

Abstract
Cytokines are critically important for the growth and development of a variety of cells. Janus kinases (JAKs) associate with cytokine receptors and are essential for transmitting downstream cytokine signals. However, the regulation of the enzymatic activity of the JAKs is not well understood. Here, we investigated the role of tyrosine phosphorylation of JAK3 in regulating its kinase activity by analyzing mutations of tyrosine residues within the putative activation loop of the kinase domain. Specifically, tyrosine residues 980 and 981 of JAK3 were mutated to phenylalanine individually or doubly. We found that JAK3 is autophosphorylated on multiple sites including Y980 and Y981. Compared with the activity of wild-type (WT) JAK3, mutant Y980F demonstrated markedly decreased kinase activity, and optimal phosphorylation of JAK3 on other sites was dependent on Y980 phosphorylation. The mutant Y980F also exhibited reduced phosphorylation of its substrates, γc and STAT5A. In contrast, mutant Y981F had greatly increased kinase activity, whereas the double mutant, YY980/981FF, had intermediate activity. These results indicate that Y980 positively regulates JAK3 kinase activity whereas Y981 negatively regulates JAK3 kinase activity. These observations in JAK3 are similar to the findings in the kinase that is closely related to the JAK family, ZAP-70; mutations of tyrosine residues within the putative activation loop of ZAP-70 also have opposing actions. Thus, it will be important to determine whether this feature of regulation is unique to JAK3 or if it is also a feature of other JAKs. Given the importance of JAKs and particularly JAK3, it will be critical to fully dissect the positive and negative regulatory function of these and other tyrosine residues in the control of kinase activity and hence cytokine signaling.
Cytokines are critical regulators of growth and development of many tissues (1). Many of these cytokines bind to receptors that are members of the hematopoietic cytokine receptor family and are capable of recruiting or activating a variety of nonreceptor protein tyrosine kinases (PTKs) to induce downstream signaling (2–4). The JAKs, in particular, have emerged as key elements in the signaling of cytokine receptors (5–7).
This family consists of four known mammalian members: JAK1, JAK2, JAK3, and Tyk2. Different JAKs associate with specific cytokine receptors and are essential for transmitting cytokine signals to downstream molecules such as the signal transducers and activators of transcription (STATs) (6). JAK3, unlike other JAKs, is preferentially expressed in hematopoietic cells (8–13) and is essential for proper development and function of the immune system (14–18). It binds to the common γ subunit (γc), a shared subunit of the receptors for IL-2 (interleukin 2), IL-4, IL-7, IL-9, and IL-15, and is activated by these cytokines. Interestingly, mutation of either γc or JAK3 results in severe combined immunodeficiency (SCID) in humans or animals (14–18), demonstrating the importance of the JAK3/γc interaction in signaling by IL-2 and other γc cytokines. This is further supported by the demonstration that IL-2 signaling is largely abrogated in the absence of JAK3 (19).
Despite their importance, the regulation of the enzymatic activity of the JAKs is not well understood. In general, most PTKs are substrates for tyrosine phosphorylation, and this is an important aspect of their regulation (20). In particular, experimental evidence has revealed that autophosphorylation of critical tyrosine residues in the kinase domain of PTKs is required for full activation of the kinase. For instance, Y416 of c-Src (21) and Y1162 of the insulin receptor kinase (IRK) (22–23) have been identified as critical sites of autophosphorylation and positive regulators of kinase activity. That is, phosphorylation of these residues usually is associated with greatly increased kinase activity and mutations in these tyrosines result in markedly decreased catalytic activity. These observations can now be understood in a structural context. The crystal structure of IRK shows that Y-1158, Y-1162, and Y-1163 sites reside within a segment termed the activation loop (23) that lies between subdomains VII and VIII of the kinase domain (24). The phosphorylation of tyrosine residues within this loop appears to function to allow access of substrates to the active site, though the exact structure of this loop may differ somewhat among different enzymes (25). In contrast, phosphorylation of other tyrosine residues in the PTKs may negatively regulate their kinase activity. An important negative regulatory site for c-Src is Y527, a site outside of the catalytic domain. This site is important for intramolecular interactions that maintain the kinase in a low basal activity state. Its phosphorylation by the kinase Csk is thought to destabilize the kinase active site, resulting in abrogation of catalytic activity (21, 26).
Although no structural information pertaining to the JAKs is currently available, we sought to understand their mechanism of activation by determining the major sites of autophosphorylation of JAK3 and subsequently generating appropriate mutants to analyze the effects of such mutations. We first used phosphopeptide mapping to demonstrate that autophosphorylation of JAK3 occurred on multiple sites. One prominently phosphorylated peptide containing tyrosine residues Y980/Y981 within the putative activation loop was identified. Mutation of both the homologous tyrosine residues, Y1054/Y1055, in Tyk2, has indicated that these sites are important in the regulation of kinase activity, although the effect of individual tyrosine mutants in the regulation of Tyk2 kinase activity was not evaluated (27). We mutated the sites individually and found that phosphorylation of Y980 and Y981 had opposing functional effects.
MATERIALS AND METHODS
Cells and Antibodies (Abs).
COS-7 cells were cultured as described previously (8, 11). The anti-IL-2Rα, (anti-Tac mAb, 7G7) was obtained from David Nelson (National Cancer Institute, Bethesda, MD). The anti-phosphotyrosine mAb 4G10 was purchased from Upstate Biotechnology (Lake Placid, New York). Anti-glutathione S-transferase (GST) was purchased from Pharmacia. The anti-JAK3 and anti-STAT5A antisera were raised as described (8, 11).
Plasmid, Mutagenesis, and Transfection.
