

Abstract
Background:
Bevacizumab has recently been approved by the US Food and Drug Administration for recurrent glioblastoma (GBM). However, patterns of relapse, prognosis, and outcome of further therapy after bevacizumab failure have not been studied systematically.
Methods:
We identified patients at Memorial Sloan-Kettering Cancer Center with recurrent GBM who discontinued bevacizumab because of progressive disease.
Results:
There were 37 patients (26 men with a median age of 54 years). The most common therapies administered concurrently with bevacizumab were irinotecan (43%) and hypofractionated reirradiation (38%). The median overall survival (OS) after progressive disease on bevacizumab was 4.5 months; 34 patients died. At the time bevacizumab was discontinued for tumor progression, 17 patients (46%) had an increase in the size of enhancement at the initial site of disease (local recurrence), 6 (16%) had a new enhancing lesion outside of the initial site of disease (multifocal), and 13 (35%) had progression of predominantly nonenhancing tumor. Factors associated with shorter OS after discontinuing bevacizumab were lower performance status and nonenhancing pattern of recurrence. Additional salvage chemotherapy after bevacizumab failure was given to 19 patients. The median progression-free survival (PFS) among these 19 patients was 2 months, the median OS was 5.2 months, and the 6-month PFS rate was 0%.
Conclusions:
Contrast enhanced MRI does not adequately assess disease status during bevacizumab therapy for recurrent glioblastoma (GBM). A nonenhancing tumor pattern of progression is common after treatment with bevacizumab for GBM and is correlated with worse survival. Treatments after bevacizumab failure provide only transient tumor control.
GLOSSARY
- CA9
= carbonic anhydrase 9;
- CI
= confidence interval;
- FDG
= [18F]fluorodeoxyglucose;
- FLAIR
= fluid-attenuation inversion recovery;
- GBM
= glioblastoma;
- HIF-1
α = hypoxia-inducible factor 1α;
- KPS
= Karnofsky performance status;
- MR
= magnetic resonance;
- OS
= overall survival;
- PFS
= progression-free survival;
- TMZ
= temozolomide;
- VEGF
= vascular endothelial growth factor;
- VEGFR
= vascular endothelial growth factor receptor.
Glioblastoma (GBM) is the most common primary brain tumor in adults; despite standard therapy with surgery, radiation therapy, and chemotherapy with temozolomide, the median survival for GBM is only 15 months.1 Essentially all patients develop recurrent or progressive disease after initial therapy, after which the median survival is approximately 6 months.2,3 GBM is a highly vascular tumor with increased expression of vascular endothelial growth factor (VEGF),4,5 a protein produced by tumor and stromal cells. VEGF binds to the family of VEGF receptors (VEGFRs) and promotes endothelial cell proliferation and migration and, consequently, tumor angiogenesis.6
Bevacizumab is a humanized monoclonal antibody that inhibits VEGF. It is approved in combination with chemotherapy for several extra-CNS cancers. Results from phase 2 clinical trials with bevacizumab for recurrent or progressive GBM have been promising,7–9 leading to US Food and Drug Administration approval. However, most patients develop progressive disease within the first year of treatment. It remains unclear how patients should be treated after disease progression on bevacizumab. Rapid clinical deterioration has been observed after discontinuing bevacizumab, presumably a consequence of withdrawing its antivasogenic edema properties.10 Consequently, some physicians continue bevacizumab but either add or change concurrently administered cytotoxic chemotherapy. However, this approach is typically ineffective.11,12 Our practice has involved discontinuing bevacizumab in favor of a different therapy. Therefore, we sought to evaluate the patterns of relapse and outcome after tumor progression on bevacizumab, as well as the effect of subsequent treatment.
METHODS
This retrospective study was approved by the institutional review board at Memorial Sloan-Kettering Cancer Center and included patients with histologically confirmed GBM who discontinued bevacizumab because of tumor progression from October 2006 to January 2009. Patients who discontinued bevacizumab because of toxicity or reasons other than tumor progression were excluded. All brain imaging studies at baseline and after starting bevacizumab were reviewed by 2 investigators (F.M.I. and A.B.L.). Response was assessed through Macdonald criteria, which are the current standard for radiographic assessments of brain tumors. They define progressive disease as either a 25% or more increase of contrast enhancing cross sectional area on sequential MRI scans or otherwise unexplained clinical deterioration.13 However, Macdonald criteria do not account for increased size of nonenhancing abnormality. Therefore, patients with nonenhancing tumor growth were considered to have progressive disease on bevacizumab only at the time of clinical worsening or if histology proved disease progression.
