and analyzed by Western blotting.
Tissue microarray construction and immunohistologic analysis
MCT-1 protein expression was interrogated on tissue microarrays of 406 human NHLs with appropriate control tissues. The lymphomas were classified according to the current WHO classification scheme (22). The selection of lymphoma samples, tissue microarray construction, and immunohistologic staining were described previously (23). Anti–MCT-1 antibody was used at a dilution of 1:800. Detection was carried out using the DakoCytomation EnVision+ System-HRP labeled polymer (DakoCytomation, Inc.). Staining was optimized on normal paraffin-embedded tonsil sections, and a cutoff of staining in >30% of lymphoma cells was assigned a positive score. This cutoff was based on the need for using a nonambiguous threshold for scoring tissue microarrays (TMA) and does not reflect differences in staining intensity between normal and neoplastic tissue or among different diagnoses. Institutional review board approval from Stanford University was obtained for these studies.
Immunohistochemistry and immunofluorescence
The paraffin sections were stained with a Vectastain Elite ABC Kit (Vector Laboratories) according to the manufacturer’s protocol. As described previously (24), the paraffin section slides were incubated overnight at 4°C with rabbit anti-human MCT-1 antibody (1:1,000). Slides were then treated with biotinylated anti-rabbit IgG and incubated with performed avidin-peroxidase complex. The sections were counterstained with hematoxylin, dehydrated, and mounted. Negative controls were included. Immunohistochemical staining was evaluated by the hematopathologist (X.F.Z.). The images were acquired with a Nikon eclipse 50i microscope (Nikon Instruments, Inc.) and formatted with an Adobe PhotoShop CS software. Human sample studies have been approved by the University of Maryland Medical School Institutional Review Board and conform to the Declaration of Helsinki.
For fluorescence double staining, the section was treated as above and first stained with mouse monoclonal anti-ERK1/2 antibody (1:200). After biotin blocking, the section was then stained with rabbit polyclonal anti–MCT-1 antibody (1:1,000). The sections were then washed with PBS and incubated with goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 568 at a concentration of 1:200 each. The stained sections were mounted using Vectashield mounting medium (Vector Labs). Control sections were similarly processed, except that the primary antisera were omitted, which yielded no staining. Images were captured using an Olympus FV1000 confocal microscope (Olympus) and processed with the Olympus Fluoview Version 1.7c software.
In vivo tumor growth in the xenograft models
Female severe combined immunodeficient mice (SCID) Beige mice were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum. SUDHL6 or Daudi cells (2 × 106) were resuspended in 100 μL PBS and then mixed with an equal volume of Matrigel. The mixture was s.c. injected into the left and right dorsal flanks of 5- to 7-wk-old female SCID mice. When the tumor reached the size of ~ 60 to 163 mm3, the drug was administered i.p. every other day at a dose of 10 mg per kilogram of body weight for a total of 20 d. Injection of the vehicle alone (5% DMSO in 0.05 mol/L PBS) was used as a control. Tumor volumes were calculated as previously performed (6). Tumors were harvested for further analysis. The significance of differences between treatment arms was determined using the Student’s t test.
Results
MCT-1 was highly expressed in 85% of human DLBCLs
We have previously identified high levels of MCT-1 protein in a limited analysis of primary B-cell NHL (4). Here, we performed immunohistochemical analyses of MCT-1 protein in 406 primary lymphoma tissue specimens using tissue microarray analysis (TMA; ref. 23). The tissue microarray included 370 B-cell lymphoma and 36 T-cell lymphoma samples. The representative views of immunohistologic scoring used in the TMA analysis are shown in Supplementary Fig. S1. As summarized in Table 1, MCT-1 is strongly expressed in 85% of 113 DLBCL samples. In 88 low-grade follicular lymphoma (FL) samples, MCT-1 staining was strongly expressed in only 6% of cases. Interestingly, 20% of high-grade (grade 3) FL showed strong positive staining for MCT-1. These data indicated that MCT-1 expression is significantly increased in DLBCL samples compared with that in low-grade FL and other NHLs, suggesting a role for MCT-1 in DLBCL development and/or progression. Importantly, either the germinal center or non–germinal center subtypes of DLBCL did not influence the high rate of MCT-1 positivity (data not shown). Although strong positive staining for MCT-1 is identified in 82% of 11 mediastinal large B-cell lymphoma and 68% of 25 peripheral T-cell lymphoma samples, respectively, the total number of those specimens examined is small and awaits larger confirmatory studies. Figure 1A (top) shows representative immunohistochemistry images of MCT-1 protein expression in a reactive lymph node and DLBCL. MCT-1 staining was predominantly detected in the cytoplasm of DLBCL lymphoma cells and, to a much lesser extent, in the normal B-cells of reactive lymph nodes and normal tonsils. These data establish that MCT-1 protein levels are highly elevated in the vast majority of DLBCL, supporting its value as a potential therapeutic target.