The JAK3 cDNA was subcloned into the expression vector pME18S (DNAX Research Institute, Palo Alto, CA) for COS-7 cell transfections and is referred to as WT JAK3 throughout. Mutagenesis of lysine residue 855 or tyrosine residues 980, 981, and 980/981 in JAK3 was performed by using the Transformer Site-Directed Mutagenesis Kit (CLONTECH) according to the manufacturer’s instructions and was confirmed by automatic DNA sequencing using PRISM Ready Reaction DyeDeoxy Terminator Cycle Sequencing Kit (Perkin–Elmer). The cytoplasmic region of human IL-2R γc subunit was subcloned into the pGEX-2T vector (Pharmacia) to express glutathione S-transferase (GST)γc fusion protein. A chimeric receptor, Tac-γc (α/γ/γ), consisting of the IL-2Rα (extracellular domain) and IL-2R-γc (transmembrane and cytoplasmic domains) was kindly provided by Warren Leonard (National Heart, Lung, and Blood Institute, Bethesda, MD) (48) and was cloned in pME18S. Murine STAT5A cDNA is a gift from Lothar Hennighausen (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD) (28). For transient transfections, COS-7 cells, at 80% confluence, were incubated with 5 μg of each cDNA construct using the DEAE–Dextran method (Promega) as described (29). In the cotransfections, a total of 10 μg of cDNA (5 μg of each cDNA) was used.
Immunoprecipitation, Immunoblotting, and Immune-Complex Kinase Assay.
Approximately 48 hr after transfection, COS-7 cells were lysed and immunoprecipitated with indicated antibodies as described (8, 11). The immunoprecipitates were washed three times with lysis buffer and then eluted from the beads with sample buffer for SDS/PAGE. For in vitro kinase assays, the JAK3 immunoprecipitates were washed once with 100 mM NaCl and 10 mM Hepes, pH 7.5, and resuspended in kinase reaction buffer (20 mM Tris, pH 7.5/5 mM MgCl2/5 mM MnCl2/1 μM ATP) containing 10 μCi [γ-32P]ATP (Amersham) with or without 2 μg of GSTγc fusion protein as an exogenous substrate as indicated. The reactions were performed at 0°C for the times indicated. The reactions were terminated by addition of lysis buffer containing 100 mM EDTA. The supernatants were incubated with Glutathione Sepharose 4B (Pharmacia) for 10 min at room temperature, and the beads were washed once with ice-cold lysis buffer to assess GSTγc phosphorylation. The JAK3 immunoprecipitates were also washed once with ice-cold lysis buffer. Beads were boiled in sample buffer, and the proteins were separated by SDS/PAGE and transferred to nitrocellulose (Schleicher & Schuell), after which immunoblotting (8, 11), autoradiography, and phosphopeptide mapping were performed. The radioactivity incorporated by JAK3 and GSTγc was quantitated using a Storm scanner (Molecular Dynamics). The levels of JAK3 and GSTγc fusion protein were also quantitated by densitometric scanning of the films.
Phosphopeptide Mapping.
Reagents for peptide mapping were purchased from J. T. Baker (Phillipsburg, NJ) and peptide mapping was performed as described previously (30). In brief, WT and mutant forms of JAK3 were expressed in COS-7 cells and immunoprecipitated using anti-JAK3 C-terminal antiserum, and in vitro kinase assays were performed for 10 min at room temperature. Samples were electrophoresed on polyacrylamide gels, transferred to nitrocellulose, and exposed to x-ray film. The band containing JAK3 was excised by using the film as a template. The excised pieces of nitrocellulose were blocked with 1% polyvinylpyrrolidone in 100 mM acidic acid for 1 hr at 37°C, washed three times with digestion buffer (1% NH4CO3, pH 8.4), and digested with 0.5 μg trypsin (Promega) overnight at 37°C. Peptides were recovered from the supernatant and dried in a vacuum centrifuge. The dried precipitates were washed and resuspended in 5 μl of H2O for analysis by high-voltage electrophoresis and thin layer chromatography (TLC). Tryptic peptides only or tryptic peptides mixed with a synthetic peptide that contains phosphorylated tyrosine residues 980 and 981 were separated in the first dimension with pH 4.72 buffer by high-voltage electrophoresis by using a HTLE-7000 electrophoretic apparatus (C.B.S. Scientific, Del Mar, CA) for 1 hr at 1.0 kV. Separation in the second dimension was performed by TLC using butanol (5%)/pyridine (2.5%)/acetic acid (2.5%)/deionized water (90%) for 6 hr. 32P-labeled peptides were visualized by autoradiography for the indicated times, and the position of the synthetic phosphopeptide was visualized by ninhydrin staining.
Electrophoretic Mobility-Shift Assays (EMSA).
COS-7 cells at 80% confluence were transfected with various plasmids using DEAE–Dextran methods. Two days later, cell extracts were prepared for EMSA as described (31). The cDNA probe, an oligonucleotide corresponding to a GAS (IFN-γ activation site)-like element found in the CD23 promoter (5′-GATC AAG ACC ATT TCT AAG AAA TCT ATC-3′), was synthesized, annealed to generate a double-stranded probe, and labeled with [γ-32P]ATP. The extracts were incubated with 32P-labeled probe for 15 min at room temperature. The DNA-binding complexes were resolved by electrophoresis in a native 4.5% polyacrylamide gel, and the results were visualized by autoradiography. For specificity controls, the extracts were incubated with unlabeled probe for 15 min at room temperature before the addition of 32P-labeled probe. For the supershift assays, the extracts were incubated with 32P-labeled probe for 15 min at room temperature, then incubated with antibody against STAT5A or with a control antibody for 30 min on ice.
RESULTS
JAK3 Contains Multiple Sites of Autophosphorylation.
