

Abstract
The Fas–Fas ligand (FasL) system plays an important role in the induction of lymphoid apoptosis and has been implicated in the suppression of immune responses. Herein, we report that gene transfer of FasL inhibits tumor cell growth in vivo. Although such inhibition is expected in Fas+ tumor cell lines, marked regression was unexpectedly observed after FasL gene transfer into the CT26 colon carcinoma that does not express Fas. Infection by an adenoviral vector encoding FasL rapidly eliminated tumor masses in the Fas+ Renca tumor by inducing cell death, whereas the elimination of Fas− CT26 cells was mediated by inflammatory cells. Analysis of human malignancies revealed Fas, but not FasL, expression in a majority of tumors and susceptibility to FasL in most Fas+ cell lines. These findings suggest that gene transfer of FasL generates apoptotic responses and induces potent inflammatory reactions that can be used to induce the regression of malignancies.
The Fas antigen is a cell surface receptor that transduces apoptotic signals into cells (1). The physiological ligand of Fas, Fas ligand (FasL), also known as CD95L or Apo-1L, is a type 2 membrane protein that can transduce this signal upon cell contact (2) or in a soluble form (3). The primary function of the Fas–FasL system is thought to be the maintenance of homeostasis in the immune system by the clonal deletion of autoreactive lymphocytes in peripheral lymphoid tissues and the elimination of expanded lymphocyte populations (3–6).
Fas is expressed in a variety of cells of lymphoid or nonlymphoid origin, as well as many malignant cell lines (1, 3). In contrast, FasL expression in normal tissues is restricted to T lymphocytes and macrophage lineages, where it suppresses cellular and humoral immunity (4–7) and has been implicated in immune tolerance in the eye (8), testis (9), and an islet transplantation model (10). FasL protein has recently been detected in human lymphoma (11), melanoma (12), hepatoma (13), and colon cancer (14), where it has been suggested that it may promote evasion of antitumor immune responses.
We recently examined the effect of FasL expression on immune responses to tumors in allogeneic recipients. Although local immune responses, including alloantibody production, were inhibited by FasL, allogeneic tumor growth was inhibited in major histocompatibility complex-disparate recipient mice (7), and significant inflammation was observed in these transplanted tumors, suggesting that FasL expression might affect tumor growth in syngeneic tumors. We therefore examined the effect of FasL gene transfer on Fas+ and Fas− tumors in syngeneic hosts.
MATERIALS AND METHODS
Cells.
Renca, a mouse renal epithelial carcinoma cell line (15), and CT26, a mouse colon carcinoma cell line (15), grown in BALB/c mice, were obtained from the American Type Culture Collection and propagated in RPMI 1640 medium supplemented with 10% fetal calf serum, l-glutamine, and antibiotics.
HepG2, a human hepatocellular carcinoma cell line, and HeLa cells, a human cervical carcinoma cell line, were obtained from the American Type Culture Collection. Human glioma cell lines (G87, G138, and G373) were a gift from P. Kish and K. Murazko (University of Michigan Medical Center, Ann Arbor). Human melanoma cell lines (M316, M342, M347, M444, M449, and M720) were a gift from A. Chang (University of Michigan Medical Center, Ann Arbor). The melanoma cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum, l-glutamine, and antibiotics. Others were grown in DMEM with 10% fetal calf serum, l-glutamine, and antibiotics.
Generation of Stably Transduced FasL-Expressing Cell Line.
The CT26 cell line that expresses FasL stably (CT26-FasL) was generated as reported (7). Briefly, the mouse FasL cDNA was inserted into a mammalian expression vector that uses the cytomegalovirus enhancer/promoter, bovine growth hormone poly(A) signal, and neomycin-resistance gene as a selection marker. CT26 cells were transfected with this plasmid by electroporation and were selected with Geneticin (1 mg/ml; GIBCO/BRL). A clone that expressed FasL at high levels was isolated by limiting dilution. As a control, the CT26 clone transfected with the plasmid backbone (CT26-neo) was prepared in the same way.
Flow Cytometric Analysis of Fas and FasL Expression.
Target cells (1 × 106 cells) were stained with anti-Fas antibody (PharMingen) or isotype control IgG followed by fluorescein isothiocyanate (FITC)-conjugated anti-IgG second antibody (PharMingen), or Fas-Fc fusion protein (7) followed by FITC-conjugated antibody to the Fc fragment of IgG (Jackson Immunoresearch) to detect the expression of Fas or FasL, respectively. Relative fluorescence intensity was measured by fluorescence-activated cell sorter (FACS) analysis of 104 cells.
Cytotoxicity Assays.