Table 1.
Immunohistologic analysis of MCT-1 expression in lymphoma subtypes
| Lymphoma subtype | No. samples | Staining |
% Positive | |
|---|---|---|---|---|
| Weak | Strong | |||
| B-cell lymphoma (n = 370) | ||||
| Precursor B-lymphoblastic | 7 | 2 | 2 | 29 |
| Follicular Lymphoma | ||||
| Grade 1 | 34 | 27 | 2 | 6 |
| Grade 2 | 54 | 48 | 3 | 6 |
| Grade 3 | 70 | 53 | 14 | 20 |
| DLBCL | 113 | 13 | 96 | 85 |
| Mediastinal large B-cell | 11 | 2 | 9 | 82 |
| Burkitt lymphoma | 3 | 1 | 0 | 0 |
| Marginal zone lymphoma | 24 | 11 | 3 | 13 |
| Mantle cell lymphoma | 18 | 9 | 1 | 6 |
| Small lymphocytic lymphoma | 31 | 27 | 0 | 0 |
| Lymphoplasmacytic lymphoma | 5 | 4 | 0 | 0 |
| T-cell lymphoma (n = 36) | ||||
| Precursor T-lymphoblastic | 11 | 8 | 0 | 0 |
| Peripheral T-cell lymphoma | 25 | 8 | 17 | 68 |
NOTE: The vast majority of DLBCL showed strong staining with a highly significant P value (1.9 × 10−15) derived using the statistical χ2 test. Percent positive is determined by number of strong staining samples over the total number of samples.
Figure 1.
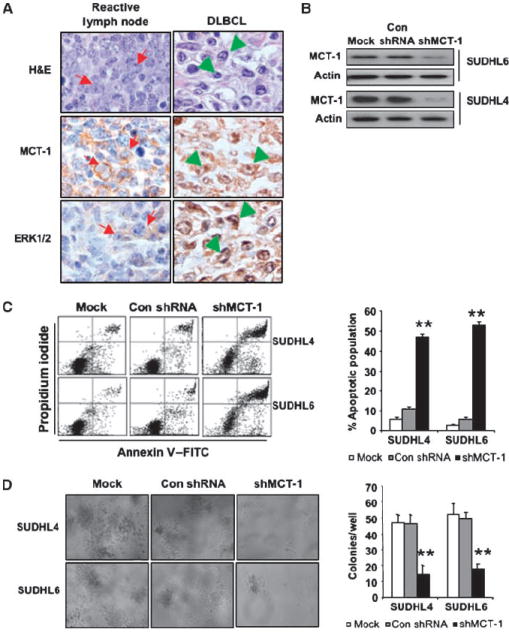
Knockdown of MCT-1 by shRNA induces apoptosis and reduces their clonogenic capacity. A, immunostaining of DLBCL shows MCT-1 and ERK1/2 expression in reactive germinal center B-cell centroblasts (arrows) and DLBCL cells (arrowheads; original magnification, ×1,000). Top, MCT-1 staining; bottom, ERK staining. B, SUDHL4 and SUDHL6 cells were transduced with a lentivirally encoded MCT-1 shRNAs (hairpin no. 3; shMCT-1) and control shRNA (Con). MCT-1 expression was monitored by Western blot analysis. Mock indicates nontransduced cells. C, at 72 h after transduction, cell apoptosis was measured via Annexin V and propidium iodide staining. Flow cytometric profile was showed in the left panel. Columns, mean of three independent experiments; bars, SD. ANOVA was determined by Student’s t test relative to control shRNA. **, P < 0.01. D, SUDHL4 and SUDHL6 cells were transduced with shMCT-1 and control shRNA. On day 9, colony formation was examined and photomicroscopy of SUDHL4 and SUDHL6 cell colonies was taken in soft-agar plates at ×10 magnification (left). Right, quantification of colony formation. The colony numbers were counted in each well of SUDHL6 and SUDHL4 cell colonies. Columns, mean of four experiments; bars, SEM.
Knocking down MCT-1 by shRNA induced DLBCL cell apoptosis and reduced their clonogenic capacity
We hypothesized that disruption of MCT-1 function would result in a diminished malignant phenotype in DLBCL cells overexpressing MCT-1. To test this hypothesis, we used a knockdown approach to inhibit MCT-1 function. SUDHL4 and SUDHL6 cells were transiently transduced with a lentivirally encoded MCT-1 shRNA (hairpin no. 3; shMCT-1) and MCT-1 expression was monitored by immunoblotting. As shown in Fig. 1B and C, when MCT-1 protein was down-regulated in SUDHL4 and SUDHL6 by shRNA, 47% of SUDHL4 and 52% of SUDHL6 cells underwent apoptosis 72 hours after transduction compared with 5% to 10% apoptosis seen in cells transduced with control shRNA. These data were confirmed by a second lentivirally encoded MCT-1 shRNA (hairpin no. 1), as shown in Supplementary Fig. S2. We next examined whether interfering with MCT-1 function by shRNA would inhibit SUDHL4 and SUDHL6 tumorigenicity using soft-agar colony formation. In Fig. 1D, we showed a statistically significant decrease in the colony formation of SUDHL4 and SUDHL6 cells when MCT-1 was knocked down. These results indicated that inhibition of MCT-1 function induced apoptosis and reduced the clonogenic capacity of the DLBCL cell lines.