To begin to delineate the sites of autophosphorylation on JAK3, we performed in vitro kinase assays, followed by phosphopeptide mapping of tryptic peptides. Fig. 1 shows that expressed WT JAK3 exhibits a number of autophosphorylated peptides (Fig. 1A Upper Left), and the most prominent spots were designated a–d. For other protein kinases, tyrosine residues between subdomains VII and VIII of the kinase domain typically are important sites of autophosphorylation. Therefore, we first determined whether the corresponding tyrosine residues of JAK3, Y980, and Y981 were autophosphorylated. To this end, both tyrosine residues were first mutated to phenylalanine individually or doubly (referred to as Y980F, Y981F, and YY980/981FF, respectively) by site-directed mutagenesis and confirmed by DNA sequencing. Next, WT and mutant forms of JAK3 were expressed in COS-7 cells, and in vitro kinase assays were performed on the immunoprecipitated proteins followed by phosphopeptide mapping of tryptic peptides. As shown in Fig. 1A (Upper Right), the phosphopeptide designated spot b was disproportionately reduced when the autophosphorylated peptides of mutant Y980F were analyzed, suggesting that spot b contains Y980. The signal in the other phosphorylated peptides of mutant Y980F was weaker as well, but to a much lesser extent than spot b. In addition, slightly residual phosphorylation of spot b was present in mutant Y980F when compared with the map of mutant YY980/981FF (Fig. 1A Lower Right), which completely lacked spot b and had somewhat reduced phosphorylation in other phosphopeptides, suggesting that Y981 is also a site of autophosphorylation and that spot b likely contains both residues. In fact, tryptic digestion of JAK3 would be predicted to yield a peptide that only contained these two tyrosine residues. To help confirm the identity of spot b, a phosphorylated peptide was synthesized and found to migrate similarly to spot b (Fig. 1B Right). Therefore, these data support the notion that Y980/Y981 are sites of autophosphorylation. It should be noted that a similar phosphopeptide mapping is obtained with JAK3 immunoprecipitated from lymphocytes (data not shown). In addition, the intensity of the signals suggested that the Y980F mutant had reduced kinase activity, whereas mutant Y981F appeared to have greater activity (Fig. 1A Lower Left).
Figure 1.
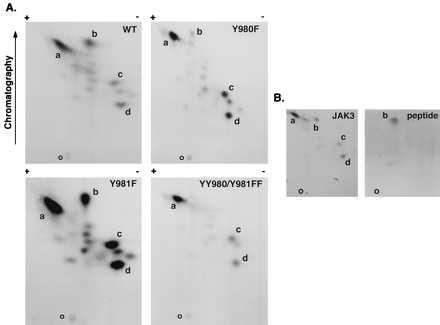
Tryptic phosphopeptide mapping of autophosphorylated JAK3. WT JAK3, mutant Y980F, mutant Y981F, and mutant YY980/981FF were expressed in COS-7 cells and immunoprecipitated using anti-JAK3 C-terminal antiserum, and in vitro kinase assays were performed for 10 min at room temperature. The phosphoproteins were subjected to SDS/PAGE, transferred to nitrocellulose, and exposed to x-ray film. The portions of the membrane containing WT and mutant JAK3 proteins were excised and digested in situ with trypsin. Eluted tryptic phosphopeptides from WT and indicated mutants (A) or eluted tryptic peptides from WT JAK3 mixed with a synthetic phosphopeptide DYpYpVVR (B) were analyzed by two-dimensional peptide mapping. The 32P-labeled phosphopeptides were visualized by autoradiography (B Left), and the position of synthetic phosphopeptide was determined by ninhydrin staining as indicated (B Right). The orientation of the positive and negative electrodes during electrophoresis is indicated along with the direction of chromatography; the origin is indicated by the letter O. The most prominent spots were designated a–d. Because of the effect of the mutations on kinase activity, the exposure time was varied to achieve similar intensities for the map of each mutant (exposure time: Y980F, 96 hr; YY980/981FF, 48 hr; WT JAK3, 16 hr; and Y981F, 6 hr).
Tyrosine 980 Positively Regulates Whereas Tyrosine 981 Negatively Regulates JAK3 Kinase Activity in Vitro.
The data provided by the phosphopeptide mapping experiments suggested that phosphorylation of the two adjacent tyrosine residues Y980 and Y981 had opposing effects. To confirm the effects of these mutations on JAK3 kinase activity, we directly compared the catalytic activity of expressed WT JAK3 with that of mutants Y980F, Y981F, and YY980/981FF using an in vitro kinase assay (Fig. 2). As shown in Fig. 2, autophosphorylation of mutant Y980F was markedly decreased (Fig. 2A, lane 3) compared with WT JAK3 (Fig. 2A, lane 1). In contrast, autophosphorylation of mutant Y981F was increased (Fig. 2A, lane 4). The phosphorylation level of mutant YY980/981FF was intermediate to levels of the two single mutants and was comparable to the phosphorylation level of WT JAK3 (Fig. 2A, lane 5). Mutation of the critical lysine within subdomain II that typically binds to ATP (K855A) inactivates JAK3, as reported for other PTKs (Fig. 2A, lane 2). This result also supports that the observed phosphorylation of JAK3 can be attributed to autophosphorylation and is independent of other PTKs, because no phosphorylation of mutant K855A is detected. Anti-JAK3 immunoblotting was used to confirm that comparable amounts of JAK3 were recovered in the immunoprecipitates from the different transfections (Fig. 2A Lower). Quantitation of the catalytic activity of the different mutants over time is shown in Fig. 2C.
Figure 2.
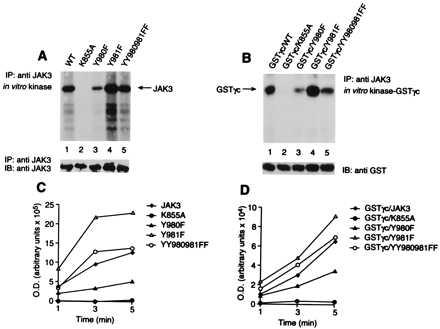
Effects of Y980 and Y981 mutation on JAK3 in vitro kinase activity. (A) JAK3 autophosphorylation. (B) γc phosphorylation. (C and D) The quantitation of phosphorylation over time. COS-7 cells were transfected with WT JAK3 (lane 1) or K855A (lane 2), Y980F (lane 3), Y981F (lane 4), and YY980/981FF (lane 5) mutant forms of JAK3. Lysates were immunoprecipitated with a JAK3 antiserum, and kinase assays were performed with GSTγc fusion protein as an exogenous substrate. (A and B) Data are from a 5-min reaction at 0°C. Immunoblotting with anti-JAK3 antiserum (A Lower) and anti-GST antiserum (B Lower) is also shown.
