

Abstract
Several unanswered questions in T cell immunobiology relating to intracellular processing or in vivo antigen presentation could be approached if convenient, specific, and sensitive reagents were available for detecting the peptide–major histocompatibility complex (MHC) class I or class II ligands recognized by αβ T cell receptors. For this reason, we have developed a method using homogeneously loaded peptide–MHC class II complexes to generate and select specific mAb reactive with these structures using hen egg lysozyme (HEL) and I-Ak as a model system. mAbs specific for either HEL-(46–61)–Ak or HEL-(116–129)–Ak have been isolated. They cross-react with a small subset of I-Ak molecules loaded with self peptides but can nonetheless be used for flow cytometry, immunoprecipitation, Western blotting, and intracellular immunofluorescence to detect specific HEL peptide–MHC class II complexes formed by either peptide exposure or natural processing of native HEL. An example of the utility of these reagents is provided herein by using one of the anti-HEL-(46–61)–Ak specific mAbs to visualize intracellular compartments where I-Ak is loaded with HEL-derived peptides early after antigen administration. Other uses, especially for in vivo tracking of specific ligand-bearing antigen-presenting cells, are discussed.
The majority of αβ receptor-bearing T cells recognizes antigen as peptide–major histocompatibility complex (MHC) class I or class II complexes displayed on the surface of antigen-presenting cells (APCs), and the biochemistry of peptide–MHC molecule interaction, the cell biology of peptide generation from intact protein antigen, and the site(s) of intracellular association of these peptides with MHC molecules have been explored in great detail (for review, see refs. 1 and 2). Despite this progress in understanding antigen processing and presentation, it is still not known exactly where MHC class II molecules are loaded with particular peptides or whether all APCs process antigen in the same way or with the same efficiency. Past studies have necessarily relied on cells that can be obtained in large numbers for biochemical analysis. Rare APCs, such as dendritic cells, could have specialized antigen-processing mechanisms not easily amenable to study by current techniques, and for many antigens and most cells, a kinetic and spatial analysis of the formation of class II complexes involving specific peptide has not been possible.
In vivo antigen presentation is also incompletely understood. It is not clear which cells capture antigen and present it in immunogenic or tolerizing form to T cells after antigen introduction by various routes. Likewise, we are only beginning to understand the dynamics of antigen-specific immune cell interactions (3). This includes such issues as where the earliest stages of antigen presentation and T cell activation occur, where T and B cells collaborate, and where CD4+ T cells interact with CD8+ T cells in help-dependent cytotoxic responses.
A major limitation in studying these issues is the lack of reagents able to identify and quantitate T cell receptor (TCR) ligands on individual cells, to detect such complexes within intact cells, or to characterize cells bearing these ligands by methods such as flow cytometry or immunohistochemistry. Nearly all assays for T cell ligands have necessarily been indirect and have relied on measuring the ability of putative ligands to activate lymphocytes of the relevant specificity. It would be much more desirable to use soluble probes for these antigenic structures. Two major approaches are available for this purpose. The first is the development of soluble versions of TCRs. Unfortunately, the affinities of all TCRs purified to date are in the range of 10−3 to 10−7 M (4), too low to stay bound to their target under the desired assay conditions. Multimerization can decrease off-rate, however, permitting TCR oligomers to serve as probes for MHC-bound antigens (5). An alternative strategy is to use antibodies. Several mAbs to MHC class II molecules associated with peptides derived from endogenous proteins have been reported (6–11). These mAbs have proven very useful, but the inability to conveniently employ the source protein as an exogenous antigen or the substantial reactivity of these mAb with cells not exposed to the nominal antigen has not permitted them to be used effectively for the types of studies mentioned above.
We report herein that immunization with APCs expressing I-Ak homogeneously loaded with a single peptide derived from hen egg lysozyme (HEL) allowed the generation of mAbs recognizing either of two distinct processed HEL determinants in the context of I-Ak. We characterize these antibodies in detail, demonstrate reactivity with a small subpopulation of self peptide–MHC class II complexes, and show that, despite this low background reactivity, they can be used for flow cytometric analysis of APCs bearing processed HEL protein, biochemical detection of peptide–MHC class II complexes formed by natural processing of native HEL, and immunofluorescent staining of intracellular compartments of MHC class II loading in individual APCs.
MATERIALS AND METHODS
Antigens.
HEL peptides containing residues 46–61 (NTDGSTDYGILQINSR) and 116–129 (KGTDVQAWIRGCRL) were synthesized by the Peptide Synthesis Facility of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Variants of HEL-(52–61) with alanine substitutions at positions 53, 56, and 57 were a gift from E. R. Unanue (Washington University, St. Louis, MO). HEL protein was purchased from Sigma.
Constructs.