MCT-1 is a direct substrate of ERK in DLBCL cells
Because a specific small molecule that selectively targets MCT-1 is currently not available, we sought to target the kinase directly upstream of MCT-1. This prompted us to examine ERK1/2 expression in DLBCL. We have analyzed ERK1/2 and MCT-1 protein levels on 12 cases of human primary DLBCL and 5 cases of human reactive lymph node by immunohistochemical staining. ERK1/2 staining was considerably stronger in DLBCL than in reactive B cells (Fig. 1A, bottom; Supplementary Fig. S3), in parallel with the pattern observed for MCT-1 in DLBCL. Pearson correlation analysis revealed that the expression level of ERK protein is strongly correlated with MCT-1 protein level in human DLBCL samples surveyed (correlation coefficient r = 0.77, P < 0.01). The correlation was also confirmed by Western blot analysis in human lymphoma cell lines. As shown in Fig. 2A, MCT-1 expression was low in Farage and PBL cells, as was ERK expression. In those cell lines (Daudi, Jurkat, and SUDHL6) with high MCT-1 expression, we found similarly elevated ERK protein levels. We next examined whether MCT-1 physically associates with ERK. As shown in the left panel of Fig. 2B, we were able to detect the reciprocal coimmunoprecipitation of endogenous MCT-1 with ERK in Jurkat and Farage–MCT-1 cells. To further confirm this interaction, in vitro GST fusion protein pull-down assays were performed. In the right panel of Fig. 2B, we showed that the GST fusion protein containing MCT-1 was able to pull down ERK. To better understand the relationship between MCT-1 and ERK in vivo, we conducted double-immunofluorescence analysis of MCT-1 and ERK in human lymph node tissues to detect their colocalization. In reactive lymph nodes, both MCT-1 and ERK have weak staining and we observed weak overlapping MCT-1 and ERK expression (Supplementary Fig. S4). In DLBCL, both MCT-1 and ERK had much stronger staining and their colocalization was much more prominent in the cytoplasm of DLBCL cells (Fig. 2C).
Figure 2.
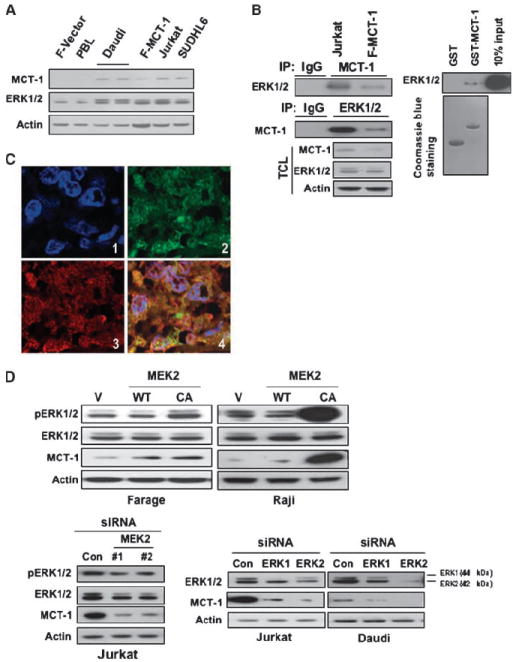
ERK positively modulates levels of MCT-1 protein. A, cell lysates were subjected to immunoblotting with the indicated antibodies. B, MCT-1 associates with ERK in cells. Left, Jurkat and Farage–MCT-1 cells were subjected to immunoprecipitation (IP) using either anti–MCT-1 or anti-ERK1/2 antibody, followed by immunoblotting with indicated antibodies. TCL, total cell lysate. Right, MCT-1 associates with ERK in vitro. GST-fused MCT-1 was immobilized onto GS beads and then incubated with Jurkat cell lysates. Immunoblotting was followed using anti-ERK1/2 antibody. C, colocalization between MCT-1 and ERK in human lymphoma tissues. Double immunofluorescent staining for MCT-1 (green) and ERK (red) at ×600 magnification indicates that there is strong overlapping in human lymphoma tissues. 1, 4′,6-diamidino-2-phenylindole (DAPI); 2, MCT-1; 3, ERK; and 4, merged image of MCT-1, ERK, and DAPI. D, overexpression of constitutively active (CA) MEK2 increased MCT-1 protein level. Farage or Raji cells were transfected with WT MEK2, CA MEK2 construct, or the vector control (V). The cell lysates were subjected to Western blot analysis (top). Bottom, knocking down of MEK2 and ERK1/2 decreased MCT-1 expression. Jurkat or Daudi cells were transfected with control siRNA or siRNAs specific for MEK2, ERK1, and ERK2. At 72 h after transfection, the expression of pERK1/2, ERK1/2, or MCT-1 was monitored by immunoblotting.