FasL-mediated cytotoxicity was assayed essentially as described (7). The tumor cells indicated (1 × 106 cells) were incubated for 2 h at 37°C with 20 μCi of sodium [51Cr]chromate (Amersham; 1 Ci = 37 GBq) in 100 μl of RPMI 1640 medium containing 10% fetal calf serum. After washing with medium, the cells were used as the target. The 51Cr-labeled target cells (1 × 104 cells) were mixed with CT26-FasL or CT26 neo (at the indicated effector/target ratios) in round-bottomed microtiter plates in a total volume of 200 μl. The plates were centrifuged at 700 rpm in a GH-3.7 rotor for 2 min and incubated for 4 h at 37°C in 5% CO2/95% air. After the centrifugation at 1200 rpm in a GH-3.7 rotor for 5 min, the supernatants were collected with the harvesting frame (Skatron, Sterling, VA) and assayed for radioactivity. The spontaneous release of 51Cr was determined by incubating the target cells with the medium alone, whereas the maximum release was determined by incubating in 0.1% Triton X-100. The specific lysis (percent) was calculated as: (experimental 51Cr release − spontaneous 51Cr release)/(maximum 51Cr release − spontaneous 51Cr release). The ratio of spontaneous 51Cr release to maximum 51Cr release was between 11.1% and 17.6%.
Adenoviral Vectors (ADVs).
The recombinant ADV encoding FasL (ADV-FasL) was prepared by homologous recombination of sub360 genomic DNA, an Ad5 derivative with a deletion in the E3 region, and a FasL expression plasmid, pAd-FasL. The pAd-FasL encodes the mouse FasL cDNA under control of the cytomegalovirus enhancer/promoter and has a deletion in the E1A and E1B region, impairing the ability of this virus to replicate and transform nonpermissive cells. The presence of FasL cDNA and absence of the E1 in this viral genome was confirmed by Southern blot analysis. The construction and propagation of ADV-FasL were performed either in a FasL-resistant clone of 293 cells that was isolated by successive FasL transfections followed by limiting dilution and exhibited low expression of Fas by FACS analysis and relative resistance to FasL stimulation in 51Cr assays or by inclusion of a caspase inhibitor, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone, in the cell culture medium (16). Cesium chloride-purified virus was dialyzed against PBS and diluted for storage in a 13% glycerol/PBS solution to yield a final concentration of 1 × 1012 viral particles per ml. All stocks were sterilized by passage through a 0.45-μm filter and evaluated for the presence of replication-competent virus. In the plaque assay, 0.9 × 102 particles corresponded to 1 plaque-forming unit on 293 cells. The ADV deleted of E1 (ADV-ΔE1) was used as a negative control and prepared as described (17).
Terminal Deoxynucleotidyltransferase-Mediated dUTP-Digoxigenin Nick-End-Labeling (TUNEL) Assay.
Cultured cells (1 × 106 cells) or fresh-frozen tissue sections were stained by the TUNEL method with FITC labeling as directed by the manufacturer (Boehringer Mannheim) and examined by FACS analysis or fluorescence microscopy (16).
Animal Experiments.
Animal experiments were carried out in accordance with both institutional and National Institutes of Health animal care regulations. Six-week-old female BALB/c, nude, severe combined immunodeficiency (SCID), and SCID-beige (CB17-origin) mice were obtained from Charles River and Taconic Farms, respectively, and kept in a specific-pathogen-free environment. Cells were harvested with trypsin, incubated in growth medium at 37°C for 1 h to recover surface molecules, washed three times with PBS, and resuspended in PBS. The cell suspensions (2 × 106 cells per 50 μl) were injected subcutaneously into the flank.
Tumor size was followed in two perpendicular dimensions by using calipers and described as the tumor cross-sectional area (mm2). For the ADV-mediated FasL gene transfer, 50 μl of viral solution (1 × 1012 particles per ml) was injected into the tumor with a 26-gauge hypodermic needle after the tumor mass was established (∼0.5 cm). One mouse of each group was sacrificed for histological examination of the tumor and major organs, and the remaining animals (Renca, n = 6; CT26, n = 4) were observed for tumor growth. For the inoculation of CT26-FasL and CT26-neo, one mouse of each group was sacrificed for examination on day 2 and the remaining mice (BALB/c, n = 5; nude, SCID, and SCID-beige, n = 3) were followed. The histology of the major organs (liver, heart, lung, kidney, spleen, and thymus) was examined by microscopic observation of hematoxylin/eosin-stained slides by an experienced pathologist (D.G.).
Immunohistochemistry.