I-Akα-ζ chain and I-Akβ-ζ chain chimeras. DNA sequences coding for the extracellular domains of Ak α and β chains (α1 and α2 and β1 and β2) were made by PCR using oligonucleotides containing XhoI and BglII restriction sites. Primer sequences were as follows: forward primer for α chain, 5′-GTCAGAGCTACTCGAGCAGAGACCAGGATGCCGTG-3′; backward primer for α chain, 5′-CTGTGACCAGATCTCCCTCAGGTTCCCAGTGTTTC-3′; forward primer for β chain, 5′-GTCAGAGCTACTCGAGTGTGCCTTAGAGATGGCTC-3′; backward primer for β chain, 5′-CTGTGACCAGATCTCCGGACTGTGCCCGCCACTC-3′. PCR products were digested with XhoI and BglII and subcloned into pCDL-SR-2B4-ζ [encoding the 2B4 TCR fused to TCR-ζ (12)] digested with the same enzymes. As a result, the Ak DNA fragments replaced the regions coding for the 2B4 TCR extracellular domains, allowing Akα and Akβ to be expressed as chimeric molecules with the ζ chain transmembrane and cytoplasmic regions (Fig. 1A).
Figure 1.
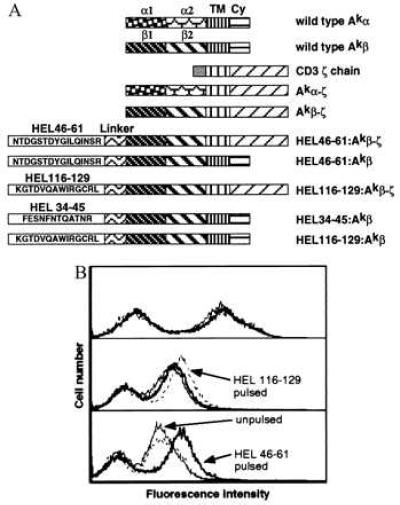
Immunization of rats with RBL expressing HEL peptide–Ak-ζ chimeras can elicit HEL–Ak-specific antibodies in serum. (A) Constructs encoding chimeric Akα-ζ and various HEL peptide–Akβ-ζ and Akβ molecules. See Materials and Methods for details on the construction. (B) Binding to HEL peptide-pulse-labeled spleen by rat antisera raised against RBL cells expressing HEL peptide–Ak-ζ molecules. 10–2.16 supernatant (Top) or serum (1:500 dilution) from rats immunized with RBL transfectants expressing HEL-(116–129)–Ak-ζ (Middle) or HEL-(46–61)–Ak-ζ (Bottom) were used to stain CBA/J mouse spleen cells pre-pulsed in medium alone (solid thin line) or with HEL-(116–129) peptide (dashed line) or HEL-(46–61) peptide (solid thick line). Binding was detected with secondary antibodies conjugated to FITC [goat anti-mouse IgG2b for 10–2.16 and goat F(ab′)2 anti-rat IgG (absorbed against mouse) for rat antisera]. Cells were analyzed by flow cytometry using propidium iodide exclusion of dead cells. The right peak corresponds to positive staining of Ak on B cells. Staining with a normal rat IgG control only gave the left peak (data not shown).
Peptide-linked Akβ chain (13) and peptide-linked Akβ-ζ chain chimeras.
HEL-(46–61)–Akβ-ζ chain and HEL-(46–61)–Akβ constructs were produced first. These constructs then served as cloning vectors for making constructs with other peptides. A DNA fragment coding for a peptide containing residues 46–61 and a linker region was inserted between codons 4 and 5 of the β1 domain with SfiI sites flanking the peptide coding region, as described (14). For making Akβ chain constructs containing other peptides, double-stranded DNA coding for the desired peptides was made by PCR and spliced into the HEL-(46–61)–Akβ-ζ chain or HEL-(46–61)–Akβ chain constructs by using SfiI sites.
Transfections.
These chimeric constructs were electroporated into RBL cells to produce stable transfectants, as described (14). G418-resistant cells were analyzed by flow cytometry 7–10 days after transfection by using a pan-Ak-specific mAb, 10–2.16 (15). Positive populations were cloned by limiting dilution and clones were checked for the ability to activate HEL-specific hybridomas.
Immunization with Transfectants Expressing Single Peptide–MHC Class II Complexes and mAb Production.
The RBL HEL peptide–Ak-ζ transfectants were used to immunize male Wistar white rats (Harlan Breeders, Indianapolis, IN). Twenty million cells in 1 ml of PBS were injected i.p. into each rat four times at 10-day intervals. Three days after the last injection, the spleens were harvested and the cells were fused with mouse myeloma Sp2/0 cells (16). The supernatants from growing hybridomas were screened using the RBL transfectants as described below. For the HEL-(46–61)–Ak fusion, 40 of 500 primary clones screened were found to react with transfectants expressing HEL peptide–Ak combinations. Only 14 of the 40 positive clones reacted uniquely with RBL expressing the HEL-(46–61)–Ak-ζ complexes. The supernatants from the 14 clones were further screened by flow cytometry using CBA/J spleen cells pulsed with either HEL-(46–61) or HEL-(116–129) peptides. Two were found to react better with the HEL-(46–61)-pulsed cells (B6G and C4H). The supernatants from the HEL-(116–129)–Ak fusion were similarly screened, and one clone (D8H) was identified that reacted uniquely with HEL-(116–129)-peptide-pulsed spleen cells. The hybridomas secreting mAbs with the desired specificities were further subcloned by limiting dilution.
Hybridoma Screening by Cell-Based ELISA.