Subsequently, we tested whether MCT-1 is an in vivo substrate of ERK. Because MEK1/2 has no known substrates besides the ERK kinase, we reasoned that selective MEK overexpression or inhibition might clarify how the mitogen-activated protein kinase pathway regulates MCT-1. Figure 2D (top) shows that MCT-1 expression was increased by overexpression of active MEK2 in Farage and Raji cells, respectively. Due to the technical difficulty in transfecting oligos into SUDHL4 and SUDHL6 cells using liposomal or electroporation methods, specific oligo siRNA for MEK2 or ERK were transfected into Jurkat and Daudi cells through electroporation. Reciprocally, knocking down MEK2 by siRNA in Jurkat cells also decreased MCT-1 expression. Furthermore, knocking down ERK1 or ERK2 expression in Jurkat and Daudi cells resulted in a decreased expression of MCT-1 (Fig. 2D, bottom). These data unequivocally linked MCT-1 protein levels with ERK signaling.
ERK inhibitor decreased MCT-1 protein level and phosphorylation and disrupted the association between MCT-1 and ERK
Because MCT-1 is a direct substrate of ERK, we envisioned disrupting MCT-1 function through ERK inhibition. Therefore, we examined whether ERK inhibitor affected MCT-1 protein levels and threonine phosphorylation. Figure 3A showed the reduction of MCT-1 and pThr-MCT-1 after small-molecule ERK inhibitor (no. 76) treatment in SUDHL6 and Farage–MCT-1 cells. Elk-1 is a previously characterized substrate of ERK and served as a positive control (25). It has previously been reported that no. 76 can block the interaction between ERK and substrate proteins by binding to ERK docking domains (18). We next determined whether the ERK inhibitor disrupted the association between MCT-1 and ERK. As shown in Fig. 3B and C, the association between MCT-1 and ERK in SUDHL6 and Farage–MCT-1 cells was decreased in the presence of no. 76 but not in the presence of the MEK inhibitor PD98059. In addition, no. 76 had minimal effect on ERK1/2 protein levels. Another MEK inhibitor, U0126, had a similar effect as PD98059 (data not shown), suggesting that ERK interactions with MCT-1 are independent of ERK phosphorylation.
Figure 3.
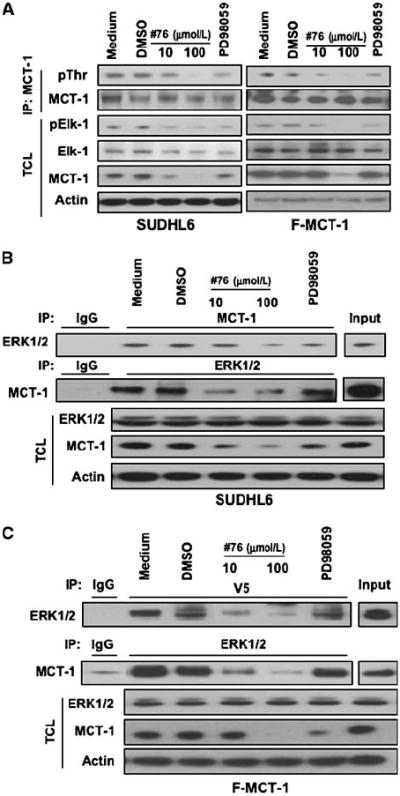
The physical association between MCT-1 and ERK1/2 is disrupted by inhibiting ERK. A, ERK inhibition reduces MCT-1 protein levels and threonine phosphorylation. SUDHL6 and Farage–MCT-1 cells were treated with ERK or MEK inhibitors in RPMI 1640 complete medium for 4 h. The levels of pElk-1, MCT-1, or ERK in the total cell lysates were monitored by immunoblotting with the indicated antibodies. The effects of inhibitors on MCT-1 threonine phosphorylation were examined by immunoprecipitation with anti–MCT-1 antibody and followed by immunoblotting with anti-phosphothreonine (pThr) antibody. B and C, SUDHL6 and Farage–MCT-1 cells were treated with ERK or MEK inhibitors in RPMI 1640 complete medium for 4 h. The cell lysates were subjected to immunoprecipitation with anti–MCT-1 or anti-ERK1/2 antibody followed by immunoblotting with anti-ERK1/2 or anti–MCT-1 antibody. Anti-V5 antibody was used in immunoprecipitation instead of MCT-1 antibody for Farage–MCT-1 cells overexpressing V5-tagged MCT-1. The expression of ERK1/2 or MCT-1 was monitored by immunoblotting of the total cell lysates. Actin was used as a loading control.