Overall survival (OS) after discontinuation of bevacizumab was calculated from the date of the tumor progression on bevacizumab until death from any cause or last contact. OS after salvage chemotherapy was calculated from the first dose of salvage chemotherapy to death from any cause or last contact. Progression-free survival (PFS) after salvage chemotherapy was calculated from the date of first dose of salvage chemotherapy until tumor progression, death from any cause, or last contact. Multivariate analyses were performed with a Cox proportional hazards regression model to identify variables that were independently predictive of OS after tumor progression on bevacizumab. Patients were grouped according to Karnofsky performance status (KPS; ≥70 and <70) because this cutoff defines the degree of independence for daily activities. Factors with p < 0.2 on univariate analyses were entered as candidate variables and multivariate analysis was performed. Calculations were performed using STATA version 10.0 (copyright 1985–2007; Stata Corporation, College Station, TX). Clinical data were updated as of February 25, 2009.
Three patients had paired tumor tissue available for analysis before bevacizumab treatment and after tumor progression on bevacizumab. Immunostains were performed in 2 of these cases from formalin-fixed paraffin-embedded tumor tissue for VEGFR2 (KDR), hypoxia-inducible factor 1α (HIF-1α), and carbonic anhydrase 9 (CA9).
RESULTS
Bevacizumab-based therapy.
Thirty-seven patients (26 men, 11 women) with a median age of 54 years discontinued bevacizumab because of tumor progression. All patients had received and failed standard treatment for GBM with radiation therapy and temozolomide before receiving bevacizumab. No patient received bevacizumab as part of the “up-front” treatment; 32 patients (86%) received a bevacizumab-based therapy at first recurrence, whereas the other 5 patients (14%) were treated with bevacizumab at the second recurrence. The most common therapies administered concurrently with bevacizumab were irinotecan (43%) and hypofractionated reirradiation (38%) (table 1). Concurrent hypofractionated radiation was part of a pilot study reported separately.14 At the time bevacizumab was discontinued, the pattern of progression was predominantly nonenhancing tumor in 13 patients (35%), new multifocal enhancement in 6 patients (16%), and progressive enhancement at the initial disease site (local recurrence) in 17 patients (46%); 1 patient was considered to have tumor progression because of clinical deterioration without imaging confirmation.