RBL stable transfectants expressing the various I-Ak constructs were added to 96-well tissue culture plates at 2 × 105 cells per well in complete medium, allowed to grow overnight at 37°C, and then were fixed with 0.5% paraformaldehyde in PBS for 15 min at room temperature. After washing with PBS, the plates were blocked with 2% BSA in PBS for 2 h at 37°C and the hybridoma supernatants added at 50 μl per well. After a 1-h incubation at 37°C, the plates were washed with PBS and incubated for 60 min at 37°C with horseradish peroxidase-conjugated goat anti-rat antibody (Jackson Immunoresearch). After washing, antibody binding was detected with 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (Sigma) in citrate buffer at pH 4.5. Plates were read at 405 nm and wells with optical density values substantially higher than the those of control wells were scored as positive.
Flow Cytometry.
Cells were stained with mouse (10–2.16; IgG2b; American Type Culture Collection TIB-93) or rat (see below) antibodies against Ak, essentially as described (17). Samples were analyzed by using a FACScan cytometer and cellquest software (Becton Dickinson) and propidium iodide to exclude dead cells.
Western Blotting.
CBA/J spleen cells were cultured in medium with 100 μM HEL protein or HEL peptides, overnight at 37°C. The cells were then washed with PBS three times, lysed in 1% Nonidet P-40/Tris⋅HCl lysis buffer at pH 8.0, and the lysate (5 × 106 cell equivalents per lane) was separated by SDS/PAGE on a 12% gel. Separated proteins were transferred onto nitrocellulose membranes, which were blocked with 5% BSA in PBS for 2 h at 37°C. After washing in PBS/0.1% Tween, B6GE1, C4H3, and D8H21 supernatants were added directly to membrane strips and incubated for 1 h at 37°C. After washing, the strips were incubated for another hour with a secondary goat anti-rat IgG antibody conjugated to horseradish peroxidase. Horseradish peroxidase was detected using chemiluminescence (ECL substrate; ICN).
Intracellular Staining.
RBL expressing I-Ak and invariant chain after gene transfer (14) were grown on poly-(l-lysine)-pretreated 13-mm diameter coverslips in 24-well plates. For antigen pulse-labeling, HEL or BSA was added to the culture medium to a final concentration of 3 mg/ml, and the cells were cultured for 6 h, washed, fixed with 0.5% paraformaldehyde in PBS for 20 min at room temperature, washed again, and permeabilized with 0.2% saponin in PBS for 30 min. Fixed cell monolayers were blocked with 2% BSA in PBS at 37°C for 1 h. Individual coverslips were stained with C4H3 supernatant or isotype-matched irrelevant antibodies, followed by biotin-conjugated mouse IgG2b anti-rat IgG2b (PharMingen) or biotin-conjugated goat anti-mouse IgG2b (Caltag, South San Francisco, CA), respectively. The biotin conjugates were revealed by using Cy3-avidin (Sigma). The cells were double stained with a mouse mAb against rat LAMP-1 (Ly1C6; IgG1; a gift from I. Mellman, Yale University, New Haven, CT) or a mouse mAb against rat trasferrin receptor (OX-26; IgG2a; PharMingen), followed by fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG1 or anti-mouse IgG2a (Caltag). Coverslips bearing the stained cells were mounted, and slides were viewed and photographed with a confocal fluorescence microscope equipped with an argon laser and dual detectors (CSLM, Molecular Dynamics).
RESULTS
Generation of mAbs to HEL Peptides Bound to I-Ak.
We reasoned that antibodies that specifically recognize antigen in its processed MHC-bound form might be best generated if one immunizes animals with MHC molecules homogeneously loaded with a single peptide. To obtain Ak molecules uniformly occupied with individual HEL peptides, we made constructs encoding Ak β molecules with amino-terminal extensions corresponding to the desired sequence (13). We also replaced the transmembrane and the cytoplasmic regions of both the α and β chains of Ak with those of the mouse TCR ζ chain because immunization with RBL cells expressing chimeric molecules of poorly immunogenic proteins fused to TCR ζ has been reported to elicit strong antibody responses to those proteins (18). As shown in Fig. 1A, two Ak β-ζ chain constructs were made, encoding two peptides corresponding to two different Ak-presented T cell determinants in HEL (19). These constructs were stably cotransfected with chimeric Ak α-ζ chain into RBL cells. Transfectants displaying HEL-(46–61)–Ak-ζ or HEL-(116–129)–Ak-ζ complexes specifically activate the cognate T cell hybridomas (data not shown), suggesting that the covalently linked complexes are folded properly.
Rats immunized with RBL stably transfected with HEL-(46–61)–Ak-ζ or HEL-(116–129)–Ak-ζ made antibodies that specifically stain a subpopulation of CBA/J spleen cells, corresponding to the B cells, much like the anti-Aβk mAb 10–2.16 (Fig. 1B). Importantly, rat serum from animals immunized with RBL transfected with HEL-(116–129)–Ak-ζ gives stronger staining of spleen cells pulsed with HEL-(116–129) peptide than of spleen cells pulsed with HEL-(46–61) peptide, whereas the antiserum raised against RBL transfected with HEL-(46–61)–Ak-ζ complexes shows a reciprocal pattern. This result indicates that immunization with the RBL transfectants elicits a substantial amount of peptide–class II complex-specific antibodies, as well as pan-reactive antibodies against I-Ak.
mAbs were generated by fusing the spleen cells from the seropositive rats with mouse myeloma SP2/0 cells. Large numbers of mAb supernatants were screened by a cell-based ELISA on 96-well plates containing RBL expressing the different covalent peptide–Ak complexes, allowing the rapid identification of peptide–Ak-reactive hybridomas. Candidate mAbs were subjected to a second round of screening by flow cytometry on peptide-loaded spleen cells and were subcloned.