Increased MCT-1 protein levels sensitized DLBCL cells to ERK inhibitor–induced apoptosis
To examine the impact of MCT-1 protein levels on the sensitivity of DLBCL cells to ERK inhibitor therapy, we examined the effect of the ERK inhibitor in DLBCL cell lines with different levels of MCT-1. To address this, we established an experimental model using several stable cell lines that were genetically modified to either overexpress or down-regulate MCT-1. Farage–MCT-1, stably overexpressing MCT-1, and Farage-Vector cells as previously described (7) were incubated with different doses of no. 76 for 48 hours. Figure 4A showed that ERK inhibitor modestly induced, in a dose-dependent manner, apoptosis in Farage-Vector cells; in contrast, Farage–MCT-1 cells were significantly more sensitive to ERK inhibitor–induced apoptosis. These data implied that MCT-1 may be a functionally relevant target of ERK and that inhibition of ERK activity would selectively induce apoptosis of DLBCL cells with elevated MCT-1 levels. To explore this further, we stably transduced SUDHL4 and SUDHL6 cells with a panel of lentivirally encoded MCT-1 shRNAs or control shRNA. Following transduction, cells were selected in puromycin-containing media for 14 days. As shown in Fig. 4B, hairpin no. 3 was the most efficient in knocking down MCT-1 expression. Stably transduced cells that were viable after knocking down MCT-1 protein were selected for further analysis. The ERK inhibitor dramatically induced apoptosis in SUDHL6 control shRNA cells in a dose-dependent manner, and SUDHL6 shMCT-1 cells were less sensitive to ERK inhibitor compared with control shRNA cells (Fig. 4C, top). Similar results were observed in SUDHL4 control shRNA and shMCT-1 cells (Fig. 4C, bottom). Cell apoptosis induced by ERK inhibitor was also documented by the cleavage of PARP in Fig. 4D. These data derived from DLBCL cells with distinct levels of MCT-1 supported our contention that MCT-1 is relevant to the phenotype observed after exposing DLBCL cells to the ERK inhibitor.
Figure 4.
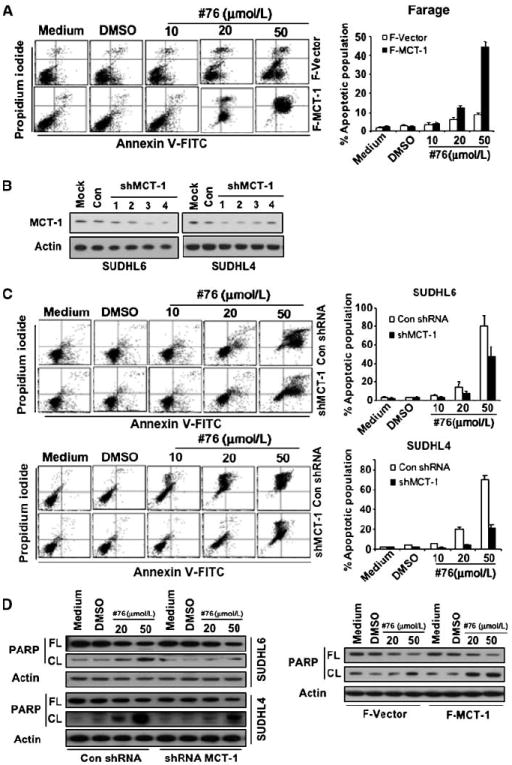
Elevated MCT-1 protein levels differentially sensitized DLBCL cells to ERK inhibition. A, Farage-Vector and Farage–MCT-1 cells were treated with different doses of no. 76. At 48 h after treatment, the cell apoptosis was measured by Annexin V and propidium iodide staining. Left, flow cytometric profile. Columns, mean of three independent experiments; bars, SD. B, SUDHL4 and SUDHL6 cells were stably transduced with a series of lentivirally encoded MCT-1 shRNAs or control shRNA. Following transduction, cells were selected in puromycin-containing medium for 14 d. MCT-1 protein level was monitored by immunoblotting. C, SUDHL6 cells stably transduced with MCT-1 shRNA no. 3 or control shRNA were treated with different doses of no. 76. At 48 h after treatment, the cell apoptosis was measured by Annexin V and propidium iodide staining. Columns, mean of three independent experiments; bars, SD. The same experiment as above was performed in SUDHL4 cells. D, PARP cleavage was detected by immunoblotting with anti-PARP antibody. FL, full length; CL, cleaved length.