Table 1 Patient characteristics and responses to bevacizumab (n = 37)
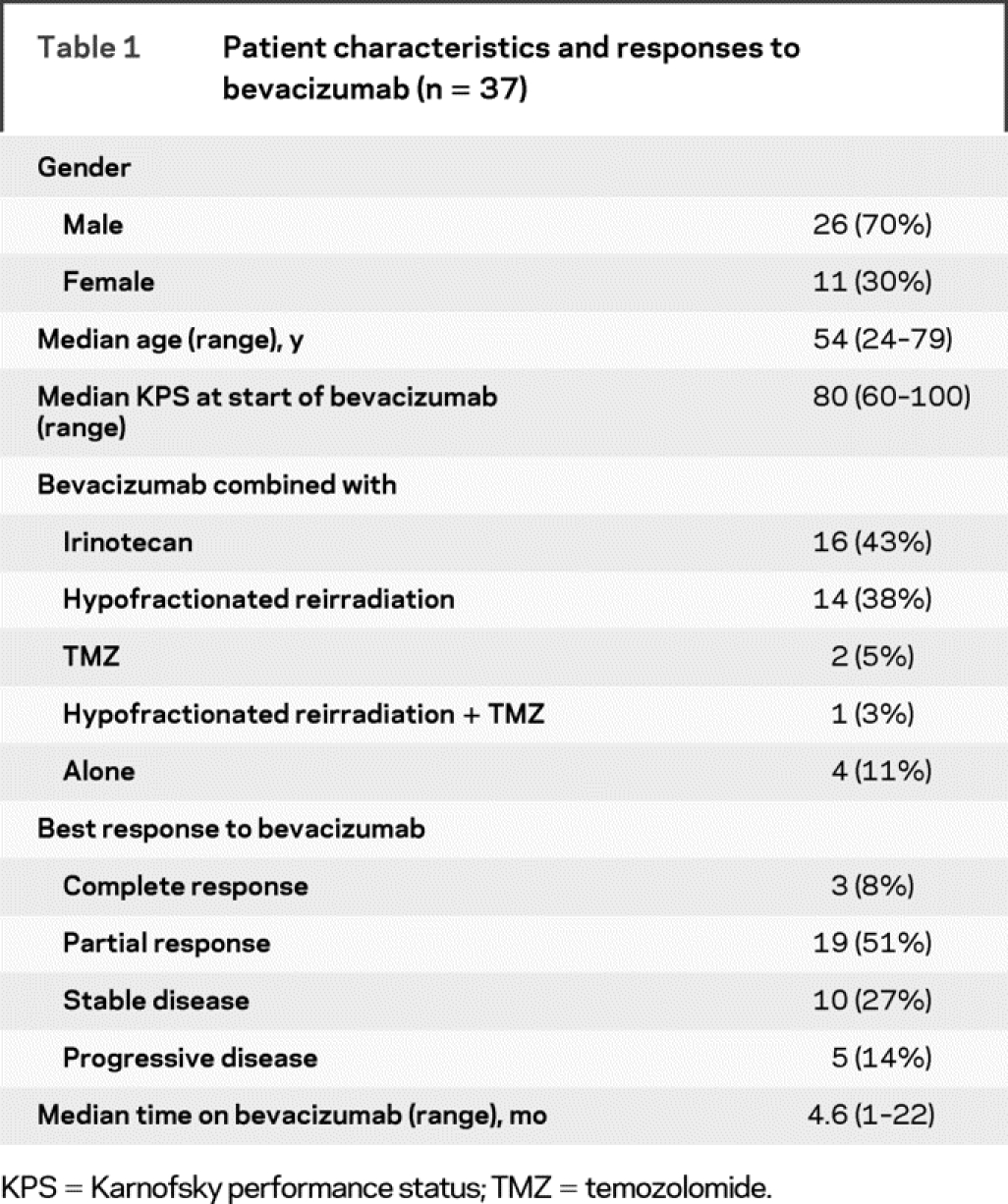
In 9 of 13 patients who had nonenhancing tumor progression, radiographic tumor progression coincided with clinical worsening and bevacizumab was discontinued at that time. Increased nonenhancing tumor was evident in 4 patients before they were classified as having progressive disease; these 4 patients continued on bevacizumab for a median of 6.9 weeks (range 4.3–14 weeks) until they developed clinical progression, and only then was bevacizumab discontinued. This reflected the ambiguity associated with interpreting MRI scans during bevacizumab. For example, in 1 patient, a progressive nonenhancing abnormality developed around the initial site of disease that did not demonstrate enhancement, increased blood flow on MR perfusion imaging, or hypermetabolism on [18F]fluorodeoxyglucose (FDG)-PET imaging (figure 1). The patient initiated corticosteroids because of the possibility that these radiographic findings represented increased edema, but without clinical benefit. Resection demonstrated highly aggressive and invasive sarcomatous disease (figure 2). Another patient with nonenhancing tumor and 2 patients with enhancing tumor progression had resection after bevacizumab failure; pathologic specimens were compatible with recurrent GBM in all patients.

Figure 1 Neuroimaging before and after bevacizumab failure
T1-weighted contrast (gadolinium)–enhanced (A and D), fluid-attenuation inversion recovery (FLAIR; B and E) magnetic resonance (MR) and [18F]fluorodeoxyglucose (FDG)-PET (C and F) before (A–C) and 3 weeks after (D–F) the last bevacizumab infusion, demonstrating increased FLAIR abnormality (E, arrow showing tumor crossing corpus callosum) without concurrent contrast enhancement of histologically proven recurrent disease (figure 2). Prebevacizumab FDG-PET demonstrated hypermetabolism (C, arrow) in the area of gadolinium enhancement that became substantially hypometabolic during bevacizumab therapy at the time of recurrence (F, arrow), which was a false negative. MR perfusion imaging (not shown) also demonstrated reduced blood flow at the type of recurrence, also a false negative. Resection demonstrated extremely high-grade tumor (figure 2, E–H).