JAK3 associates with the common γ subunit, γc. One of the earliest events in cytokine-dependent signal transduction is phosphorylation of the receptor subunits, and it is believed that JAKs are the principal mediators of this event. To ascertain that the effects of mutation of Y980 and Y981 on autophosphorylation of JAK3 correlated with the effects on phosphorylation of its relevant substrates, we first asked if the various mutations of JAK3 affected the level of γc phosphorylation. To this end, we measured the in vitro phosphorylation of a GSTγc fusion protein that contains a cytoplasmic region of human IL-2R γc subunit (Fig. 2B). As expected, phosphorylation of γc was observed when the fusion protein was incubated with WT JAK3 (Fig. 2B, lane 1) but not with the mutant K855A (Fig. 2B, lane 2). Consistent with the data in Fig. 2A, substantially reduced phosphorylation was observed upon incubation with the mutant Y980F (Fig. 2B, lane 3), whereas greater phosphorylation was observed with the mutant Y981F (Fig. 2B, lane 4). The level of γc phosphorylation with the mutant YY980/981FF was again found to be intermediate to levels of the two single mutants and comparable to the levels of WT JAK3 (Fig. 2B, lane 5). The membrane was also immunoblotted to demonstrate that comparable amounts of GSTγc were recovered from each sample (Fig. 2B Lower). Phosphorylation of γc was also examined over time, and quantitative analysis of each sample is shown in Fig. 2D.
To confirm that the JAK3 mutants behaved similarly in cells as they did in vitro, we first examined their phosphorylation levels in situ. The different constructs were expressed in COS-7 cells, immunoprecipitated, electrophoresed, and immunoblotted with anti-phosphotyrosine antibodies (Fig. 3A). Except for the mutant K855A (Fig. 3A, lane 2), the WT JAK3 and mutants were all phosphorylated (Fig. 3A Upper). Compared with WT JAK3 (Fig. 3A, lane 1), mutant Y980F exhibited markedly decreased levels of phosphorylation (Fig. 3A, lane 3), whereas mutant Y981F demonstrated increased level of autophosphorylation (Fig. 3A, lane 4). The opposing effects mediated by mutation of Y980 and Y981 were confirmed once again, with the phosphorylation of mutant YY980/981FF being comparable to that of WT JAK3 (Fig. 3A, lane 5). This result further confirms that the observed phosphorylation of JAK3 can be attributed to autophosphorylation and is independent of JAK1 and other PTKs in COS-7 cells because no phosphorylation of mutant K855A is observed. The membrane was stripped and anti-JAK3 immunoblotting was used to ensure that comparable amounts of JAK3 were recovered in the immunoprecipitates from the different transfections (Fig. 3A Lower). These results indicate that alteration in the regulation of JAK3 activity in intact cells by the various mutants paralleled what was seen in vitro. These results support the conclusion that phosphorylation of Y980 of JAK3 is a required step for efficient phosphorylation of its substrate, γc.
Figure 3.
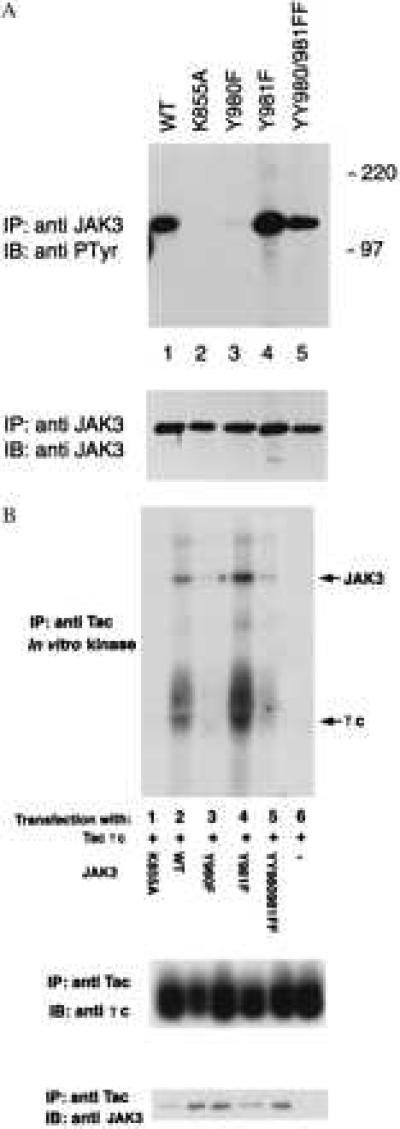
(A) Opposing effects of mutation of Y980 and Y981 on JAK3 phosphorylation in COS-7 cells. COS-7 cells were transfected with cDNAs (5 μg) encoding: WT JAK3 (lane 1), K855A (lane 2), Y980F (lane 3), Y981F (lane 4), and YY980/981FF (lane 5). Two days after transfection, cell lysates were precipitated with a JAK3 C-terminal antiserum. Samples were analyzed by SDS/PAGE, transferred to nitrocellulose, and subjected to immunoblotting with antiphosphotyrosine mAb (4G10, Upper) or anti-JAK3 antiserum (Lower). (B) JAK3 Y980 is required for efficient γc phosphorylation. COS-7 cells were cotransfected with 5 μg Tac-γc cDNA and 5 μg cDNAs encoding: K855A (lane 1), JAK3 (lane 2), Y980F (lane 3), Y981F (lane 4), and YY980/981FF (lane 5), or transfected with 5 μg Tac-γc cDNA only (lane 6), and immunoprecipitated with a mAb anti-Tac (7G7). In vitro kinase assays were performed on the immunoprecipitates at room temperature for 5 min. Samples were analyzed by SDS/PAGE and transferred to nitrocellulose, subjected to autoradiography (Top) and immunoblotting with mAb anti-γc (Middle), and anti-JAK3 antiserum (Bottom).