In vivo antitumor efficacy of the ERK inhibitor
We subsequently examined whether no. 76 inhibits lymphoma growth in SCID mice. As shown in Fig. 5A, 10 mg/kg of no. 76 inhibited tumor growth in both Daudi and SUDHL6 xenograft models. At this dose, no lethal toxicity or significant weight loss was observed among treated animals. To confirm that the growth inhibition effect was correlated with MCT-1, we performed both immunoblotting and immunohistochemistry on xenografts. Figure 5B showed that no. 76 dramatically decreased MCT-1 protein levels but had minimal effect on ERK and p-ERK protein levels in Daudi and SUDHL6 xenografts. Similar data were obtained through examining MCT-1 protein levels by immunohistochemistry assays in xenografts (Fig. 5C). These data showed that the ERK inhibitor effectively inhibits lymphoma growth in vivo by interfering with ERK signaling and subsequently reducing MCT-1 levels and function.
Figure 5.
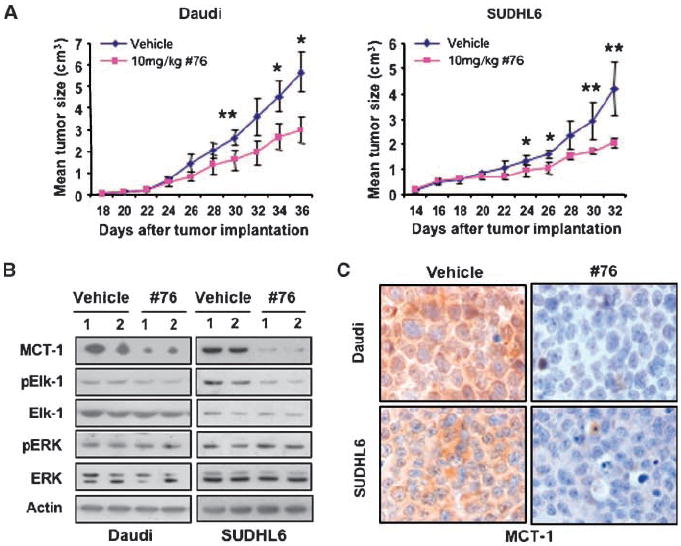
The ERK inhibitor has significant in vivo antitumor activity in human lymphoma xenograft models. A, 10 mice bearing s.c. tumors of either Daudi or SUDHL6 cells were divided into vehicle and no. 76 groups, with five mice in each group. The average tumor volume of each group with SD is shown as a function of time (*, P < 0.05; **, P < 0.01). Antitumor efficacy of ERK inhibitor was shown in Daudi and SUDHL6 xenograft models. B, in vivo mechanism of action of the ERK inhibitor in Daudi and SUDHL6 xenograft models. Two separate pooled protein extracts from both Daudi and SUDHL6 xenograft tumors were subjected to Western blot analysis using the indicated antibodies. C, immunostaining with anti–MCT-1 antibody shows that exposure to ERK inhibitor decreases MCT-1 protein level in Daudi and SUDHL6 xenograft models (original magnification, ×1,000).
Discussion
DLBCL is the most common lymphoid malignancy in adults, accounting for ~ 30,000 new cases each year (1). In addition to de novo DLBCL, 25% to 60% of FL, a low-grade NHL, will transform to an aggressive DLBCL (1, 26, 27). Defining the critical pathways that underlie the pathogenesis of DLBCL is an important step toward targeted therapy with the potential for greater efficacy and less toxicity. Using a high-throughput approach, we were able to show significantly elevated levels of a potential target, the MCT-1 oncogene in the vast majority (85%) of DLBCLs. In contrast, we did not observe significant levels of MCT-1 protein in most of the indolent lymphoid malignancies examined, including FL and chronic lymphocytic leukemia.
Disruption of MCT-1 function in DLBCL is hypothesized to result in an attenuated malignant phenotype. In this study, knocking down MCT-1 using shRNA in several DLBCL cell lines dramatically induced cell apoptosis. This provided the first direct genetic evidence that specific disruption of MCT-1 was capable of killing lymphoma cells with high endogenous levels of MCT-1. Previously, using a dominant-negative approach, we showed that a MCT-1 deletion mutant resulted in an altered translational profile and decreased anchorage-independent colony formation (8). We reasoned that in MCT-1 highly expressing DLBCLs, lymphoma cells might be more dependent on MCT-1 compared with low-level expressing cells. This phenomenon, “oncogene addiction,” first described by Weinstein, postulates that cancer cell growth and survival can often be impaired by the inactivation of a single oncogene and provides a rationale for molecular targeted therapy (28).