Figure 2 Histopathology before and after bevacizumab failure
Tumor tissue at recurrence before (A–D) and after (E–H) treatment with bevacizumab for patient shown in figure 1. (A) Persistent glioma with treatment effect (right) and necrosis (left) in a glioblastoma after radiation therapy and temozolomide but before bevacizumab (hematoxylin & eosin, ×25). (B) Persistent tumor is devoid of nuclear hypoxia-inducible factor 1α (HIF-1α) expression. (C) Vascular endothelial growth factor receptor 2 (VEGFR2) expression is confined to endothelial cells in tumor vasculature. (D) Carbonic anhydrase 9 (CA9) is not expressed in persistent tumor. (E) Recurrence resected 3 weeks after last bevacizumab demonstrating sarcomatous, spindle cell morphology with mitotic figures (arrows) and necrosis at right (hematoxylin & eosin, ×40). (F) Region of recurrent tumor demonstrating increased nuclear expression of HIF-1α. (G) VEGFR2 expression remains confined to endothelial cells in tumor vasculature. (H) Up-regulated CA9 expression in recurrent tumor is found neighboring regions of necrosis. (B–D and F–H: immunoperoxidase with hematoxylin counterstain, all ×40.)
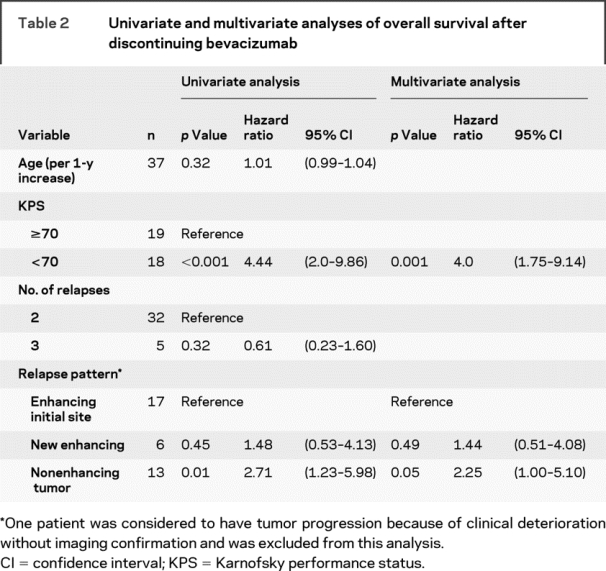
The median OS after tumor progression on bevacizumab among all 37 patients was 4.5 months (95% confidence interval [CI] 2.1–5.9 months). Thirty-four patients died, and the median follow-up of 3 surviving patients was 6 months. Lower KPS and a nonenhancing pattern of recurrence were associated with shorter OS after discontinuing bevacizumab (table 2).
Table 2 Univariate and multivariate analyses of overall survival after discontinuing bevacizumab
Postbevacizumab salvage therapy.
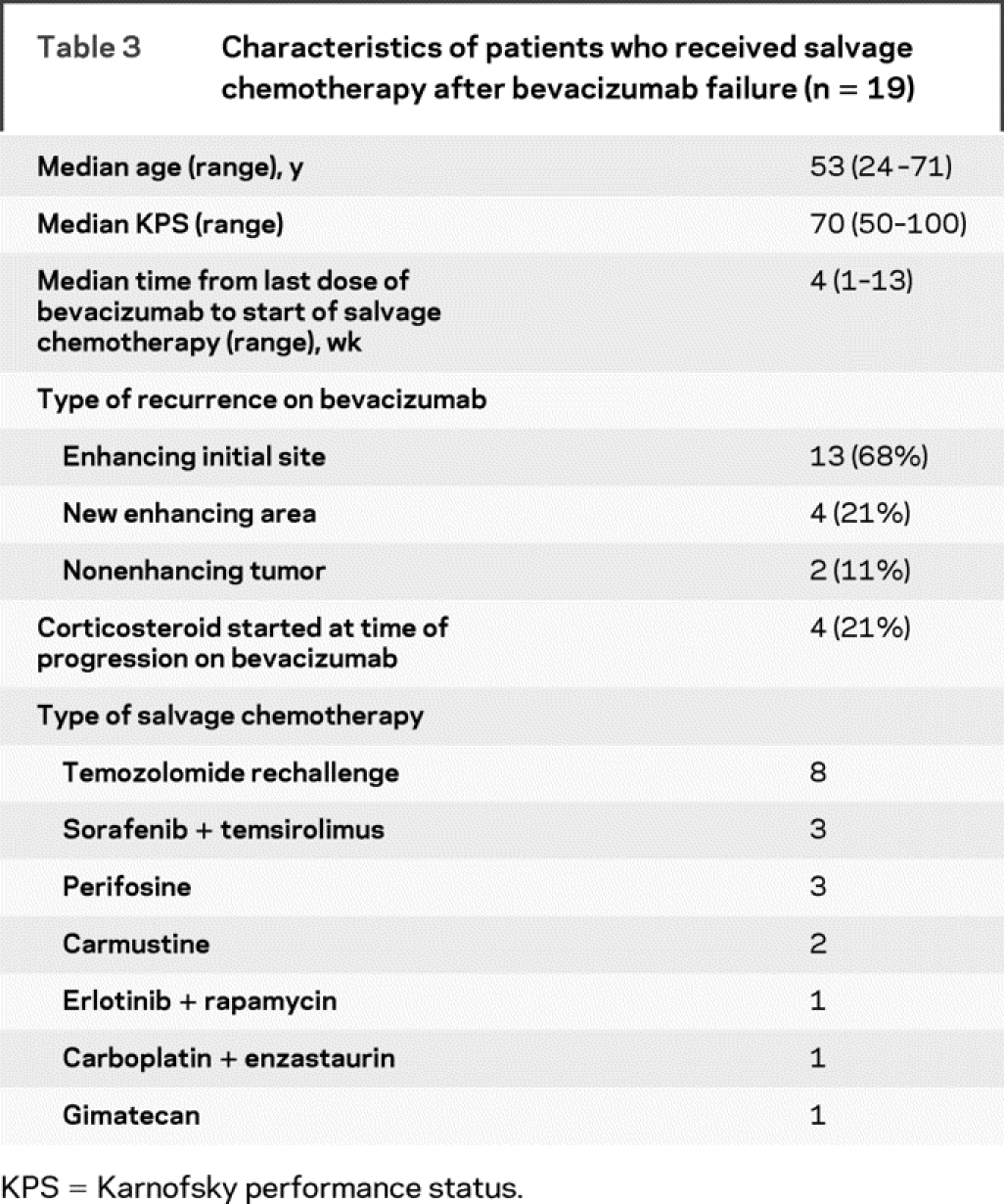