These results were also confirmed in a separate experiment in which JAK3 and Tac-γc were coexpressed in COS-7 cells (Fig. 3B). Tac-γc was immunoprecipitated by using mAb anti-Tac (7G7), and its ability to be phosphorylated in vitro by associated JAK3 was analyzed. The results were in agreement with data provided in Fig. 2.
Effect of JAK3 Mutations on STAT5A Phosphorylation and Activation.
The ligand-induced phosphorylation of receptor subunits by JAKs is thought to create a docking site that is recognized by STAT SH2 domains (5); subsequently, the STATs are also phosphorylated by JAKs. IL-2 has been shown to activate STAT5, and phosphorylation of the IL-2Rβ cytoplasmic domain is thought to provide docking sites for STAT5A or STAT5B, which subsequently become phosphorylated by JAK1, JAK3, or both (32–35). To assess whether the activating or inhibitory JAK3 mutants would have a corresponding effect on STAT activation, WT or mutant versions of JAK3 were coexpressed with the STAT5A in COS-7 cells. The cell lysates were immunoprecipitated with STAT5A antiserum, and the immune complexes were immunoblotted by using anti-phosphotyrosine mAb (Fig. 4 Upper). As expected (36), phosphorylation of STAT5A was observed when it was coexpressed with WT JAK3, but no phosphorylation of STAT5A was observed when expressed alone (Fig. 4, lane 5) or when STAT5A was coexpressed with the catalytically inactive mutant, K855A (Fig. 4, lane 2). Consistent with what was observed using γc, the phosphorylation of STAT5A was markedly decreased upon cotransfection with Y980F (Fig. 4, lane 3) as compared with WT JAK3 (Fig. 4, lane 1), whereas STAT5A phosphorylation was markedly increased with mutant Y981F (Fig. 4, lane 4). To ensure that equivalent levels of STAT5A were expressed, the membrane was stripped and immunoblotted with anti-STAT5A antiserum. As shown in Fig. 4 (Lower), similar amounts of protein were detected in each sample. The cell lysates were also immunoblotted using anti-JAK3 antiserum, and comparable amounts of protein were observed in each sample (data not shown). These results strongly support the contention that phosphorylation of JAK3 on Y980 is important for optimal phosphorylation of substrates like STAT5A.
Figure 4.
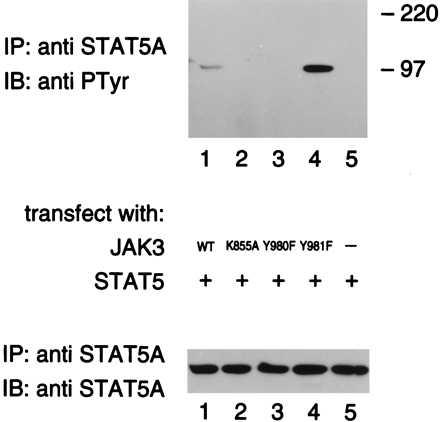
Autophosphorylation of Y980 is necessary for STAT5A phosphorylation. COS-7 cells were cotransfected with STAT5A cDNA (5 μg) and the cDNAs (5 μg) encoding: WT JAK3 (lane 1), K855A (lane 2), Y980F (lane 3), and Y981F (lane 4), or transfected with 5 μg STAT5A cDNA only (lane 5). Two days later, cells were lysed, immunoprecipitated with anti-STAT5A antiserum, and immunoblotted with mAb 4G10 (Upper) and anti-STAT5A antiserum (Lower).
Upon phosphorylation, STATs translocate to the nucleus and bind specific DNA elements to activate transcription (6, 7). To confirm the effect of the different JAK3 mutants on STAT5A activation, we analyzed STAT5A DNA-binding activity by using EMSA. We again transfected COS-7 cells with various JAK3 mutants along with the STAT5A cDNA. As shown in Fig. 5, no protein binding to the DNA probe was detected in COS-7 cells transfected with STAT5A only, STAT5A with mutant K855A, or in a mock transfection (Fig. 5, lanes 1, 3, and 7). When STAT5A was cotransfected with WT JAK3, a DNA complex was observed (Fig. 5, lane 2). However, much less DNA-binding activity was observed when STAT5A was cotransfected with mutant Y980F (Fig. 5, lane 4), whereas greater DNA-binding activity was observed with mutant Y981F (Fig. 5, lane 5). The double mutant, YY980/981FF, yielded a slightly reduced effect on DNA-binding activity (Fig. 5, lane 6) compared with WT JAK3 (Fig. 5, lane 1). To confirm that binding of STAT5 to the CD23-GAS element was specific, unlabeled probe was added and it efficiently competed the signal (Fig. 5, lanes 8–12). Supershift analysis was also used to confirm that the DNA complex comprised STAT5A (Fig. 5, lanes 13–20). The complexes were only supershifted when incubated with anti-STAT5A antiserum (lanes 14–16), and no supershift was observed when the complex was incubated with the control Ab (lanes 18–20). These results confirmed that alterations in activity of JAK3 measured in vitro and in cells correlated with the phosphorylation and activation of STAT5A. That is, phosphorylation of Y980 of JAK3 is critical for optimal kinase activity and likely is necessary for normal STAT5A activation. In contrast, phosphorylation of Y981 of JAK3 likely down-regulates kinase activity, interfering with its ability to phosphorylate relevant substrates.
Figure 5.