Fresh-frozen tissues of CT26-FasL and CT26-neo tumor on day 2 were fixed with acetone. The section was first incubated with anti-Ly-6G (GR-1) monoclonal antibody (RB6–8C5) (PharMingen) or an isotype control rat IgG. Biotinylated anti-rat IgG2b second antibody (PharMingen) was added followed by the addition of preformed avidin biotinylated horseradish peroxidase complex. The signal was visualized by incubation in peroxidase substrate. The RB6–8C5 antibody reacts mainly with neutrophils and, to a lesser extent, with activated monocytes/macrophage (18).
RESULTS
To examine the effect of FasL expression in a Fas+ tumor, the Renca renal epithelial carcinoma cell line was studied. Another cell line, the Fas− CT26 colon carcinoma, was studied for comparison. The Renca cell line, in contrast to CT26, expressed Fas and was susceptible to lysis by FasL in vitro in a chromium release assay, and this lysis of Renca cells was inhibited by a Fas-Fc fusion protein (Fig. 1A). To facilitate gene transfer into established tumors in vivo, an ADV encoding mouse FasL, ADV-FasL, was used. Because producer line 293 cells express Fas and are susceptible to FasL, a FasL-resistant clone of 293 cells was selected and used to produce this vector. ADV-FasL was also prepared in normal 293 cells by inclusion of a known peptide inhibitor of caspases, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. ADV-FasL infection in vitro resulted in the expression of FasL in both cell lines, though expression was lower in CT26 cells than in Renca cells (Fig. 1B). Analysis using the TUNEL assay, which detects DNA strand breaks in cells undergoing apoptosis, revealed that the majority of ADV-FasL infected Renca cells were apoptotic 24 h after infection in vitro but CT26 cells exhibited no apoptosis (Fig. 1C). Two days after ADV-FasL infection, ≥95% of Renca cells were nonviable, in contrast to the ADV-ΔE1 vector control, whereas ADV-FasL infection did not affect the viability of CT26 cells in vitro (Fig. 1D).
Figure 1.
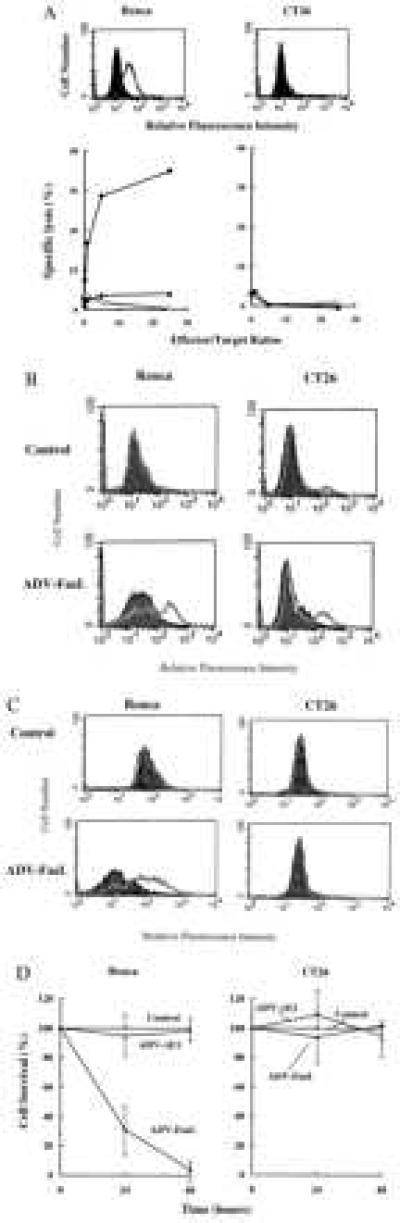
Infection by ADV-FasL induces apoptotic death in Fas+ Renca but not Fas− CT26 cells in vitro. (A) Fas expression and sensitivity to FasL in Renca and CT26 cells. Renca and CT26 cells (1 × 106 cells of each strain) were stained with anti-mouse Fas antibody followed by FITC-conjugated second antibody and analyzed with FACS (Upper). Sensitivity of Renca, but not CT26, cells to FasL exposure as determined by a chromium release assay in vitro is shown (Lower). Renca and CT26 target cells were labeled with 51Cr and mixed with CT26-FasL (•) or CT26-neo (○) effector cells at various ratios. FasL-induced cytotoxicity for Renca cells was neutralized by adding 5 μg of Fas-Fc fusion protein (7) in the medium (▴). The cytotoxicity caused by FasL was measured by 51Cr release after 4 h of coincubation. (B) FasL expression after adenoviral gene transfer in Renca and CT26 cells in vitro. Renca and CT26 cells were infected with ADV-FasL at multiplicity of infection (MOI) of 103 particles per cell. FasL was detected at 24 h after infection with a Fas-Fc fusion protein (open curve) compared with a control supernatant (shaded curve) followed by FITC-conjugated anti-IgGFc second antibody and analyzed by FACS. (C) Apoptosis after ADV-FasL infection in Renca but not in CT26 cells in vitro. Cells were infected with ADV-FasL (MOI = 103), and apoptotic cells were analyzed by TUNEL staining (open curve) compared with controls (shaded curve) at 24 h after infection. (D) Time course of viability after ADV-FasL infection. Renca or CT26 cells were infected with ADV-ΔE1 (triangles) or ADV-FasL (squares) at an MOI of 1 × 103. At 24 and 48 h after transfection, the cells were harvested and the number of surviving cells was counted by trypan-blue staining. The shown data represent the average (±SD) of three wells, and the percentage cell survival was defined as the relative number of viable cells compared with controls (circle).