Nineteen patients (including 2 of 4 who underwent surgery after discontinuing bevacizumab) received salvage chemotherapy. The characteristics of the 19 patients who received salvage therapy are shown in table 3. The salvage chemotherapies were heterogeneous. The median time from the last dose of bevacizumab to start of salvage chemotherapy was 4 weeks. After a median follow-up of 13 months, all 19 patients had tumor progression and 16 died. The median PFS for the patients who received salvage chemotherapy was 2 months (95% CI 1.2–3.3 months); the 6-month PFS rate was 0%, with no patients censored for PFS. The median OS was 5.2 months (95% CI 3.3–8.4 months) among these 19 patients.
Table 3 Characteristics of patients who received salvage chemotherapy after bevacizumab failure (n = 19)
In addition, 15 patients received supportive care only after discontinuing bevacizumab; 2 others were lost to follow-up. The median survival of these 17 patients after tumor progression on bevacizumab was 2 months (95% CI 1.3–3.3 months). Patients who did not receive salvage treatment had lower KPS (median KPS of 60 compared with median of 70, K-sample equality of median test with corrected Pearson χ2, p = 0.015) and more frequent nonenhancing pattern of progression (65% vs 11%, Fisher exact test, p = 0.002) compared with patients who received salvage therapy. One patient recently underwent resection of recurrent tumor without starting salvage chemotherapy yet.
Immunohistochemistry.
Sequential tumor samples were analyzed from 2 patients at diagnosis, after initial therapy before bevacizumab, and from resection after bevacizumab. Both patients had a partial response to bevacizumab, lasting 7 and 3 months. These patients underwent surgical resection 4 and 6 weeks after the last dose of bevacizumab. Before bevacizumab, VEGFR2 staining was present but restricted to tumor blood vessels in 1 patient and was negative in the other patient. There was no increase in VEGFR2 expression after bevacizumab therapy. Both patients had marked increase in the hypoxia markers HIF-1α and CA9 at the time of bevacizumab failure compared with the tumor tissue before bevacizumab exposure (figure 2).