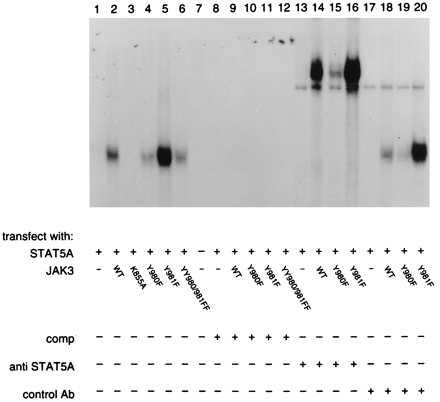
Regulation of STAT5A DNA binding by mutation of JAK3 residues Y980 and Y981. COS-7 cells were transfected with 5 μg STAT5A cDNA only (lanes 1, 8, 13, and 17) or cotransfected with 5 μg STAT5A and 5 μg cDNAs encoding: WT JAK3 (lanes 2, 9, 14, and 18), K855A (lane 3), Y980F (lanes 4, 10, 15, and 19), Y981F (lanes 5, 11, 16, and 20), or YY980/981FF (lanes 6 and 12), or mock transfected (lane 7). Two days later, cells were harvested and nuclear extracts were prepared. Protein determination was measured using BCA protein assay reagent (Bio-Rad). EMSA was performed using 5 μg protein and a labeled probe from the GAS-like element in the CD23 promoter (lanes 1–7). Competition with unlabeled probe is shown in lanes 8–12. Supershift analysis with anti-STAT5A antiserum (lanes 13–16) compared with a control antibody (lanes 17–20) is also shown.
DISCUSSION
In this report, we investigated the role of tyrosine phosphorylation of JAK3 in regulating its kinase activity. We first provide evidence that JAK3 has multiple autophosphorylation sites and that both Y980 and Y981, within the putative activation loop of JAK3, are phosphorylated. We found that Y980 is required for efficient phosphorylation of Y981 and other tyrosines within JAK3. Phosphorylation of 980 was also critical for phosphorylation of γc and STAT5A. In contrast, mutation of Y981 increases JAK3 activity and thus may be a negative regulatory site.
It is well recognized that JAK activation is associated with its own phosphorylation on tyrosine residues in the JAK/STAT signal transduction pathway, but it is less clear precisely how JAKs become activated (5–7). Upon ligand binding, JAK activation, which occurs as a results of receptor dimerization/oligomerization, is thought to involve auto- or trans-phosphorylation by another JAK. This leads to kinase activation, resulting in phosphorylation of the receptors and subsequently, recruitment and phosphorylation of STATs. For the homodimeric receptors like the growth hormone receptor, receptor dimerization effectively results in the apposition of two or more JAK2 molecules, which rapidly become transphosphorylated and activated. The mechanism of activation of different JAKs associated with heteroligomeric receptors is more complicated. In the case of the IFN receptors, two different JAKs are required for signaling. JAK1 and Tyk2 are phosphorylated in response to IFN-α, whereas JAK1 and JAK2 are phosphorylated in response to IFN-γ. In mutant cell lines, which lack either JAK1 or JAK2, the remaining kinase is not phosphorylated, and hence activation in response to IFN-γ does not occur (37). Likewise, tyrosine phosphorylation of Tyk2 in response to IFN-α is dependent on the presence of JAK1, and the reverse is also true (38). These data favor a model in which cross-phosphorylation by at least two JAKs is essential for signaling. This is further supported by the observations in which kinase-negative mutants of JAK1 sustained neither IFN-α nor IL-6 response. However, kinase-negative JAK1 did support substantial IFN-γ-induced gene expression (39). In addition, phosphorylation of JAK2 also did not require enzymatically active JAK1, suggesting an enzymatic role for JAK2 in IFN-γ signaling. Interestingly, kinase-negative JAK2 cannot sustain IFN-γ-inducible gene expression in mutant cells lacking JAK2. For IFN-γ response, therefore, the initial phosphorylation of JAK1 and JAK2 appears to be mediated by JAK2, whereas phosphorylation of IFN-γ receptor is carried out by JAK1 (39), indicating a functional non-equivalence of the JAKs in signaling.
For the γc-containing receptors, the mechanism by which JAK1 and JAK3 activation occurs is not well understood. Analysis of IL-2 signaling in B cell lines lacking JAK3 has demonstrated that IL-2-induced phosphorylation of IL-2β, JAK1, and STAT5 all require the presence of JAK3 (19). In contrast, it has been suggested that JAK1 activation is not required for activation of JAK3 and STAT5 in the human T cell line (40). These findings strongly suggest that phosphorylation of JAK3 is important in IL-2 signaling, and the importance of JAK1 remains to be determined.
Although our understanding of JAK activation is still rather incomplete, activation of PTK by phosphorylation of critical tyrosine residues within the kinase domain is a well recognized mechanism of positive regulation in virtually all PTKs. For the IRK, cis-autophosphorylation of Y1162 cannot occur because both substrate- and ATP-binding sites are blocked by Phe-1151. Trans-autophosphorylation of Y1162 is thought to dislodge the autoinhibitory loop, thus permitting the accessibility of both substrate and Mg-ATP to the catalytic site (23, 26). Our data as well as others indicate that overexpression of JAK3 results in its activation (36). In our studies, this results in autophosphorylation of Y980 and other tyrosines. Physiologically, JAK1 interacts with IL-2Rβ and JAK3 interacts with IL-2Rγ, and therefore, receptor chain pairing leads to kinase juxtaposition, presumably leading to mutual phosphorylation and activation during IL-2 signaling (41–43). However, JAK3 is also recruited to the receptor complex after ligand binding. Therefore, the physiological Y980 kinase might be JAK1 or JAK3, because it too can also bind the IL2-Rβ (11, 41). We also showed that phosphorylation of Y980 is required for efficient phosphorylation of Y981 and other tyrosine residues of JAK3. Our findings that mutation of Tyr-980 to Phe had markedly decreased kinase activity and an even greater decrease in phosphorylation of its substrates suggest that phosphorylation of Y980 is analogous to Src-Y416 and IRK-Y1162 and positively regulates kinase activity. Furthermore, no phosphorylation of mutant K855A was observed in COS-7 cells, suggesting that JAK1 and other endogenous PTKs cannot phosphorylate JAK3 without activation.