To characterize the response of Renca cells to FasL expression in vivo, tumors were inoculated subcutaneously into the flanks of syngeneic BALB/c mice. After nodules were established (∼0.5 cm), tumors were injected with PBS, ADV-ΔE1, or ADV-FasL. Previous studies with similar titers of ADVs resulted in infection of 90–95% of cells in established Renca tumors (19). After injection of ADV-FasL, Renca tumors began to regress at 24 h, and no tumor was detectable 24 h after the second injection 2 days later (Fig. 2A). Mice were observed until day 19 with no recurrence of the malignancy, a finding confirmed histologically at the time of necropsy. Injection of ADV-ΔE1 at the same dose had no effect on tumor growth (Fig. 2A). Histological analysis at 24 h after the first injection of ADV-FasL revealed massive cell death in tumor tissue (Fig. 2B), and cells were markedly positive by TUNEL staining (Fig. 2B Insets).
Figure 2.
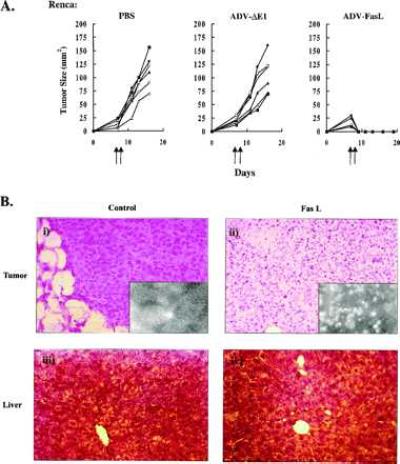
Regression and histologic analysis of Renca tumors in vivo after ADV-FasL gene transfer. (A) Growth of Renca tumors after adenovirus injection. Direct injection of PBS (n = 6), ADV-ΔE1 (n = 6), or ADV-FasL (n = 6) into established Renca tumors. Renca cells (2 × 106 cells) were inoculated into BALB/c mice. After the tumor (∼0.5 cm) was established, nodules were directly injected on days 7 and 8 (arrows). All ADVs were injected with 50 μl of a suspension containing 1 × 1012 particles per ml by using a 26-gauge hypodermic needle. Tumor size was measured in two perpendicular dimensions (a and b) and the tumor cross-sectional was calculated as (π/4)ab. (B) Histological comparison of Renca tumors treated with ADV-ΔE1 or ADV-FasL. (i) Hematoxylin/eosin-stained paraffin section shows Renca cells treated with ADV-ΔE1 growing without any immune or inflammatory response after 24 h. They are negative for TUNEL staining (Inset). (ii) In Renca tumors treated with ADV-FasL, the majority of tumor cells were destroyed and TUNEL-positive (Inset). (iii) Hematoxylin/eosin staining of liver from a mouse bearing the Renca tumor treated with ADV-ΔE1 (Left) or ADV-FasL (Right). No pathological changes were observed. (×400; Inset, ×600.)
ADV-FasL exerted a similar potent antitumor effect in CT26 tumors (Fig. 3A). This response was unexpected because this cell line was not sensitive to FasL in vitro (Fig. 1A). After the two injections of ADV-FasL on successive days, the tumor mass showed nearly complete regression. In this tumor, in contrast to Renca cells, recurrence was observed when injections were discontinued, probably because this line is less infectable by the ADVs (Fig. 1B). To characterize the effects of FasL in CT26 further, a subline that stably expressed FasL was derived. Stably transduced lines that express FasL (CT26-FasL) or vector-transduced cells (CT26-neo) were isolated (7) and inoculated subcutaneously into BALB/c mice. CT26-FasL cells failed to form tumors in syngeneic recipients, in contrast to CT 26-neo control cells (Fig. 3B).
Figure 3.