Specificity of mAbs to HEL Peptides Bound to I-Ak.
Although many supernatants reacted with the desired covalent peptide–MHC class II combination, only two mAbs (designated C4H3 and B6GE1; both IgG2b) from the rat immunized with the HEL-(46–61)–Ak-ζ transfectants and one (D8H21; IgG2a) from the rat immunized with HEL-(116–129)–Ak-ζ were obtained that appear to have the desired specificity for noncovalently associated peptide–Ak complexes. B6GE1 specifically stains RBL transfected with Ak–HEL-(46–61) but not with Ak–HEL-(116–129), whereas the opposite is true of D8H21 (Fig. 2A). Neither mAb stains RBL transfected with a control construct, Ak–HEL-(34–45) (Fig. 2A). C4H3 behaves much like B6GE1 but appears to cross-react slightly with Ak–HEL-(116–129) (Fig. 2A); however, this cross-staining is only a few percent of that seen with the cognate mAb–ligand pair. This staining is not the result of recognition of the linker peptide because C4H3 does not stain cells expressing Ak–HEL-(34–45) (Fig. 2A). It appears to reflect low-affinity cross-recognition of the Ak–HEL-(116–129) complex because staining of both the Ak–HEL-(46–61) and Ak–HEL-(116–129) transfectants can also be seen with B6GE1 and D8H21 at very high mAb concentrations (data not shown).
Figure 2.

Rat mAbs specifically stain HEL peptide–Ak complexes. (A) Staining of RBL transfected with single peptide–Ak constructs. RBL transfected with covalent Ak–HEL peptide constructs or with wild-type I-Ak were stained with the indicated mAb supernatants (solid thick lines) or isotype-matched irrelevant controls (thin dotted lines) followed by appropriate secondary antibodies conjugated to FITC [mouse F(ab′)2 anti-rat IgG for rat mAbs or rat F(ab′)2 anti-mouse IgG for 10–2.16]. Cells were analyzed by flow cytometry and propidium iodide exclusion of dead cells. (B) Staining of spleen cells pulsed with HEL or HEL peptides in vitro. CBA/J spleen cells were incubated overnight in medium alone (thin dotted lines) or with the indicated concentrations of HEL or HEL peptides (thick solid lines) and were stained with the indicated rat mAb supernatants. mAb binding was detected as in Fig. 1B.
A second category of degenerate recognition is the staining of RBL transfected with wild-type I-Ak and not exposed to HEL antigen (Fig. 2A, compare solid and dotted lines). This staining cannot be attributed to peptide-independent low-affinity binding to the Ak molecule itself because it is not seen with RBL transfected with the control construct Ak–HEL-(34–45) or, for B6GE1, with Ak–HEL-(116–129), despite the fact that both those transfectants express more Ak molecules (as measured by 10–2.16 staining) than the transfectant expressing wild-type I-Ak (Fig. 2A). Similarly, each mAb also stains to some extent a subpopulation of HEL-unexposed spleen cells corresponding to B lymphocytes (Fig. 2B, dotted lines). This staining requires expression of I-Ak (Fig. 3A). Finally, the antibody bound to HEL-unexposed cells does not wash off at a substantially faster rate than it does from cells bearing authentic Ak–HEL-(46–61) complexes (data not shown), indicating that the staining is not likely to reflect low-affinity binding to most I-Ak molecules from which much of the mAb has eluted during sample processing. Thus, these data suggest that these mAbs cross-react with I-Ak occupied with a specific minor subset of naturally processed peptides derived either from serum proteins or proteins synthesized by the RBL cells and/or mouse splenic B cells.
Figure 3.
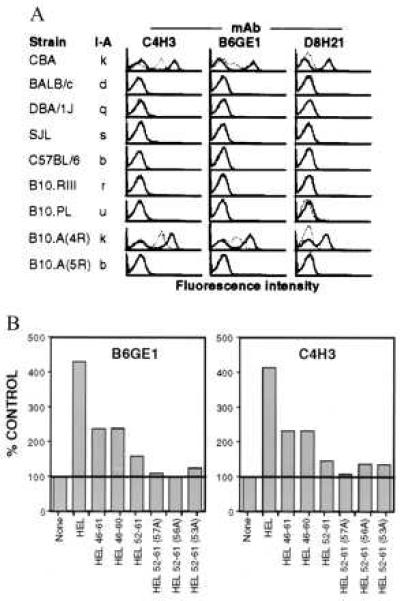
Specificity of HEL–Ak-specific mAbs. (A) Staining by HEL–Ak-specific mAbs is restricted by I-Ak. Spleen cells from the indicated strains of mice were stained with HEL–Ak-specific mAbs after overnight pulsing at 37°C with (bold line) or without (dashed line) 100 μM of the indicated peptides. Staining was performed as in Fig. 2B. (B) Fine specificity of HEL–Ak-specific mAbs. CBA/J spleen cells were incubated overnight in medium alone or with the indicated peptides at 100 μM. Cells were double-stained with B6GE1 or C4H3 [revealed with biotin-mouse F(ab′)2 anti-rat IgG and phycoerythrin-streptavidin] and FITC-RA3–6B2 (anti-B220). Data are the mean fluorescence values of 1 × 104 B220+ cells normalized to the mean fluorescence obtained with anti-I-Akβ and are expressed as a percentage of the values obtained with samples incubated in medium alone (control). The actual mean fluorescence values for the latter were as follows: B6GE1, 181.52; C4H3, 425.5.