Like JAK3, ZAP-70 has duplicated tyrosine residues (Y492/Y493) in the putative activation loop of the kinase domain. Initial phosphorylation of Y493 by Lck in turn permits autophosphorylation of ZAP-70 at other tyrosine residues including Y492 (44–46). Mutation at Y493 results in reduced basal kinase activity and the inability to be activated by Lck. In contrast to the effect of Y493 phosphorylation, the effect of Y492 phosphorylation evidently is different (44–46). Phosphorylation of Y492 has been shown to inhibit kinase activity and antigen receptor function. Mutation of ZAP-70 Y492 resulted in much higher basal kinase activity, which could be stimulated further by phosphorylation on Y493. Thus, like ZAP-70, phosphorylation of adjacent tyrosine residues in the activation loop of JAK3 may have distinct functional effects. Interestingly, despite their similar structures, ZAP-70 and Syk did not appear to be regulated in an identical manner (47). Mutation of tyrosine residues in Tyk2 (Y1054 and Y1055) resulted in reduced basal catalytic activity and abolished the ligand-dependent activation of this protein (27). Mutation of tyrosines in the activation loop of JAK2 has been reported recently (49). These results clearly show that consequences of mutation of the comparable tyrosine residues in JAK2 and JAK3 are different. This study showed that mutation of Y1007 of JAK2 abolished kinase activity; mutation of both sites also reportedly blocked kinase activity. Therefore, the JAKS may be regulated in subtly different ways.
Understanding the precise mechanisms involved in regulating enzymatic activity of JAK3 is important, as it may provide important insights that may facilitate the design of pharmacological antagonists. JAK3 provides an attractive target for the generation of novel immunosuppressive drugs for several reasons, perhaps the most compelling of which is that JAK3 deficiency results in SCID in human and animals (14–18) but does not cause pathology in other organs and tissues. The limited tissue expression combined with its nonredundant function therefore make it an excellent pharmacological target. Ideally, such a compound would exploit the unique features of JAK3, and the opposing effects of the adjacent tyrosine residues within the action loop may be one of those features that could be exploited. Ultimately though, it will be important to fully delineate the phosphorylation sites within JAK kinases to understand precisely how the positive and negative regulation of JAK3 and other JAKs are effected and to identify potential binding sites for associated signaling proteins. In view of the importance of JAK3 for lymphoid cells, understanding the regulation of its enzymatic activity and the mechanisms by which it signals is of great interest not only for the generation of antagonists but also because this understanding should provide clues into the molecular basis of lymphocyte activation and immunoregulation.
Acknowledgments
We thank Drs. Julian Watts for advice concerning phosphopeptide mapping, John Ryan for technical assistance with EMSAs, and Lothar Hennighausen and Xiuwen Liu for kindly providing the STAT5A cDNA. We are also grateful to Alan Cheng for critical reading of this manuscript and to Mrs. Anka Hymel and Mr. George Poy for DNA sequencing.
ABBREVIATIONS
- EMSA
electrophoretic mobility-shift assay
- IFN
interferon
- γc
common γ subunit
- IL-2R
interleukin 2 receptor
- IRK
insulin receptor kinase
- JAK
Janus kinase
- GST
glutathione S-transferase
- PTK
protein tyrosine kinase
- SCID
severe combined immunodeficiency
- STATs
signal transducer and activators of transcription
- TLC
thin layer chromatography
- WT
wild type
References
- 1.Callard R, Gearing A. The Cytokine Facts Book. San Diego: Academic; 1994. [Google Scholar]
- 2.Bazan J F. Immunol Today. 1990;11:350–354. doi: 10.1016/0167-5699(90)90139-z. [DOI] [PubMed] [Google Scholar]
- 3.Cosman D. Cytokine. 1993;5:95–106. doi: 10.1016/1043-4666(93)90047-9. [DOI] [PubMed] [Google Scholar]
- 4.Kishimoto T, Taga T, Akira S. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 5.Ihle J N. Nature (London) 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 6.Schindler C, Darnell J E J. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 7.Johnston J A, Bacon C M, Riedy M C, O’Shea J J. J Leukocyte Biol. 1996;60:441–452. doi: 10.1002/jlb.60.4.441. [DOI] [PubMed] [Google Scholar]
- 8.Kawamura M, McVicar D W, Johnston J A, Blake T B, Chen Y Q, et al. Proc Natl Acad Sci USA. 1994;91:6374–6378. doi: 10.1073/pnas.91.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musso T, Johnston J A, Linnekin D, Varesio L, Rowe T K, O’Shea J J, McVicar D W. J Exp Med. 1995;181:1425–1431. doi: 10.1084/jem.181.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tortolani P J, Johnston J A, Bacon C M, McVicar D W, Shimosaka A, Linnekin D, Longo D L, O’Shea J J. Blood. 1995;85:3444–3451. [PubMed] [Google Scholar]
- 11.Johnston J A, Kawamura M, Kirken R A, Chen Y Q, Blake T B, Shibuya K, Ortaldo J R, McVicar D W, O’Shea J J. Nature (London) 1994;370:151–153. doi: 10.1038/370151a0. [DOI] [PubMed] [Google Scholar]
- 12.Gurniak C B, Berg L J. Blood. 1996;87:3151–3160. [PubMed] [Google Scholar]
- 13.Rane S G, Reddy E P. Oncogene. 1994;9:2415–2423. [PubMed] [Google Scholar]