DISCUSSION
Increased nonenhancing tumor with decreased or stable enhancing disease is a common pattern of GBM progression on bevacizumab and occurred in 35% of our patients. This pattern differs from that typically observed, in which worsening enhancement at the initial site of disease occurs in 90% to 95% of patients who do not receive bevacizumab.15,16 Others report analogous results.11 In our patients, this pattern was associated with shorter survival, which may be explained by more extensive disease causing lower performance status. It could also reflect a change in tumor biology, which is supported by our finding that nonenhancing tumor progression was a negative prognostic factor independent of performance status. Anti-VEGF therapy can facilitate co-option of normal vasculature and tumor invasion, potentially leading to a more aggressive tumor phenotype.17–19 In addition, nonenhancing progression may delay the determination of failure in the absence of worsening enhancement.13 Clearly, traditional tumor evaluation with MRI and FDG-PET are insufficient to assess fully the response to antiangiogenic therapy (figure 1). Anti-VEGF therapy decreases contrast leakage and produces a rapid decrease in enhancement that does not correlate with decreased tumor size.20 Decrease in contrast enhancement within 24 hours of treatment initiation has been demonstrated with cediranib (AZD2171), a pan-VEGF receptor tyrosine kinase inhibitor,20 and within days of bevacizumab therapy.21 New response criteria are needed and clinical trials using anti-VEGF therapy will need to incorporate imaging techniques that allow evaluation of tumor response independent of the effects of these drugs on restoration of the blood-brain barrier.22
The molecular basis of bevacizumab failure is poorly understood. Multiple mechanisms are likely involved, including activation of other angiogenesis pathways, increase in tumor invasiveness independent of angiogenesis, and increase in the recruitment of vascular progenitor cells from the bone marrow.23,24 In our patients with paired tumor tissue, there was a marked increase of hypoxia markers HIF-1α and CA9 after bevacizumab failure. Hypoxia is a well-known promoter of angiogenesis, tumor survival, invasion, and resistance to therapy.25 Moreover, several patient series have shown that hypoxia marker CA9 is associated with poor survival in gliomas.26–28 Although rapid GBM growth could have worsened tumor hypoxia in our patients, it is also possible that the increase in tumor hypoxia was caused by the antiangiogenic effect of bevacizumab. In fact, an experimental GBM model has shown that genetic or pharmacological inhibition of VEGF promotes tumor hypoxia and a more invasive and aggressive tumor behavior.24 Moreover, hypoxia is known to induce recruitment of bone marrow derived cells that promote an alternative pathway for GBM angiogenesis and growth in animal models.19 A pathologic study of human gliomas that progressed on bevacizumab showed an increased number of bone marrow derived cells in these tumor specimens.29
Salvage chemotherapy after tumor progression on bevacizumab provided only transient tumor control; 19 patients treated with a variety of drug regimens had an estimated 6-month PFS rate of 0%. This is similar to a 6-month PFS rate of 2% from a study in which patients with tumor progression continued bevacizumab but changed the concurrent cytotoxic chemotherapy, mostly from irinotecan to carboplatin,11,12 and data demonstrating that initiating irinotecan after disease progression on bevacizumab monotherapy are ineffective.7 Therefore, it may be reasonable to reserve bevacizumab for treatment of later GBM recurrences because effective postbevacizumab therapies are lacking. This is especially true for patients without significant enhancing tumor or vasogenic edema and younger patients with good performance status.7,30
It is notable that the median OS of patients who received salvage treatment was 5.2 months, which is not shorter than that of historic controls treated in clinical trials that were deemed ineffective for recurrent GBM.2,3 This may suggest that selected patients with good performance status after tumor progression on bevacizumab may be appropriate clinical trial candidates despite our null 6-month PFS rate for the therapy immediately after bevacizumab. However, our results and those of others11,12 suggest that bevacizumab may negatively bias 6-month PFS for the next salvage therapy. It is possible that PFS is not an appropriate surrogate for OS after bevacizumab failure. Alternatively, bevacizumab withdrawal itself may increase tumor enhancement that may be interpreted as tumor growth, and experimental data have suggested that anti-VEGF therapy withdrawal can lead to rapid tumor vasculature regrowth.31 Possible solutions to avoid this negative bias include mandating a washout period from prior bevacizumab therapy, which has a half-life of 3 weeks, for inclusion in a clinical trial of drugs that do not directly target VEGF/VEGFR signaling. This is similar to the 5 days of stable or decreasing corticosteroid dose typically required because of the potentially confounding effects of corticosteroids on contrast-enhanced brain imaging before initiating a clinical trial. Alternatively, stratification for prior bevacizumab therapy could be required in future clinical trials for recurrent GBM.