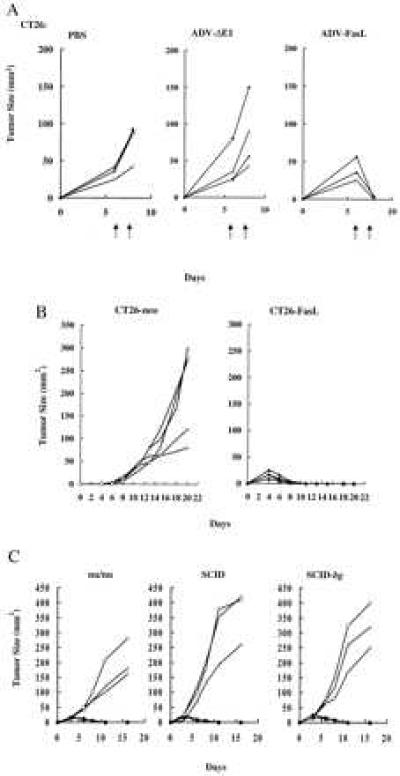
Regression of CT26 tumors after adenoviral-mediated or stable FasL gene expression. (A) Growth of CT26 tumor after adenovirus injection. CT26 cells (2 × 106 cells) were inoculated and injected with PBS, ADV-ΔE1, or ADV-FasL as in Fig. 2A (each at n = 4) on days 6 and 7 (arrows). (B) FasL expression induced rejection of CT26 cells in BALB/c recipient mice. The vector-transduced control CT26 line (CT26-neo) or the FasL-expressing CT26 line (CT26-FasL; n = 5) were inoculated subcutaneously into BALB/c mice (2 × 106 cells). Tumor cross-sectional area was calculated from the two-dimensional measurements with a caliper. (C) Rejection of CT26-FasL cells in immunodeficient mice. Nu/nu, SCID, and SCID-beige (SCID-bg) mice were inoculated with 2 × 106 CT26-FasL cell (solid symbols, each at n = 3) or CT26-neo (open symbols, each at n = 3), and tumor growth was monitored.
To determine whether regression of CT26 cells expressing FasL was dependent on immune cells, these lines were inoculated into immunodeficient mouse strains, including nude (20), SCID (21), or SCID-beige (22) mice, which are deficient in T, B, and natural killer cell activities. Surprisingly, all immunodeficient mice rejected CT26-FasL but not CT26-neo cells (Fig. 3C). Histologic analysis revealed a response that differed from FasL gene transfer into Renca cells. Destruction of tumor cells was observed, but it was associated with a significant accumulation of inflammatory cells consisting of neutrophils and monocytes around CT26-FasL cells in BALB/c mice within 48 h after inoculation, compared with controls (Fig. 4A Upper). Analysis of injected sites in SCID-beige mice also revealed that CT26-FasL tumors contained intense inflammatory infiltrates (Fig. 4A Lower), similar to the response observed in immunocompetent BALB/c mice. The identity of neutrophils in these lesions was confirmed morphologically (Fig. 4A Insets) and by immunostaining with a monoclonal antibody, anti-Ly-6G, that reacts with these cells (Fig. 4B). This result suggested that tumor regression did not require T, B, or natural killer cells responses and was instead mediated by inflammatory cells of mononuclear and granulocytic origin. Interestingly, inflammation was observed in the abdominal muscle layer beneath the injection point in ADV-FasL-treated mice but not in ADV-ΔE1 treated ones. No pathological changes or toxicities were observed elsewhere, for example, in the histologic sections of liver (Fig. 2B Lower) or other major organs (heart, lung, kidney, spleen, and thymus). This finding is in contrast to those found after intravenous injection of anti-Fas antibody, which has been reported to cause fulminant hepatitis (23). It is likely that ADV-FasL would exhibit similar toxicity if administrated systemically, but direct injection into the subcutaneous tumor mass appears to be localized and does not give rise to such toxicity in this model.
Figure 4.
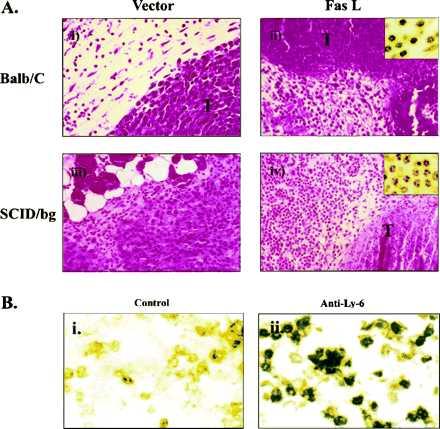
Histological examination of the CT26-neo and CT26-FasL cells inoculated into BALB/c mice by hematoxylin/eosin staining. (A) (i) CT26-neo cells in BALB/c mice are shown 2 days after inoculation. The tumor cells (T) were growing intact. (ii) In CT26-FasL cells inoculated in BALB/c mouse, tumor cells were largely nonviable and an inflammatory cell infiltrate (Inset) was observed. (iii) CT26-neo cells in SCID-beige mouse. (iv) CT26-FasL cells in SCID-beige mouse. Infiltration of neutrophils (Inset) around the destroyed cancer cells was observed. (×400; Inset, ×1000.) (B) Immunostaining of inflammatory cells in a CT26-FasL tumor. The CT26-FasL tumor was dissected on day 2 and stained with an isotype control IgG (i) or anti-Ly-6G antibody (RB6–8C5) specific for neutrophils and activated monocytes/macrophages (ii) (18). A large percentage of the infiltrating cells in the CT26-FasL tumor were Ly-6G positive. (×400.)