Because a specific and effective small-molecule inhibitor that selectively targets MCT-1 is currently not available, we sought to target the kinase directly upstream of MCT-1. We found that MCT-1 colocalized with ERK and that ERK was also readily detectable in human DLBCL, which is consistent with other reports that the ERK1/2 pathway is constitutively expressed/activated in some NHL cell lines (29). Furthermore, we have shown that knocking down ERK1/2 expression using a specific siRNA decreased MCT-1 protein levels. Conversely, an increase in ERK phosphorylation resulted in a marked elevation of MCT-1 protein. Combined with our previous data that MCT-1 was phosphorylated by ERK directly in vitro (12), we have established that MCT-1 is a direct substrate of ERK in vivo. Consequently, targeting the MEK/ERK signaling pathway is likely to be a useful therapeutic approach in DLBCL that exhibit high levels of MCT-1 protein. By using a novel small-molecule ERK inhibitor, we were able to show that the ERK inhibitor dramatically induced apoptosis of DLBCL cells and that elevated MCT-1 protein levels differentially sensitized DLBCL cells to inhibiting ERK. Significantly, treatment with the ERK inhibitor had marked antitumor activity in a human lymphoma xenograft established from DLBCL cell lines with elevated MCT-1 protein levels. These data suggested that MCT-1 is indeed a relevant target of ERK inhibition. In addition to the interruption of MCT-1 function, it is likely that ERK inhibition also regulates other proteins in DLBCL because ERK is known to phosphorylate a number of targets, including p90Rsk-1, c-Myc, and Elk-1 (30). Nevertheless, our above data, derived from syngeneic cells with distinct levels of MCT-1 protein displaying differential sensitivity to ERK inhibition, strongly supports our contention that MCT-1 is relevant to the phenotype observed after exposing DLBCL cells to the ERK inhibitor. The observation that knocking down MCT-1 induces apoptosis in lymphoma cells and recently published work demonstrating that a dominant-negative MCT-1 protein also affects the survival of lymphoma cells support the biological importance of interfering with MCT-1 function (8). It is therefore reasonable to consider MCT-1 an important target of ERK and that its functional disruption contributes at least in part to the phenotype observed by ERK inhibition.
The mechanism by which the ERK inhibitor no. 76 modifies MCT-1 activity is still unclear. One possible scenario is that the ERK inhibitor binds to the docking domain of ERK, which blocks the binding of MCT-1 to ERK. This is consistent with our findings that ERK inhibitor disrupted the association between MCT-1 and ERK. Here, we show that the ERK inhibitor decreased MCT-1 protein levels and phosphorylation. Thus, physical interaction between MCT-1 and ERK contributes to MCT-1 protein stability and biological activity. We also showed that knockdown of MEK2 by siRNA reduced MCT-1 protein levels. The MEK inhibitors (PD98059 and U0126) showed similar results as no. 76 but were somewhat less effective, suggesting that inhibition of ERK is more effective in disrupting MCT-1 function compared with inhibition of MEK. Our data also imply that ERK and MCT-1 can interact regardless of whether ERK is active or not. However, phosphorylation also contributes to protein stability of MCT-1 (12). Thus, MEK inhibitors only interfere with the phosphorylation of MCT-1, whereas ERK docking domain inhibitors reduce both protein interaction and subsequent phosphorylation. An important finding in this study was that no significant toxicity or weight loss was observed among treated animals at the dose of 10 mg/kg. This lack of apparent toxicity makes targeting specific ERK substrates an attractive clinical approach.
In summary, our findings reveal a functional linkage between MEK/ERK signaling and the oncogenic activity of MCT-1. We identified in a large-scale screen of NHL elevated levels of the MCT-1 protein in 85% of DLBCL. We showed for the first time that specifically knocking down MCT-1 was able to induce apoptosis in a number of DLBCL cell lines as well as suppress colony formation. Furthermore, we established that MCT-1 is a substrate of ERK and that ERK modulated both its protein levels and activity. By using a highly specific small molecule interfering with ERK function, we were able to show a significant degree of antitumor activity in an in vivo human lymphoma xenograft model established from DLBCL cell lines with elevated MCT-1 protein levels. Together, these data provide a rationale for the therapeutic use of ERK inhibitors in MCT-1–overexpressing lymphoma and support the development of novel small inhibitory molecules directed toward MCT-1 as a promising approach in DLBCL.
Supplementary Material
Acknowledgments
Grant support: Merit Review Award from the Department of Veterans Affairs (R.B. Gartenhaus) and NIH grant CA120215 (P. Shapiro).