This study carries the inherent limitations of any retrospective study. Moreover, criteria for discontinuing bevacizumab were not standardized among treating physicians, and salvage treatments varied. However, this study adds information on the limitation of the current response criteria for GBM that analyzes only contrast-enhancing tumor and clinical status and the outcome after tumor progression on bevacizumab. Finally, paired tumor tissue and radiographic analyses suggest that bevacizumab therapy may change the GBM biology, but further studies are required.
AUTHOR CONTRIBUTIONS
Statistical analyses were conducted by F.M. Iwamoto and A.B. Lassman.
ACKNOWLEDGMENT
The authors thank Judy Lampron for her expert editorial assistance.
DISCLOSURE
Dr. Iwamoto and Dr. Beal report no disclosures. Dr. Abrey receives royalties from publishing Fast Facts, Brain Tumor (Healthcare Press Inc., 2001); honoraria from Genentech, Inc. and Physicians’ Education Resource; and research support from Genentech, Inc., Schering-Plough Corp., AstraZeneca, Novartis, Pfizer Inc., and the Brain Tumor Society (Co-PI). Dr. Gutin serves on a scientific advisory board for and receives research support from Genentech, Inc.; and serves as a Principal Editor for Neurosurgery. Dr. Rosenblum serves on the editorial boards of Acta Neuropathologica, the American Journal of Surgical Pathology, Brain Pathology, the Journal of Neuropathology & Experimental Neurology; and receives research support as an ACNS Reviewer from Consortium Agreement with National Childhood Foundation, Children’s Oncology Group [COG ACNS0122, COG ACNS0232, and COG ACNS0621]. Dr. Reuter serves on the editorial boards of the American Journal of Surgical Pathology, Modern Pathology, the Journal of Urologic Pathology, Urologic Oncology, Advances in Anatomic Pathology, Molecular Urology, Prostate Cancer and Prostate Diseases, Pathology, Annals of Diagnostic Pathology, the International Journal of Clinical and Experimental Pathology, Kidney Cancer Journal, the Journal of Clinical Oncology, and the Journal of Urology; receives royalties from publishing Sternberg’s Diagnostic Surgical Pathology (Lippincott Wilkins & Williams, 2004); has received non–industry-sponsored speaker honoraria; and receives research support from NIH/NCI [2 P50 CA92629-06 (Codirector), 2 P30 CA008748-27(Codirector of Pathology Core), 1N01 CM-62206 (Coinvestigator), 5 R01 CA121327-02 (Coinvestigator), and 5 R01 CA076423-09 (Coinvestigator)]. Dr. DeAngelis serves on the editorial board of Neurology; served on a scientific advisory board for Genentech, Inc.; receives royalties from publishing The Neurologic Complications of Cancer (Oxford University Press, 2008); and has received research support from the NIH [UO1 CA-105663-01 (Participating Member in the NABTC)]. Dr. Lassman serves on the editorial board of the Journal of Neuro-Oncology; served on scientific advisory boards for Schering-Plough, Inc., Sigma-Tau Pharmaceuticals, Inc., Bristol-Myers Squibb, ImClone Systems, Genentech, Inc. and Eisai, Inc.; served as consultant for Enzon Pharmaceuticals, Inc., the National Comprehensive Cancer Network, Genentech, Inc., Cephalon, Inc., IDBM Consulting, Ltd., Leerink Swan, LLC, and Gerson Lehrman Group; served on the speakers’ bureau of Schering-Plough; receives honoraria from Physicians’ Education Resource, the Robert Michael Educational Institute, and Medical Communications Media; and receives research support from Keryx Biopharmaceuticals (PI), Sigma-Tau Pharmaceuticals (PI), the Radiation Therapy Oncology Group (Investigator), and the National Brain Tumor Foundation (PI).