These data with transfected RBL cells and normal spleen cells show that the mAbs are not uniquely reactive with the covalently bound constructs used in their elicitation. To address their specificity for HEL peptides or processed HEL associated noncovalently with Ak, H-2k spleen cells were incubated with these antigens and stained with the various mAbs. B cell staining with B6GE1 or C4H3 is much brighter for cells incubated with HEL-(46–61) peptide as compared with cells exposed to HEL-(116–129) peptide, and the converse is true for staining with D8H21 (Fig. 2B, solid lines). More importantly, all three mAbs specifically stain spleen cells incubated with intact HEL, demonstrating that they recognize naturally processed antigen (Fig. 2B). Staining of HEL-incubated spleen cells with the HEL-(46–61)-specific mAbs is always higher than that of peptide-exposed cells, whereas the same is not true of staining by D8H21 (Fig. 2B). This difference presumably relates to the relative efficiencies of the distinct pathways involved in processing these two HEL determinants (20). Thus, despite the low-level degeneracy seen with the highly expressed covalent HEL peptide constructs or the binding to what seems to be a small number of naturally generated peptide–Ak complexes, these mAbs show strong reactivity with the cognate synthetic or naturally processed HEL peptide bound to Ak.
Staining by B6GE1 and C4H3 is not influenced by deletion of a single amino acid at the carboxyl terminus of HEL-(46–61) and both mAbs also stain cells pulsed with HEL-(52–61), the core epitope recognized by T cells raised against HEL-(46–61)–Ak (21) (Fig. 3B). Binding to the HEL-(52–61) core epitope by B6GE1 is largely abrogated or substantially diminished by alanine substitutions at positions 53, 56, and 57, the three primary T cell epitopic residues in HEL-(46–61)–Ak (22). Binding by C4H3 is less sensitive to these mutations (Fig. 3B). These data indicate that B6GE1 and, to a lesser extent, C4H3 have fine specificities similar to those of many T cells recognizing HEL-(46–61)–Ak.
Biochemical Identification of Processed Antigen–MHC Class II Complexes with Peptide–Ak-Specific mAbs.
We next evaluated the potential unique applications of these mAbs to the study of antigen processing. Lysates from CBA spleen cells pulsed with either native HEL or HEL peptides were separated by SDS/PAGE without sample boiling and Western blotted onto nitrocellulose. The membrane-immobilized antigens were then probed with various antibodies. A rabbit antiserum specific for the cytoplasmic tail of Akα (23) binds to compact SDS-stable dimers, corresponding to a subset of peptide-loaded Ak molecules (1), and to free α chain, regardless of antigen pulsing (Fig. 4). mAbs C4H3 and B6GE1 recognize compact dimer bands in lysates from HEL-(46–61)-pulsed cells but not in lysates from cells pulsed with HEL-(116–129), but D8H21 only binds to the compact dimer band in lysates from the latter (Fig. 4). More importantly, both C4H3 and B6GE1 also bind to compact dimers formed after processing of native HEL and C4H3 can also immunoprecipitate compact dimers from lysates of HEL-pulsed spleen cells (Fig. 4 and data not shown). The failure of D8H21 to detect compact dimers in native HEL-pulse-labeled samples by Western blot (Fig. 4) or by immunoprecipitation (data not shown) may be due to the less-efficient formation of complexes involving this subdominant determinant (19, 20), consistent with flow cytometric analysis (see Fig. 2B), or to the formation of complexes containing the naturally processed determinant that do not resist SDS denaturation (24). None of the mAbs recognize free α or β chain, as expected from their specificity (Fig. 4). These results demonstrate that both mAbs against Ak:HEL-(46–61) can be used to analyze biochemically the loading of class II molecules with a single epitope derived from natural processing of an exogenous model antigen.
Figure 4.
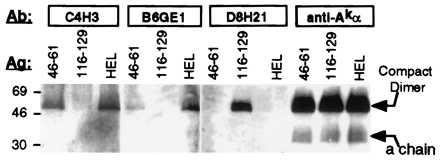
HEL-dependent binding to Ak compact dimers by HEL–Ak-specific mAbs. CBA/J spleen cells were pulsed with the indicated peptides and lysed, and the lysates were separated by SDS/PAGE and transferred to nitrocellulose by Western blotting. The membrane was probed with the indicated rat mAbs or with a rabbit antiserum against Aα. Compact dimer was identified as a band migrating at around 60 kDa in unheated samples.