These results suggested that FasL gene transfer can exert antitumor effects through two different mechanisms, either by induction of apoptosis through Fas–FasL engagement in Fas+ tumors or through its ability to induce inflammation that is independent of Fas signaling to Fas− tumor cells. To examine the expression of Fas and FasL in human cancer, a panel of malignant cell lines were examined for Fas expression and susceptibility to FasL (Fig. 5A). FACS analysis revealed that HepG2, HeLa, three glioblastoma cell lines, and three of six melanoma cell lines expressed Fas antigen. All of these Fas+ cell lines, with the exception of one glioblastoma (Fig. 5A, G373), were susceptible to lysis by FasL. In contrast to recent descriptions of tumor tissue (11–14), none of the human melanomas expressed FasL (Fig. 5B) both by FACS analysis and a Fas-dependent 51Cr lysis assay using Jurkat cells as target. The detection of Fas and susceptibility to FasL stimulation in several independent human tumor cell lines, together with its effect in Fas+ and Fas− tumors, suggests that FasL gene transfer may be applicable to a variety of human tumors.
Figure 5.
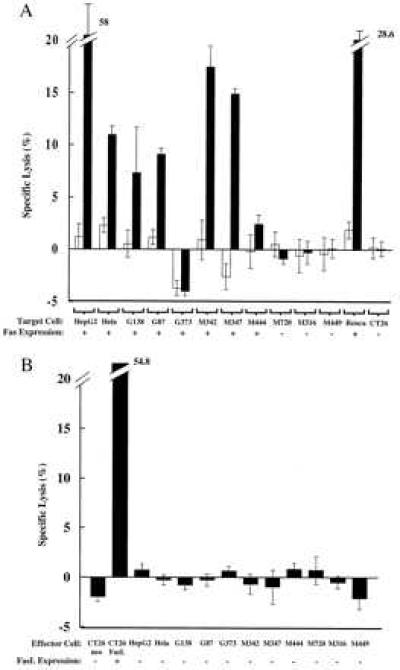
Expression of Fas and susceptibility to FasL in human malignancies. (A) HepG2 cells (HepG2), Hela cells (Hela), three glioma cell lines (G87, G138, and G373), and six melanoma cell lines (M316, M342, M347, M444, M449, and M720) established from human malignancies were labeled with 51Cr and coincubated with CT26-neo (open bars) or CT26-FasL (solid bars) at an effector/target ratio of 5 for 4 h, as described (7), and data represent mean ± SD from three assays. Expression of Fas in each cell line was examined by FACS analysis using anti-human Fas antibody (PharMingen) or isotype-control mouse IgG followed by FITC-conjugated anti-mouse IgG second antibody (PharMingen). For comparison, similar analysis with Renca cells and CT26 cells is presented (at the right). (B) Expression of FasL and cytotoxicity to Jurkat cells in human malignancies. A lysis assay was performed using 51Cr-labeled Jurkat target cells, which are susceptible to FasL. The indicated tumor cells were cocultivated at an effector/target ratio of 5 for 4 h. FACS analysis was performed with a Fas-Fc fusion protein and FITC-conjugated anti-mouse IgGFc second antibody (Jackson Immunoresearch).
DISCUSSION
The role of FasL in tumor growth and immune tolerance is more complex than previously suspected. In this study, we find that FasL expression on the surface of tumor cells promotes tumor regression through apoptosis or inflammation rather than enhanced tumor growth through its effects on immune suppression (7, 11–14). The mechanism of antitumor effect was dependent on Fas expression of the tumor cell. In the case of Fas+ tumors, there appeared to be direct induction of apoptosis. In cells resistant to lysis by FasL, tumor regression was induced through an independent mechanism involving FasL-induced inflammation and involved a potential “bystander effect” in vivo, because we have also observed tumor regression in CT26 cells infected with ADV-FasL in vivo in which infection is not complete.
The mechanism of the proinflammatory effect of FasL remains to be elucidated. Our data using SCID-beige mice suggest that neutrophils, monocytes, or both play a role in this process. The histologic findings suggested that the neutrophil was involved as an effector cell in this process. Monocytes produce a variety of chemokines and cytokines, including interleukin 8, a potent chemoattractant for neutrophils (24), and they have been reported susceptible to FasL stimulation (7, 25, 26). It is therefore likely that FasL expression on tumor cells induced apoptosis of infiltrating monocytes, causing release of these inflammatory mediators that promote further inflammation, neutrophil migration, and tumor destruction.