Address correspondence and reprint requests to Dr. Andrew B. Lassman, Department of Neurology, Memorial Sloan-Kettering Cancer Center, 1275 York Ave., New York, NY 10065 lassmana@mskcc.org
Disclosure: Author disclosures are provided at the end of the article.
Presented in part at the American Society of Clinical Oncology Annual Meeting, May 30–June 3, 2008, Chicago, IL.
Received March 13, 2009. Accepted in final form July 7, 2009.
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–996. [DOI] [PubMed] [Google Scholar]
- 2.Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 1999;17:2572–2578. [DOI] [PubMed] [Google Scholar]
- 3.Lamborn KR, Yung WK, Chang SM, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol 2008;10:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamszus K, Ulbricht U, Matschke J, Brockmann MA, Fillbrandt R, Westphal M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin Cancer Res 2003;9:1399–1405. [PubMed] [Google Scholar]
- 5.Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992;359:845–848. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003;9:669–676. [DOI] [PubMed] [Google Scholar]
- 7.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 2009;27:740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vredenburgh JJ, Desjardins A, Herndon JE II, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 2007;13:1253–1259. [DOI] [PubMed] [Google Scholar]
- 9.Vredenburgh JJ, Desjardins A, Herndon JE II, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 2007;25:4722–4729. [DOI] [PubMed] [Google Scholar]
- 10.Ananthnarayan S, Bahng J, Roring J, et al. Time course of imaging changes of GBM during extended bevacizumab treatment. J Neurooncol 2008;88:339–347. [DOI] [PubMed] [Google Scholar]
- 11.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 2008;70:779–787. [DOI] [PubMed] [Google Scholar]
- 12.Quant EC, Norden AD, Drappatz J, et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol Epub 2009 Mar 30. [DOI] [PMC free article] [PubMed]
- 13.Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 1990;8:1277–1280. [DOI] [PubMed] [Google Scholar]
- 14.Gutin PH, Iwamoto FM, Beal K, et al. Safety and efficacy of bevacizumab with hypofractionated stereotactic irradiation for recurrent malignant gliomas. Int J Radiat Oncol Biol Phys 2009;75:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaspar LE, Fisher BJ, Macdonald DR, et al. Supratentorial malignant glioma: patterns of recurrence and implications for external beam local treatment. Int J Radiat Oncol Biol Phys 1992;24:55–57. [DOI] [PubMed] [Google Scholar]
- 16.Hochberg FH, Pruitt A. Assumptions in the radiotherapy of glioblastoma. Neurology 1980;30:907–911. [DOI] [PubMed] [Google Scholar]
- 17.Kunkel P, Ulbricht U, Bohlen P, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res 2001;61:6624–6628. [PubMed] [Google Scholar]
- 18.Rubenstein JL, Kim J, Ozawa T, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000;2:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du R, Lu KV, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008;13:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007;11:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pope WB, Lai A, Nghiemphu P, Mischel P, Cloughesy TF. MRI in patients with high-grade gliomas treated with bevacizumab and chemotherapy. Neurology 2006;66:1258–1260. [DOI] [PubMed] [Google Scholar]
- 22.Sorensen AG, Batchelor TT, Wen PY, Zhang WT, Jain RK. Response criteria for glioma. Nat Clin Pract Oncol 2008;5:634–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15:220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA 1997;94:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haapasalo JA, Nordfors KM, Hilvo M, et al. Expression of carbonic anhydrase IX in astrocytic tumors predicts poor prognosis. Clin Cancer Res 2006;12:473–477. [DOI] [PubMed] [Google Scholar]
- 27.Korkolopoulou P, Perdiki M, Thymara I, et al. Expression of hypoxia-related tissue factors in astrocytic gliomas: a multivariate survival study with emphasis upon carbonic anhydrase IX. Hum Pathol 2007;38:629–638. [DOI] [PubMed] [Google Scholar]
- 28.Sathornsumetee S, Cao Y, Marcello JE, et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol 2008;26:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer I, Cunliffe CH, Bollo RJ, et al. High-grade glioma before and after treatment with radiation and Avastin: initial observations. Neuro Oncol 2008;10:700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nghiemphu PL, Liu W, Lee Y, et al. Bevacizumab and chemotherapy for recurrent glioblastoma: a single-institution experience. Neurology 2009;72:1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mancuso MR, Davis R, Norberg SM, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 2006;116:2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]