Identification of Intracellular Compartments in Which Processed HEL–Ak Complexes Form.
Although bulk biochemical analysis in conjunction with organelle fractionation has proven very powerful in elucidating the steps involved in MHC class II antigen loading (25–29), published studies examining these events at the single cell level after acute antigen exposure are lacking. To view directly the site(s) of formation of HEL peptide–Ak complexes, we used C4H3 for intracellular immunofluorescent staining of Ak-expressing RBL cells that had been incubated with native HEL (Fig. 5). C4H3 stains cells pulsed with BSA very weakly if at all (Fig. 5 Upper Left) but gives bright staining of HEL-pulsed cells in a vesicular pattern that colocalizes primarily with endosomal organelles containing large amounts of the lysosomal marker LAMP-1 (Fig. 5 Upper Right). This result is in accord with prior studies showing the major site of HEL-(46–61) generation and Ak binding to be in late endocytic/lysosomal organelles (30, 31). Such colocalization is not seen when the cells incubated with HEL are double-stained with antibodies to the transferrin receptor (to identify early endosomes) and with C4H3 (Fig. 5 Lower Right). These results indicate that C4H3 can be used to detect Ak molecules recently loaded with HEL peptides in the endocytic compartments of single cells and to distinguish which subset of endocytic vesicles contains these complexes.
Figure 5.
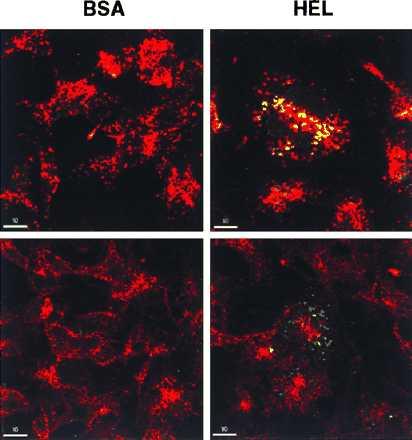
Visualization of compartments involved in intracellular association of the HEL-(46–61) determinant with Ak. RBL stably transfected with wild-type I-Ak and invariant chain were incubated for 6 h in medium containing BSA or HEL at 3 mg/ml, as indicated. Cells were washed, fixed, permeabilized, and stained with the indicated mAbs. C4H3 binding was revealed with FITC (green); LAMP-1 or transferrin receptor was revealed with Cy3 (red). Images were acquired with a ×63 objective lens. Excitation was at 468–514 nm and emission was collected at 540 ± 15 nm for FITC and 580 ± 15 nm for Cy3.
DISCUSSION
The conventional way to detect specific MHC–peptide complexes relies on the activation of T cells bearing relevant TCRs. However, such functional assays cannot be used to detect the intracellular formation and trafficking of peptide–MHC complexes in intact APCs, to identify TCR-ligand-bearing APCs in tissue sections, to phenotype these APCs in cell suspensions, to determine the number of cells in a given population that bear specific peptide–MHC molecule complexes on their surface, or to quantitate the surface level of specific peptide–MHC molecule complexes on these cells. mAbs that specifically recognize peptide–MHC complexes provide a means to overcome these limitations.
Antibodies that specifically recognize antigen–MHC class II complexes have been reported. These include mAbs against a mouse I-Eα peptide–I-Ab complex (Y-Ae) (6, 8), against myelin basic protein peptide–class II complexes of rat and human (7, 11), and against invariant chain CLIP–MHC class II (9, 10). Some of these antibodies have proven very useful for studying antigen processing and presentation (9, 28, 32) but suffer from several limitations for tracking antigen-bearing cells in vivo or for studying intracellular compartments for antigen processing in single cells. The mAb to myelin basic protein–class II complexes have significant background reactivity with endogenous peptide–MHC combinations. In some strains of mice, the Eα, and in all nonmutant mice, the CLIP peptides are from a self protein that is coexpressed in MHC class II+ cells in vivo, rather than from foreign proteins that can be administered acutely by various routes. This makes it difficult to use these reagents to study the kinetics of appearance of ligand-bearing APCs in vivo in normal animals. They are also of limited use for determining either the initial site(s) of formation or subsequent trafficking of peptide-loaded MHC class II molecules in intact cells, although the Y-Ae reagent has proved valuable through immunofluorescence and immunoelectron microscopy studies in addressing the steady-state intracellular distribution of specific complexes (32).
In contrast, we have generated mAbs against MHC class II molecules bound to peptides derived from a model soluble foreign antigen that can be administered to mice in vivo or to APCs in vitro in a route-, dose-, and time-controlled manner. HEL has been extensively characterized as a model antigen (33). In addition, several other reagents exist for studying HEL processing and presentation that make it a powerful system for studying lymphocyte biology. These reagents include mutants of HEL, natural species variants, and transgenic mice that have a monoclonal repertoire of anti-HEL T cells or B cells (34, 35). All these advantages should make the mAb described herein important tools for studying antigen presentation at the cell biological and organismal levels. These same considerations prompted an independent group to generate a similar mAb to Ak–HEL-(48–62) using a somewhat different immunization approach (36). These investigators have shown the value of this reagent for biochemical measurement of the efficiency of class II loading during active processing of a protein antigen (36).