Neutrophils have been shown to be susceptible to lysis by FasL (27) and are likely directly stimulated by its expression on tumor cells (28). Recently, Seino et al. (28) also reported the inflammatory effect of FasL by using other transformed cell lines that stably express FasL. Though the relation to Fas expression and the feasibility of ADV-mediated gene transfer was not addressed, they suggested that neutrophils mediated the FasL-induced proinflammatory effect because treatment with the anti-Ly-6G antibody inhibited the inflammatory response, consistent with the findings reported herein (28). In a transplantation model, Kang et al. also recently reported inflammation associated with FasL expression in pancreatic β cell transplants (29).
Although inflammation caused by FasL has the potential to affect normal tissue, no significant histological changes were observed in the major organs of treated mice, presumably because FasL expression was localized at the injection site, and the expression levels at these distant organs were lower. Importantly, no abnormalities were observed in the liver, which is susceptible both to ADV infection (30) and to anti-Fas antibody administration (23). Thus, it is likely that this inflammatory response requires a high concentration of FasL expression and is limited as long as vector expression is localized to the site of injection.
We have recently shown that expression of Fas ligand in malignant cells in an allogeneic transplantation model prolongs survival of allogeneic CT26 cells in mice previously challenged with CT26-FasL (7). Though delayed, allogeneic tumors were rejected at later times. Infiltration of inflammatory cells was noted in the study, but this response was also observed in vector-transfected control cells and could not be attributed to FasL expression rather than allogeneic transplant rejection. It is important to note that the previous study (7) differs significantly from this report. The mechanism of allogeneic cell rejection and the effects of Fas ligand expression differ substantially from those which affect syngeneic tumor growth. Allogeneic rejection is mediated largely by T cells, and this T cell response, in addition to the unexpected finding of abrogation of the alloantibody response, was markedly diminished by FasL expression (7). In syngeneic tumor responses, there is no allogeneic response that induces profound inflammation, and the effect of FasL as a proinflammatory gene product could, therefore, be discerned. Thus, the mechanisms of rejection are quite different in these models, and the present study establishes the potential of FasL gene transfer to enhance syngeneic tumor rejection.
Previous reports have suggested that FasL induces immune suppression (7) in testis (9), eye (8), transplantation (10), and malignancy (12), but we find that the effects of FasL are not simply immunosuppressive, and it may potentially induce proinflammatory responses that lead to substantial antitumor responses. Possibly, it could be more effective if used in combination with prior treatment by any number of immune stimulatory genes or proteins (e.g., allogeneic major histocompatibility complex proteins, superantigens, cytokines, or lymphokines) that might facilitate recognition and elimination of minimal residual disease by the immune system that may not otherwise be eliminated by FasL gene transfer. These findings also suggest that it is doubtful that FasL expression on malignant cells is responsible for decreased antitumor responses, as implied in recent studies where FasL has been described in tumors (11–14). Our analysis of FasL expression in melanomas showed no expression by FACS analysis or by its ability to induce Fas-mediated cell lysis (Fig. 5B). In vivo studies have also not convincingly demonstrated FasL expression on the surface membranes of most melanomas (12), for example. Although it is conceivable that melanoma cells can be induced to express FasL under special conditions achieved in vivo, it is more likely that proteases released in tumors cause release of soluble FasL from lymphoid and mononuclear cells that can be found in tumor tissue and serum of patients with advanced malignancy. Extracellular matrix-associated soluble FasL may exert different, presumably immunosuppressive, effects that may not be generally proinflammatory in vivo. Because the effects of FasL on the immune response are local and transient (7), the immunosuppressive effect of FasL is unlikely to represent an impediment to its development for gene transfer studies in humans. The ability to generate such potent inflammatory and apoptotic antitumor responses suggests that gene transfer of FasL may compensate for locally suppressive immune effects on tumor recognition and provide a useful molecular genetic intervention for malignancy.
Acknowledgments
We thank Ms. Donna Gschwend and Ms. Nancy Barrett for manuscript preparation, Ling Ling Xu for preparation of histological specimens, and other members of the Nabel laboratory for their helpful advice and comments. This work was supported in part by grants from the National Institutes of Health (NIH RO1 AI36606 and POI CA59327, G.J.N.).