- 14.Macchi P, Villa A, Gillani S, Sacco M G, Frattini A, Porta F, Ugazio A G, Johnston J A, Candotti F, O’Shea J J, Vezzoni P, Notarangelo L D. Nature (London) 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 15.Russell S M, Tayebi N, Nakajima H, Riedy M C, Roberts J L, Aman M J, Migone T S, Noguchi M, Markert M L, Buckley R H, O’Shea J J, Leonard W J. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 16.Thomis D C, Gurniak C B, Tivol E, Sharpe A H, Berg L J. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 17.Nosaka T, van Deursen J M, Tripp R A, Thierfelder W E, Witthuhn B A, McMickle A P, Doherty P C, Grosveld G C, Ihle J N. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 18.Park S Y, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, Saito T. Immunity. 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 19.Oakes S A, Candotti F, Johnston J A, Chen Y-Q, Ryan J J, Taylor N, Liu X, Hennighausen L, Notarangelo L D, Paul W E, Blaese R M, O’Shea J J. Immunity. 1996;5:605–615. doi: 10.1016/s1074-7613(00)80274-5. [DOI] [PubMed] [Google Scholar]
- 20.Courtneidge S A. Semin Cancer Biol. 1994;5:239–246. [PubMed] [Google Scholar]
- 21.Superti-Furga G, Courtneidge S A. Bioassays. 1995;17:321–330. doi: 10.1002/bies.950170408. [DOI] [PubMed] [Google Scholar]
- 22.Wilden P A, Kahn C R, Siddle K, White M F. J Biol Chem. 1992;267:16660–16668. [PubMed] [Google Scholar]
- 23.Hubbard S R, Wei L, Ellis L, Hendrickson W A. Nature (London) 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 24.Hunter T. Methods Enzymol. 1991;200:3–37. doi: 10.1016/0076-6879(91)00125-g. [DOI] [PubMed] [Google Scholar]
- 25.Morgan D O, De Bondt H L. Curr Opin Cell Biol. 1994;6:239–246. doi: 10.1016/0955-0674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 26.Xu W, Harrison S C, Eck M J. Nature (London) 1997;385:595–601. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 27.Gauzzi M C, Velazquez L, McKendry R, Mogensen K E, Fellous M, Pellegrini S. J Biol Chem. 1996;271:20494–20500. doi: 10.1074/jbc.271.34.20494. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Robinson G W, Gouilleux F, Groner B, Hennighausen L. Proc Natl Acad Sci USA. 1995;92:8831–8835. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Short Protocols in Molecular Biology. New York: Wiley; 1992. [Google Scholar]
- 30.Watts J D, Affolter M, Krebs D L, Wange R L, Samelson L E, Aebersold R. J Biol Chem. 1994;269:29520–29529. [PubMed] [Google Scholar]
- 31.Ryan J J, McReynolds L J, Keegan A, Wang L H, Garfein E, Rothman P, Nelms K, Paul W E. Immunity. 1996;4:123–132. doi: 10.1016/s1074-7613(00)80677-9. [DOI] [PubMed] [Google Scholar]
- 32.Hou J, Schindler U, Henzel W J, Wong S C, McKnight S L. Immunity. 1995;2:321–329. doi: 10.1016/1074-7613(95)90140-x. [DOI] [PubMed] [Google Scholar]
- 33.Johnston J A, Bacon C M, Finbloom D S, Rees R C, Kaplan D, et al. Proc Natl Acad Sci USA. 1995;92:8705–8709. doi: 10.1073/pnas.92.19.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedmann M C, Migone T S, Russell S M, Leonard W J. Proc Natl Acad Sci USA. 1996;93:2077–2082. doi: 10.1073/pnas.93.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaffen S L, Lai S Y, Ha M, Liu X, Hennighausen L, Greene W C, Goldsmith M A. J Biol Chem. 1996;271:21381–21390. doi: 10.1074/jbc.271.35.21381. [DOI] [PubMed] [Google Scholar]
- 36.Lin J X, Mietz J, Modi W S, John S, Leonard W J. J Biol Chem. 1996;271:10738–10744. [PubMed] [Google Scholar]
- 37.Watling D, Guschin D, Muller M, Silvennoinen O, Witthuhn B A, Quelle F W, Rogers N C, Schindler C, Stark G R, Ihle J N, Kerr I M. Nature (London) 1993;366:166–170. doi: 10.1038/366166a0. [DOI] [PubMed] [Google Scholar]
- 38.Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur A G, Barbieri G, Witthuhn B A, Schindler C, Pellegrini S, Wilks A F, Ihle J N, Stark G R, Kerr I M. Nature (London) 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- 39.Briscoe J, Rogers N C, Witthuhn B A, Watling D, Harpur A G, Wilks A F, Stark G R, Ihle J N, Kerr I M. EMBO J. 1996;15:799–809. [PMC free article] [PubMed] [Google Scholar]
- 40.Higuchi M, Asao H, Tanaka N, Oda K, Takeshita T, Nakamura M, Van Snick J, Sugamura K. Eur J Immunol. 1996;26:1322–1327. doi: 10.1002/eji.1830260622. [DOI] [PubMed] [Google Scholar]
- 41.Russell S M, Johnston J A, Noguchi M, Kawamura M, Bacon C M, et al. Science. 1994;266:1042–1045. doi: 10.1126/science.7973658. [DOI] [PubMed] [Google Scholar]
- 42.Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu Z J, Oishi I, Silvennoinen O, Witthuhn B A, Ihle J N, Taniguchi T. Science. 1994;266:1045–1047. doi: 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- 43.Boussiotis V A, Barber D L, Nakarai T, Freeman G J, Gribben J G, Bernstein G M, D’Andrea A D, Ritz J, Nadler L M. Science. 1994;266:1039–1042. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- 44.Chan A C, Dalton M, Johnson R, Kong G H, Wang T, Thoma R, Kurosaki T. EMBO J. 1995;14:2499–2508. doi: 10.1002/j.1460-2075.1995.tb07247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong G, Dalton M, Wardenburg J B, Straus D, Kurosaki T, Chan A C. Mol Cell Biol. 1996;16:5026–5035. doi: 10.1128/mcb.16.9.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wange R L, Guitian R, Isakov N, Watts J D, Aebersold R, Samelson L E. J Biol Chem. 1995;270:18730–18733. doi: 10.1074/jbc.270.32.18730. [DOI] [PubMed] [Google Scholar]
- 47.Chu D H, Spits H, Peyron J F, Rowley R B, Bolen J B, Weiss A. EMBO J. 1996;15:6251–6261. [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura Y, Russell S M, Mess S A, Friedmann M, Erdos M, Francois C, Jacques Y, Adelstein S, Leonard W J. Nature (London) 1994;369:330–333. doi: 10.1038/369330a0. [DOI] [PubMed] [Google Scholar]
- 49.Feng J, Witthuhn B A, Matsuda T, Kohlhuber F, Kerr I M, Ihle J N. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]