Interestingly, in addition to staining specific HEL peptide–Ak complexes, C4H3 and B6GE1 were found to also weakly stain cells expressing I-Ak in an HEL-independent manner. This background staining is unlikely to be due to low-affinity binding to Ak alone because the two mAbs do not stain RBL cells transfected with Ak–HEL-(34–45) (Fig. 2A) but do react with cells expressing I-Ak bound to a variety of naturally processed self peptides (Fig. 2 and data not shown). This suggests that both mAbs have a substantial affinity for I-Ak molecules loaded with a small subset of self peptides, among which may be the homologous regions of mouse and rat lysozymes (unpublished observations). However, it is important to note that the HEL-dependent increase in mAb binding that we report herein using a variety of techniques cannot be attributed to changes in the levels of any such self peptides. In all experiments, control cells were treated in the same way as those exposed to HEL, to account for manipulation-induced changes in APC activation that might alter this self-peptide background.
Low but significant staining of cells not exposed to the cognate foreign antigen source, as seen herein with mAbs specific for HEL peptide–MHC class II combinations, has also been observed with antibodies directed against other peptide–MHC class II and also peptide–MHC class I complexes (7, 11, 37, 38), including the mAb to Ak–HEL-(48–62) described by Dadaglio et al. (36). It is thus interesting to speculate that most mAbs specific for a given foreign peptide–MHC complex and possessing an experimentally useful affinity will be likely to cross-react with complexes of the same MHC loaded with at least one self peptide. This phenomenon sets a lower limit of sensitivity for the use of these reagents. Nevertheless, the absolute level of the background staining with our reagents can be estimated to be 100 to a few hundred complexes per cell (unpublished observations), thus permitting detection of cells bearing physiologically relevant levels of complexes (a few hundred or greater). Furthermore, in several types of assays, binding to self peptide–Ak complexes is very low or undetectable, such as in Western blots (Fig. 4) and microscope-based immunofluorescence (Fig. 5), due either to a distinct affinity requirement for detection or a lower sensitivity as compared with flow cytometry.
An area in which these reagents clearly have an advantage over current techniques is the study of antigen-loading compartments in individual APCs and the identification of APCs in vivo that bear ligands recognizable by particular T cells. By immunofluorescence, we were able to stain intracellular compartments with C4H3 in HEL-pulsed cells but not in cells pulsed with a control protein (Fig. 5). The ability to visualize antigen loading compartments in a system where the addition of antigen can be carefully controlled in amount, form, and time of exposure should be enormously powerful for studying antigen processing in different APC populations.
Finally, these mAbs are proving extremely valuable for the in vivo analysis of cell interactions underlying adaptive immune responses. Using immunohistochemistry and flow cytometry, we have been able to both identify and localize within lymphoid tissues cells that process and present HEL after in vivo antigen administration (17). This latter application promises to greatly facilitate our understanding of how T cell immune responses are initiated and the identity of the critical presenting cells after various routes/modes of antigen administration.
Acknowledgments
We are grateful to Dr. E. R. Unanue (Washington University, St. Louis, MO) for HEL peptides. We thank Drs. F. Castellino and B. Lucas for helpful discussions and Drs. J. M. Austyn, F. Castellino, and D. H. Margulies for reading the manuscript. G.Z. and C.R.S. were supported by Visiting Fellowships from the Fogarty International Center.
ABBREVIATIONS
- MHC
major histocompatibility complex
- TCR
T cell receptor
- HEL
hen egg lysozyme
- APC
antigen-presenting cell
- FITC
fluorescein isothiocyanate
References
- 1.Germain R N. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 2.Cresswell P. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 3.Goodnow C C, Cyster J G. Curr Biol. 1997;7:219–223. doi: 10.1016/s0960-9822(06)00105-9. [DOI] [PubMed] [Google Scholar]
- 4.Margulies D H, Plaksin D, Khilko S N, Jelonek M T. Curr Opin Immunol. 1996;8:262–270. doi: 10.1016/s0952-7915(96)80066-5. [DOI] [PubMed] [Google Scholar]
- 5.Plaksin D, Polakova K, McPhie P, Margulies D H. J Immunol. 1997;158:2218–2227. [PubMed] [Google Scholar]
- 6.Rudensky A, Rath S, Preston H P, Murphy D B, Janeway C J. Nature (London) 1991;353:660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- 7.Aharoni R, Teitelbaum D, Arnon R, Puri J. Nature (London) 1991;351:147–150. doi: 10.1038/351147a0. [DOI] [PubMed] [Google Scholar]