ABBREVIATIONS
- ADV
adenoviral vector
- FasL
Fas ligand
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP-digoxigenin nick end labeling
- FITC
fluorescein isothiocyanate
- SCID
severe combined immunodeficiency
- FACS
fluorescence activated cell sorter
References
- 1.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 2.Suda T, Takahashi T, Golstein P, Nagata S. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 3.Nagata S, Golstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 4.Brunner T, Mogil R J, LaFace D, Yoo N J, Mahboubi A, Echeverri F, Martin S J, Force W R, Lynch D H, Ware C F, Green D R. Nature (London) 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 5.Dhein J, Walczak H, Baumler C, Debatin K, Krammer P H. Nature (London) 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 6.Ju S, Panka D J, Cui H, Ettinger R, El-Khatib M, Sherr D H, Stanger B Z, Marshak-Rothstein A. Nature (London) 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 7.Arai H, Chan S Y, Bishop D K, Nabel G J. Nat Med. 1997;3:843–848. doi: 10.1038/nm0897-843. [DOI] [PubMed] [Google Scholar]
- 8.Griffith T, Brunner T, Fletcher S, Green D, Ferguson T. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 9.Bellgrau D, Gold D, Selawry H, Moore J, Franzusoff A, Duke R. Nature (London) 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- 10.Lau H T, Yu M, Fontana A, Stoeckert C J., Jr Science. 1996;273:109–112. doi: 10.1126/science.273.5271.109. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, Hatake K, Drummond A H, Nagata S. Nat Med. 1996;2:317–322. doi: 10.1038/nm0396-317. [DOI] [PubMed] [Google Scholar]
- 12.Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French L E, Schneider P, Bornand T, Fontana A, Lienard D, Cerottini J C, Tschopp J. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 13.Strand S, Hofmann W J, Hug H, Muller M, Otto G, Strand D, Mariani S M, Stremmel W, Krammer P H, Galle P R. Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 14.O’Connell J, O’Sullivan G C, Collins J K, Shanahan F. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fearon E R, Pardoll D M, Itaya T, Golumbek P, Levitsky H I, Simons J W, Karasuyama H, Vogelstein B, Frost P. Cell. 1990;60:397–403. doi: 10.1016/0092-8674(90)90591-2. [DOI] [PubMed] [Google Scholar]
- 16.Chinnaiyan A M, Woffendin C, Dixit V M, Nabel G J. Nat Med. 1997;3:333–337. doi: 10.1038/nm0397-333. [DOI] [PubMed] [Google Scholar]
- 17.Ohno T, Gordon D, San H, Pompili V J, Imperiale M J, Nabel G J, Nabel E G. Science. 1994;265:781–784. doi: 10.1126/science.8047883. [DOI] [PubMed] [Google Scholar]
- 18.Jutila M A, Kroese F G M, Jutila K L, Stall A M, Fiering S, Herzenberg L A, Berg E L, Butcher E C. Eur J Immunol. 1988;18:1819–1826. doi: 10.1002/eji.1830181125. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Perkins N D, Ohno T, Nabel E G, Nabel G J. Nat Med. 1995;1:1052–1056. doi: 10.1038/nm1095-1052. [DOI] [PubMed] [Google Scholar]
- 20.Giovanella B C, Fogh J. Adv Cancer Res. 1985;44:69–120. doi: 10.1016/s0065-230x(08)60026-3. [DOI] [PubMed] [Google Scholar]
- 21.Bosma G C, Custer R P, Bosma M J. Nature (London) 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 22.Roder J, Duwe A. Nature (London) 1979;278:451–453. doi: 10.1038/278451a0. [DOI] [PubMed] [Google Scholar]
- 23.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Nature (London) 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 24.Baggiolini M, Dewald B, Waltz A. Interleukin 8 and Related Chemotactic Cytokines. New York: Raven; 1992. pp. 247–263. [Google Scholar]
- 25.Richardson B C, Lalwani N D, Johnson K J, Marks R M. Eur J Immunol. 1994;24:2640–2645. doi: 10.1002/eji.1830241111. [DOI] [PubMed] [Google Scholar]
- 26.Ashany D, Song X, Lacy E, Nikolic-Zugic J, Friedman S M, Elkon K B. Proc Natl Acad Sci USA. 1995;92:11225–11229. doi: 10.1073/pnas.92.24.11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liles W C, Kiener P A, Ledbetter J A, Aruffo A, Klebanoff S J. J Exp Med. 1996;184:429–440. doi: 10.1084/jem.184.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seino K, Kayagaki N, Okumura K, Yagita H. Nat Med. 1997;3:165–170. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 29.Kang S M, Schneider D B, Lin Z, Hanahan D, Dichek D A, Stock P G, Baekkeskov S. Nat Med. 1997;3:738–743. doi: 10.1038/nm0797-738. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Wilson J M. J Immunol. 1995;155:2564–2570. [PubMed] [Google Scholar]