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol. 1998;16:2780–95. doi: 10.1200/JCO.1998.16.8.2780. [DOI] [PubMed] [Google Scholar]
- 2.Abramson JS, Shipp MA. Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach. Blood. 2005;106:1164–74. doi: 10.1182/blood-2005-02-0687. [DOI] [PubMed] [Google Scholar]
- 3.Prosniak M, Dierov J, Okami K, et al. A novel candidate oncogene, MCT-1, is involved in cell cycle progression. Cancer Res. 1998;58:4233–7. [PubMed] [Google Scholar]
- 4.Shi B, Hsu HL, Evens AM, Gordon LI, Gartenhaus RB. Expression of the candidate MCT-1 oncogene in B- and T-cell lymphoid malignancies. Blood. 2003;102:297–302. doi: 10.1182/blood-2002-11-3486. [DOI] [PubMed] [Google Scholar]
- 5.Hsu HL, Shi B, Gartenhaus RB. The MCT-1 oncogene product impairs cell cycle checkpoint control and transforms human mammary epithelial cells. Oncogene. 2005;24:4956–64. doi: 10.1038/sj.onc.1208680. [DOI] [PubMed] [Google Scholar]
- 6.Levenson AS, Thurn KE, Simons LA, et al. MCT-1 oncogene contributes to increased in vivo tumorigenicity of MCF7 cells by promotion of angiogenesis and inhibition of apoptosis. Cancer Res. 2005;65:10651–6. doi: 10.1158/0008-5472.CAN-05-0845. [DOI] [PubMed] [Google Scholar]
- 7.Reinert LS, Shi B, Nandi S, et al. MCT-1 protein interacts with the cap complex and modulates messenger RNA translational profiles. Cancer Res. 2006;66:8994–9001. doi: 10.1158/0008-5472.CAN-06-1999. [DOI] [PubMed] [Google Scholar]
- 8.Mazan-Mamczarz K, Hagner P, Dai B, Corl S, Liu Z, Gartenhaus RB. Targeted suppression of MCT-1 attenuates the malignant phenotype through a translational mechanism. Leuk Res. 2009;33:474–82. doi: 10.1016/j.leukres.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 10.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 11.Wendel HG, Silva RL, Malina A, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandi S, Reinert LS, Hachem A, et al. Phosphorylation of MCT-1 by p44/42 MAPK is required for its stabilization in response to DNA damage. Oncogene. 2007;26:2283–9. doi: 10.1038/sj.onc.1210030. [DOI] [PubMed] [Google Scholar]
- 13.Duesbery NS, Webb CP, Vande Woude GF. MEK wars, a new front in the battle against cancer. Nat Med. 1999;5:736–7. doi: 10.1038/10457. [DOI] [PubMed] [Google Scholar]
- 14.Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000;96:1655–69. [PubMed] [Google Scholar]
- 15.Zheng B, Fiumara P, Li YV, et al. MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003;102:1019–27. doi: 10.1182/blood-2002-11-3507. [DOI] [PubMed] [Google Scholar]
- 16.Dai Y, Landowski TH, Rosen ST, Dent P, Grant S. Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood. 2002;100:3333–43. doi: 10.1182/blood-2002-03-0940. [DOI] [PubMed] [Google Scholar]
- 17.Dai Y, Chen S, Pei XY, et al. Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood. 2008;112:2439–49. doi: 10.1182/blood-2008-05-159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hancock CN, Macias A, Lee EK, Yu SY, Mackerell AD, Jr, Shapiro P. Identification of novel extracellular signal-regulated kinase docking domain inhibitors. J Med Chem. 2005;48:4586–95. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 19.Mansour SJ, Candia JM, Gloor KK, Ahn NG. Constitutively active mitogen-activated protein kinase kinase 1 (MAPKK1) and MAPKK2 mediate similar transcriptional and morphological responses. Cell Growth Differ. 1996;7:243–50. [PubMed] [Google Scholar]
- 20.Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–19. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Kim O, Yang J, Qiu Y. Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J Biol Chem. 2002;277:30066–71. doi: 10.1074/jbc.M201713200. [DOI] [PubMed] [Google Scholar]
- 22.Jaffe ESHN, Stein H, Vardiman JW. Pathology and genetics of tumors of the hematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. [Google Scholar]
- 23.Natkunam Y, Lossos IS, Taidi B, et al. Expression of the human germinal center-associated lymphoma (HGAL) protein, a new marker of germinal center B-cell derivation. Blood. 2005;105:3979–86. doi: 10.1182/blood-2004-08-3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai B, Kim O, Xie Y, et al. Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its over-expression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 2006;66:8058–64. doi: 10.1158/0008-5472.CAN-06-1364. [DOI] [PubMed] [Google Scholar]
- 25.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 26.Horning SJ, Rosenberg SA. The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. N Engl J Med. 1984;311:1471–5. doi: 10.1056/NEJM198412063112303. [DOI] [PubMed] [Google Scholar]
- 27.Elenitoba-Johnson KS, Jenson SD, Abbott RT, et al. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Proc Natl Acad Sci U S A. 2003;100:7259–64. doi: 10.1073/pnas.1137463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
- 29.Jazirehi AR, Vega MI, Chatterjee D, Goodglick L, Bonavida B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemo-sensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res. 2004;64:7117–26. doi: 10.1158/0008-5472.CAN-03-3500. [DOI] [PubMed] [Google Scholar]
- 30.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.