- 8.Murphy D B, Rath S, Pizzo E, Rudensky A Y, George A, Larson J K, Janeway C A., Jr J Immunol. 1992;148:3483–3491. [PubMed] [Google Scholar]
- 9.Morkowski S, Goldrath A W, Eastman S, Ramachandra L, Freed D C, Whiteley P, Rudensky A Y. J Exp Med. 1995;182:1403–1413. doi: 10.1084/jem.182.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eastman S, Deftos M, DeRoos P C, Hsu D H, Teyton L, Braunstein N S, Hackett C J, Rudensky A. Eur J Immunol. 1996;26:385–393. doi: 10.1002/eji.1830260218. [DOI] [PubMed] [Google Scholar]
- 11.Puri J, Arnon R, Gurevich E, Teitelbaum D. J Immunol. 1997;158:2471–2476. [PubMed] [Google Scholar]
- 12.Engel I, Ottenhoff T H M, Klausner R D. Science. 1992;256:1318–1321. doi: 10.1126/science.1598575. [DOI] [PubMed] [Google Scholar]
- 13.Kozono H, White J, Clements J, Marrack P, Kappler J. Nature (London) 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 14.Zhong G, Castellino F, Romagnoli P, Germain R N. J Exp Med. 1996;184:2061–2066. doi: 10.1084/jem.184.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oi V T, Jones P P, Goding J W, Herzenberg L A, Herzenberg L A. Curr Top Microbiol Immunol. 1978;81:115–120. doi: 10.1007/978-3-642-67448-8_18. [DOI] [PubMed] [Google Scholar]
- 16.Hochschwender M S, Langeberg L K, Schneider D W, Lindstrom J M. Production of Rat × Mouse Hybridomas for the Study of the Nicotinic Acetylcholine Receptor. New York: Plenum; 1985. [Google Scholar]
- 17.Zhong G, Reis e Sousa C, Germain R N. J Exp Med. 1997;186:673–682. doi: 10.1084/jem.186.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callan C F M, Reyburn H T, Bowness P, Ottenhoff T H, Engel I, Klausner R D, Bell J I, McMichael A. Proc Natl Acad Sci USA. 1993;90:10454–10458. doi: 10.1073/pnas.90.22.10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Momburg F, Fuchs S, Prexler J, Busch R, Post M, Hämmerling G J, Adorini L. J Exp Med. 1993;178:1453–1458. doi: 10.1084/jem.178.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong G, Romagnoli P, Germain R N. J Exp Med. 1997;185:429–438. doi: 10.1084/jem.185.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen P M, Matsueda G R, Haber E, Unanue E R. J Immunol. 1985;135:368–373. [PubMed] [Google Scholar]
- 22.Allen P M, Matsueda G R, Evans R J, Dunbar J B, Jr, Marshall G R, Unanue E R. Nature (London) 1987;327:713–715. doi: 10.1038/327713a0. [DOI] [PubMed] [Google Scholar]
- 23.Sant A J, Hendrix L R, Coligan J E, Maloy W L, Germain R N. J Exp Med. 1991;174:799–808. doi: 10.1084/jem.174.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germain R N, Rinker A., Jr Nature (London) 1993;363:725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- 25.Amigorena S, Drake J R, Webster P, Mellman I. Nature (London) 1994;369:113–120. doi: 10.1038/369113a0. [DOI] [PubMed] [Google Scholar]
- 26.West M A, Lucocq J M, Watts C. Nature (London) 1994;369:147–151. doi: 10.1038/369147a0. [DOI] [PubMed] [Google Scholar]
- 27.Qiu Y, Xu X, Wandinger-Ness A, Dalke D P, Pierce S K. J Cell Biol. 1994;125:595–605. doi: 10.1083/jcb.125.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudensky A Y, Maric M, Eastman S, Shoemaker L, DeRoos P C, Blum J S. Immunity. 1994;1:585–594. doi: 10.1016/1074-7613(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 29.Castellino F, Germain R N. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 30.Harding C V, Collins D S, Slot J W, Geuze H J, Unanue E R. Cell. 1991;64:393–401. doi: 10.1016/0092-8674(91)90647-h. [DOI] [PubMed] [Google Scholar]
- 31.Harding C V, Geuze H J. J Immunol. 1993;151:3988–3998. [PubMed] [Google Scholar]
- 32.Morkowski S, Raposo G, Kleijimeer M, Geuze H J, Rudensky A Y. Eur J Immunol. 1997;27:609–617. doi: 10.1002/eji.1830270306. [DOI] [PubMed] [Google Scholar]
- 33.Sercarz E E, Lehmann P V, Ametani A, Benichou G, Miller A, Moudgil K. Annu Rev Immunol. 1993;11:729–766. doi: 10.1146/annurev.iy.11.040193.003501. [DOI] [PubMed] [Google Scholar]
- 34.Ho W Y, Cooke M P, Goodnow C C, Davis M M. J Exp Med. 1994;179:1539–1549. doi: 10.1084/jem.179.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodnow C C, Crosbie J, Adelstein S, Lavoie T B, Smith-Gill S J, Brink R A, Pritchard-Briscoe H, Wotherspoon J S, Loblay R H, Raphael K, Trent R J, Basten A. Nature (London) 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 36.Dadaglio G, Nelson C A, Deck M B, Petzold S J, Unanue E R. Immunity. 1997;6:727–738. doi: 10.1016/s1074-7613(00)80448-3. [DOI] [PubMed] [Google Scholar]
- 37.Duc H T, Rucay P, Righenzi S, Halle P O, Kourilsky P. Int Immunol. 1993;5:427–431. doi: 10.1093/intimm/5.4.427. [DOI] [PubMed] [Google Scholar]
- 38.Porgador A, Yewdell J W, Deng Y, Bennink J R, Germain R N. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]